
Abstract
Precise breakpoint mapping of balanced chromosomal rearrangements is crucial to identify disease etiology. Ten female patients with X-autosome balanced translocations associated with phenotypic alterations were evaluated, by mapping and sequencing their breakpoints. The rearrangements’ impact on the expression of disrupted genes, and inferred mechanisms of formation in each case were assessed. For four patients that presented one of the chromosomal breaks in heterochromatic and highly repetitive segments, we combined cytogenomic methods and short-read sequencing to characterize, at nucleotide resolution, breakpoints that occurred in reference genome gaps. Most of rearrangements were possibly formed by non-homologous end joining and have breakpoints at repeat elements. Seven genes were found to be disrupted in six patients. Six of the affected genes showed altered expression, and the functional impairment of three of them were considered pathogenic. One gene disruption was considered potentially pathogenic, and three had uncertain clinical significance. Four patients presented no gene disruptions, suggesting other pathogenic mechanisms. Four genes were considered potentially affected by position effect and the expression abrogation of one of them was confirmed. This study emphasizes the importance of breakpoint-junction characterization at nucleotide resolution in balanced rearrangements to reveal genetic mechanisms associated with the patients’ phenotypes, mechanisms of formation that originated the rearrangements, and genomic nature of disrupted DNA sequences.
Similar content being viewed by others

Introduction
Balanced chromosome abnormalities (BCAs) are known to be a class of structural variations that changes the orientation and/or localization of a chromosome segment without gain or loss of genomic material. Although most carriers are phenotypically normal, around 6% of them are thought to have an associated disease phenotype [1], commonly presenting with multiple congenital abnormalities and/or intellectual disability [2]. The precise breakpoint mapping is crucial for genotype-phenotype correlations, annotation of disease associated genes [3] and can provide information about mutational signatures at junction points [4]. In females with X-autosome balanced translocation there is a preferential inactivation of the normal X-chromosome. Thus, gene disruption in the rearranged X chromosome may cause complete absence of functional copies of this gene [5]. In this study, we evaluated ten female patients with balanced X-autosome translocations and abnormal phenotype. We sequenced their junction points at base-pair resolution and inferred the mechanisms of formation in each case. We demonstrated that the nucleotide resolution characterization of breakpoints in BCAs with one of the breakpoints at heterochromatic and highly repetitive regions (e.g., centromeres or short arms of acrocentric chromosomes) is feasible by combining cytogenomic methods and short-read sequencing.
Materials and methods
Enrollment
We studied ten female patients with reciprocal X-autosome balanced translocations and eight female controls with age ranging from 7 to 51 years were selected for gene expression investigation. All samples used in this study were collected after written appropriate informed consent and approval of the local ethics committee (CONEP 36019314.9.0000.5505, CEP 0028/2015). Of note, clinical and cytogenomic evaluation for patients 1, 4, 5, 6, 7, 8, and 10 had previously been reported [6,7,8,9]. However, most rearrangements had not been mapped to sequence resolution, except for patients 6 and 8 who were previously described in clinical studies [8, 9]. The clinical description of all patients, the molecular characterization of their rearrangements and their expression profiles are described in detail in Supplementary Case Reports.
Banding, molecular cytogenetic evaluation and X-inactivation pattern evaluation
Five hundred and fifty resolution G-banding karyotype was performed from lymphocyte cultures. DNA samples were isolated from peripheral blood using Gentra Puregene Kit (Qiagen-Sciencesm). Chromosomal microarrays were performed using the Affymetrix Genome-Wide Human SNP-Array 6.0 (Affymetrix Inc.) or Human Genome CGH array 4 × 44k (Agilent Technologies). X-chromosome inactivation studies were performed by Human Androgen Receptor Assay, replication banding with BrdU and/or 5-ethynyl-2′-deoxyuridine (EdU) incorporation assay [10].
Breakpoint mapping
Two strategies were used to map the breakpoints: array painting followed by WGS, and array-CGH in a family member carrier of the same rearrangement but in unbalanced form.
Array painting, FISH, and whole genome sequencing
Breakpoints were ascertained by microdissection and amplification of the derivative chromosomes followed by array-CGH as previously described [11]. FISH using BAC (bacterial artificial chromosome) probes, which mapped across the breakpoints, was applied on 12 out of 16 breakpoints, to validate their locations (Supplementary Table S1). In order to reach a higher resolution, the patients had their breakpoints addressed by shallow WGS. For the WGS, 2 µg of genomic DNA was sheared using Covaris and 550 bp fragments were targeted to maximize the physical coverage. The sequencing library was prepared using Tru-Seq DNA PCR-free Sample Prep Kit (Illumina) to avoid PCR duplicates, and 100 bp paired-end reads were sequenced on the HiSeq 2500 platform (Illumina). Sequence-control, software real-time analysis, and CASAVA software v1.8.2 (Illumina) were used for image analysis and base calling. Burrows-Wheeler Aligner (BWA-MEM) [11] with default parameters was used to map the data to hg38. We reached 4.8× coverage and a sequence yield of 15.3 Gb on average. The mean insert size was 606 bp, alignment rate 97.8%, proper pair coverage 94.5%, duplicate rate 4.8%, and chimera rate 0.3%. The BAM file (Binary Alignment/Map format) was submitted to BreakDancerMax version 1.4.4 analysis [12] using the –t option to detect interchromosomal junctions. Based on the breakpoint localization according to array painting and/or BreakDancerMax analysis, BAM files were filtered for aligned reads adjacent to the breakpoints. Filtered BAM files were visualized in Integrative Genomics Viewer (IGV) [13] in search of chimeric inserts and split-reads. Patient 6’ rearrangement was evaluated only by WGS (Table 2).
Array-CGH and sequencing
Patient 6 transmitted her rearrangement as an unbalanced translocation to her daughter. The child’s rearrangement was characterized by chromosomal microarray analysis (CytoScan ® 750 K Array, Affymetrix and 8 × 60 K customized slide Agilent Technologies) as previously described [9].
Junction point sequencing
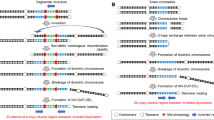
The junction points were further characterized by Sanger sequencing. Different sets of primers were designed around the breakpoints and long-range PCR was performed as previously described [14]. Primers are detailed in Supplementary Table S2. To amplify junctions between two non-heterochromatic regions, the primers were designed for the sequence flanking the breakpoints according to reference genome. However, four patients presented one breakpoint in non-heterochromatic region and the other breakpoint at heterochromatic region, including reference genome gaps, e.g., 9q12 and 21p13, or at regions with highly repetitive sequence, e.g., chromosome 9 centromere and 21p11.1. In order to design primers flanking the breakpoints in these four heterochromatic regions, we used IGV to visualize the BAM files from the WGS, inspecting the reads mapped around the breakpoint in the non-heterochromatic region. We selected the reads that were part of chimeric inserts and searched for the sequence of their mates in the raw sequencing data (FASTQ files). We manually de novo assembled the heterochromatic breakpoint region sequence combining the sequence of these reads obtained from the FASTQ files. The primers were designed based on this de novo assembly to amplify the junction point. The strategy used to Sanger sequence the junction point in these heterochromatic regions is schematically described in Fig. 1.

Strategy used to Sanger sequence the junction-points of translocations with breakpoints at heterochromatic regions. a IGV screenshot of whole genome sequencing pair-end reads of patient 8, visualizing split-reads (dark red) and chimeric inserts (red) in Xq13.3 breakpoint region. b Numbered reads from panel a were selected and the sequence of their mates were obtained from the FASTQ files, to assembly the sequence of the autosomal breakpoint region, which is located at chromosome 9 centromeric region. Reads in dark blue are the remaining portion of the split-reads 1 and 2 (dark red in panel a) observed in IGV. c At the top, Sanger sequencing electropherogram of the der(X) junction point. The breakpoint is indicated by an arrowhead. Below, alignment of the Sanger sequencing of the der(X) junction point, read 1, read 2, and the read 6’s mate sequences from panel a to chromosomes X and 9 references. Sequences from chromosome X and 9 are in red and blue, respectively. A deletion of 2 bp in patient 8’s chromosome X is indicated in bold in the X-chromosome reference sequence
Chromosomal rearrangements were fully described in Supplemental Case Reports following HGVS nomenclature [15]. All gene disruptions validated by Sanger sequencing were submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/), as well as their clinical consequences to the patients (accessions SCV000854423 to SCV000854429).
Classification of disrupted genes pathogenicity
The genes disrupted by the breakpoints were classified in three categories: pathogenic, potentially pathogenic, or uncertain clinical significance. As pathogenic, we considered genes that presented three criteria: (I) no description of a loss of function genetic variant in general population databases, (II) previous association with the patient’s phenotype in at least two other individuals, and (III) clear relation to the development, function or maintenance of the affected tissue or cell type. Potentially pathogenic genes matched only two of these criteria and genes with uncertain clinical significance, one or none of them.
Prediction of topological associated domains (TADs) disruption
TADs were addressed in human embryonic stem cells [16] and lymphoblastoid cell lines [17] using the Encode browser for Hi-C data visualization. Genes strongly associated with the patient’s clinical features and encompassed by disrupted TADs were considered likely affected by position effect and further investigated. Additionally, positions of predicted regulatory elements were obtained from RegulomeDB chromatin state annotations [18], which are based on the Roadmap Epigenomics Consortium integrative analysis of human epigenomes from different tissues and cell types [19].
Reverse transcription quantitative PCR (RT-qPCR)
Whole blood RNA was isolated from the patients and controls using the PAXgene BloodRNA MDx Kit (Qiagen-Siences). The cDNA was synthetized using High Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific). TaqMan assays (Thermo Fisher Scientific) were selected for all the genes disrupted at the breakpoints and for genes potentially affected by position effect. GAPDH and ACTB gene expression were used as internal controls. Assays are detailed in Supplementary Table S3.
Results
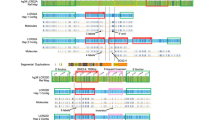
G-banding karyotype and chromosomal microarray indicated that all patients presented BCAs, without cryptic genomic imbalances. As expected, X-chromosome inactivation studies showed a skewed X-inactivation pattern, with the normal X-chromosome preferentially inactivated. Patients 1 to 9 had at least one of the two junction points characterized at the nucleotide level. The breakpoint definition for patient 3 is illustrated in Fig. 2, whereas results for the other patients are fully described in the Supplementary Case Reports and summarized in Table 1.

Patient 3’s breakpoint characterization at the nucleotide level and rearrangement’s impact on the expression of disrupted genes. a Partial G-banding karyotype and b ideogram of the chromosomes involved in her translocation. c, d Sanger sequencing electropherograms of the der [7] (c) and der(X) (d) junction points. Black dashed box on der[7] shows microhomologies between chromosomes X and 11 breakpoint regions. Black dashed box on der(X) shows nucleotide insertions at the junction-point. Below, alignment of the junction points to chromosome X and 11 reference sequences. e RT-qPCR expression levels of APOOL in whole blood of n = 8 Brazilian female controls and patient 3. Note APOOL unmodified expression in the patient when compared to controls
Breakpoint mapping
Molecular cytogenetic methods were effective in mapping 12 out of 16 breakpoints with a resolution range of 3.5 to 427.7 kb (Table 2). Given the inability of array-based methods to evaluate heterochromatic and highly repetitive regions, the autosomal breaks of patients 7 to 10 could not be mapped by array painting. The latter, however, mapped the non-heterochromatic breakpoints in these four patients, allowing the identification of inserts spanning the junction points in the further WGS analysis (Fig. 2).
The analysis of the WGS data with the BreakDancerMax software only detected junctions between non-heterochromatic regions, as it was effective in mapping both breakpoints in patients 1 to 6. BAM file chimeric inserts and/or split-reads were visualized on IGV and the breakpoints at non-heterochromatic regions were mapped with a higher resolution, ranging from 2 to 297 bp (Table 2). On the other hand, none of the four patients with one of the breakpoints in a heterochromatic region (patients 7 to 10) had their chromosomal junctions detected by the BreakDancerMax algorithm (Table 2). The four highly repetitive breakpoint regions could be manually de novo assembled (Fig. 2).
Sanger sequencing validation of the junction points
All 12 junctions between two non-heterochromatic regions were successfully amplified and sequenced (Table 2). For patients 7 to 10, in accordance to the nature of heterochromatic and highly repetitive regions, the blat search of sequences from the autosomal breakpoint regions returned pericentromeric regions and short arm of acrocentric chromosomes (Supplementary Table S4). Four out of eight junctions between a heterochromatic and a non-heterochromatic region could be Sanger sequenced (patients 7 to 9, Table 2). From the four junctions involving heterochromatic regions in which Sanger sequencing was inefficient, two are from patient 10. This patient is carrier of a translocation between Xp11.4 and 21p13, with the autosomal breakpoint in a gap in the reference genome. The few bases of the amplicons obtained from her both derivative chromosomes aligned to Xp11.4. However, the electropherogram base calling was interrupted before spanning the interchromosomal junctions, indicating that her junction points were successfully PCR amplified, but the highly repetitive nature of 21p13 sequence prevented the full sequencing of the amplicon (Supplementary Case Reports).
Mechanisms of chromosomal rearrangement formation
The sequencing of the junction points allowed the delineation of the disrupted sequences’ nature for 16 non-heterochromatic breakpoint regions (Table 3). Sixty-nine percent (11/16) of these breakpoints were located at repeat elements, e.g., LINEs, SINEs, LTRs, and STR. Five out of the six breakpoints at LINE sequences were in the X-chromosome. Only patients 1 and 3 presented both breakpoints at repeat elements.
The mutational signatures of the junction points were used to infer about the mechanisms of formation (Table 3). Microhomology at the junction points was observed in five patients (1, 3, 4, 6, and 8). In two cases (patients 2 and 5), nucleotides deletion and the insertion of few base pairs suggested the Non-Homologous End Joining (NHEJ) mechanism [20]. For patient 3, the presence of a duplication of about 200 bases accompanied by microhomology indicated the Microhomology Mediated Break Induced Repair (MMBIR) [21]. For three patients (1, 4, and 6), either NHEJ or MMBIR could be attributed to the formation of the translocations. For patients 7 to 10 the mechanism of formation could not be determined since their heterochromatic breakpoints could not be confidently mapped.
Expression and phenotypic outcome of disrupted genes
The disruption of six coding sequences and one promoter region were observed at the breakpoints in six out of ten patients (Table 1). Five genes/promoters were disrupted at the X-chromosomal breakpoint: NEXMIF, ZDHHC15, AMMECR1, APOOL, and IL1RAPL1 (Table 1). These first three genes had the complete absence of expression in the patient (see Supplementary Case Reports for details). Two autosomal genes were found as disrupted: ZNF611 and NEDD4L. The former presented a reduced expression and the later, a higher expression in the patient (Table 1, Supplementary Case Reports). Patient 1 presented fusion genes formed at the junction points with the theoretical fusion transcripts being in phase. The experimental evidences of these fusion genes and fusion transcripts are detailed in Supplementary Case Reports.
The functional impairments of NEXMIF, IL1RAPL1, and AMMECR1 genes were considered as pathogenic. NEDD4L gene disruption was considered potentially pathogenic, and ZNF611, APOOL, and ZDHHC15 were classified as genes with uncertain clinical significance. The interpretation of the disrupted genes pathogenicity is fully described in Supplementary Case Reports. Four subjects (patients 5, 6, 9, and 10) presented no gene or promoter disruption at the breakpoints, suggesting that more complex mechanisms are responsible for their phenotypes. None of the four patients with premature ovarian failure had a disrupted gene that could be considered as candidate for their ovarian phenotype.
Disruption of TADs
In four patients, genes harbored at the disrupted TAD matched the criteria to be considered potentially affected by position effect. None of these patients have gene disruptions that explain their phenotypic outcomes. In patient 10, the X-chromosome breakpoint is localized ~20 kb downstream TSPAN7 (MIM# 300096) gene sequence (Fig. 3a), a well-known neurodevelopment gene [22] involved in excitatory synapse development [23]. The TSPAN7 gene expression was abrogated in whole blood from patient 10 (Fig. 3b), suggesting that her neurodevelopment delay was caused by TSPAN7 loss of function via position effect. Concordantly, the TAD disrupted by patient 10’s X-chromosome breakpoint harbors TSPAN7 and predicted enhancers active in brain and blood cells. In patient 6, chromosome 4 breakpoint is 19 kb downstream to NKX3-2 (MIM# 602183) and disrupted the TAD that contains this gene and predicted chondrocyte enhancers. NKX3-2 is a homeobox gene that plays a role in the development of skeleton including cranial bones [24]. Abnormal expression of this gene has been previously associated with oculo-auriculo-vertebral spectrum [25], condition present in patient 6. The NKX3-2 gene is not expressed in whole blood.

Predicted position effect on genes neighboring the breakpoints. a HiC data visualization from ENCODE [17] of X-chromosome 37.1-41.2 Mb (hg19) region showing TAD (dashed line) that harbors patient 10’s X-chromosome breakpoint (black bar). This breakpoint is 20 kb downstream TSPAN7 gene (arrow). b RT-qPCR expression levels of TSPAN7 in whole blood of n = 8 Brazilian female controls and patient 10. Note the absence of TSPAN7 expression in the patient. c HiC data visualization from ENCODE [18] of X-chromosome 72.1-77.6 Mb (hg19) region showing TAD (dashed line) that harbors X-chromosomal breakpoint of patients 4 and 7 (black bars) and a ~850 kb duplication (gray bar) previously described [31], and the TAD (dashed line) that harbors FGF16 gene (arrow)
The other patients with position effect as a likely pathogenic mechanism present with ovarian failure. Patient 3’s X-chromosome breakpoint disrupts a TAD that harbors POF1B (MIM# 300603) gene, which encodes a cytoplasmic protein involved in regulating epithelial polarity and in the formation of epithelial permeability barriers [26]. Point mutations in POF1B have been associated with primary amenorrhea [27]. In patient 5, chromosome 2 breakpoint is 123 kb upstream FHL2 (MIM# 602633) gene, which is highly expressed in ovarian granulosa cells and related to DAX1 upregulation, required for ovarian development [28]. POF1B and FHL2 gene expression study in whole blood showed an unmodified expression level between patients and controls (data not shown), which does not exclude long-range regulatory changes, since the chromatin topology and interactions might differ between blood and ovarian cells.
Two other translocations associated with premature ovarian failure (patients 4 and 7) did not match the criteria that we established to indicate “position effect”; however, their X-chromosome breakpoint disrupted the same TAD, which also harbors a ~850 kb duplication previously associated with premature ovarian failure [29] (Fig. 3c). The neighboring TAD harbors FGF16, which acts as a key regulator in early oocyte development in teleost fishes, tilapia and chicken [30,31,32], is expressed in the human ovary and promotes cell proliferation and invasion in ovarian cancer [33]. The FGF16 gene is not expressed in whole blood.
Discussion
We performed a high-resolution breakpoint characterization on a very selective and rare cohort: molecularly confirmed balanced X-autosome translocations in female patients with phenotypic alterations. Illumina short-read sequencing is considered the current gold-standard method for routine genome-wide breakpoint mapping in structural variants [34]. However, the alignment of repeated DNA sequences has always been a technical limitation. To enrich the library for the breakpoint regions, with DNA segments that harbor the junction points, a PCR-based library preparation is required, which affects the coverage and the alignment quality of the highly repetitive and CG-rich genomic regions [2]. In our study, the usage of PCR-free library preparation method to perform the shallow WGS, with no targeted genomic capture, was crucial to address breakpoints in heterochromatic regions with minimized costs. Future dissemination of long-read sequencing might improve the ability to characterize highly repetitive DNA segments of the human genome [35]; however, these methods are still not implemented in routine diagnosis.
Currently, standard sequencing-based methods are ineffective on the high-resolution breakpoint definition in BCAs with one of the chromosomal breaks at highly repetitive regions regardless of the coverage, tool or algorithm used for the breakpoint calling, since these DNA segments cannot be confidently mapped (e.g., centromeres or the short arm of acrocentric chromosomes). It is expected that ~7–8% of BCAs’ breakpoints are inaccessible by short-read sequencing [36]. Patients with these inaccessible breakpoints are frequently removed from high-resolution breakpoint mapping studies on subjects with BCA, leading to biased conclusions about the frequency of formation mechanisms of the rearrangements and the genomic nature of the DNA sequences that they disrupt. Even though our cohort has four out of ten subjects with breakpoints at heterochromatic regions, we were able to identify all the 20 breakpoint regions. Additionally, the Sanger sequencing validation was efficient for most of the junction points analyzed. These analyses demonstrate the feasibility of identifying breakpoints at highly repetitive DNA segments, including gaps in the reference genome, using a cost-effective approach. The combination of cytogenomic methods with low-coverage WGS was crucial for characterizing junction points involving heterochromatic regions, since in these cases the tracking of WGS chimeric inserts and split-reads was based on the array painting results.
The junction point sequencing allowed insights about the formation mechanism of these chromosomal rearrangements. The mutational signatures of NHEJ, the major mechanism in BCAs’ formation [37], were compatible with the junction points of six patients. For non-recurrent translocations associated with human congenital anomalies, it has been described that breakpoint regions are enriched for repeat elements [36]. In our study, 11 out of 16 non-heterochromatic breakpoints occurred at repeat elements. LINE sequences were the most frequent repeat element disrupted in our cohort and four out of nine X-chromosome breakpoints Sanger-validated disrupted a LINE element. This pattern can indicate a possible role of LINE sequences in the mutational mechanism of rearrangements involving the X-chromosome or just reflect the X-chromosome composition. Further studies are required to clarify the relation between LINE sequences and the formation of chromosomal rearrangements involving the X-chromosome.
It has been demonstrated that there is an enrichment of genes related to developmental disorders at the breakpoints of BCA associated to congenital abnormalities [36]. In our cohort, two genes (NEXMIF and IL1RAPL1) were determined as causative of intellectual disability [6] and one gene (NEDD4L) is potentially associated with neurodevelopmental disorders. In the four patients with no gene disruption at their breakpoints, more complex pathogenic mechanisms await clarifications, such as disruption of regulatory interactions, enhancer adoption, modification on the chromatin landscape, among others. In six patients, position effect affecting breakpoint neighboring genes was pointed as a putative pathogenic genetic mechanism and the regulatory interactions that act on these candidate genes (TSPAN7, NKX3-2, POF1B, FHL2, and FGF16) require further functional analysis. Four of these six patients present ovarian failure phenotype and, concordantly, premature ovarian failure has been associated with position effect in females with balanced X-autosome translocations [38]. It is worth mentioning that high-resolution chromosomal architecture data is only publicly available for a limited variety of human cell types and tissues, which does not include the developing ovary. During the development of the female gonad, the reorganization of the chromatin contacts caused by these chromosomal rearrangements might alter gene expression and function.
In this work, six out of ten patients had their breakpoints revised after the detailed molecular breakpoint definition. This emphasizes the importance of breakpoints mapping at nucleotide resolution in balanced chromosomal rearrangements to reveal the pathogenic mechanism and improve the genotype-phenotype correlation. This study demonstrates the feasibility to molecularly characterize breakpoints at heterochromatic regions associating cytogenomic methods and short-read next-generation sequencing in a cost-effective manner. This characterization of BCAs with chromosomal breaks at highly repetitive DNA segments can provide new insights into their mechanisms of formation and the properties of the genomic regions disrupted in these rearrangements, describing new genes associated with human diseases.
Web resources
Web Resources: 1000 Genomes: http://www.internationalgenome.org/. DECIPHER: https://decipher.sanger.ac.uk/. DGV (Database of Genomic Variants): http://dgv.tcag.ca/dgv/app/home. ENCODE (Encyclopedia of DNA Elements): https://www.encodeproject.org/. ExAC (The Exome Aggregation Consortium): http://exac.broadinstitute.org/. GO (Gene Ontology): http://www.geneontology.org/. GTEx (Genotype-Tissue Expression): http://www.gtexportal.org/. IMPC (International Mouse Phenotyping Consortium): http://www.mousephenotype.org/. OMIM (Online Mendelian Inheritance in Man): http://www.omim.org/.UCSC Blat Search: https://genome.ucsc.edu/cgi-bin/hgBlat
References
Warburton D. De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am J Hum Genet. 1991;49:995–1013.
Schluth-Bolard C, Labalme A, Cordier MP, Till M, Nadeau G, Tevissen H, et al. Breakpoint mapping by next generation sequencing reveals causative gene disruption in patients carrying apparently balanced chromosome rearrangements with intellectual deficiency and/or congenital malformations. J Med Genet. 2013;50:144–50.
Bugge M, Bruun-Petersen G, Brondum-Nielsen K, Friedrich U, Hansen J, Jensen G, et al. Disease associated balanced chromosome rearrangements: a resource for large scale genotype-phenotype delineation in man. J Med Genet. 2000;37:858–65.
Nilsson D, Pettersson M, Gustavsson P, Forster A, Hofmeister W, Wincent J, et al. Whole-Genome Sequencing of Cytogenetically Balanced Chromosome Translocations Identifies Potentially Pathological Gene Disruptions and Highlights the Importance of Microhomology in the Mechanism of Formation. Hum Mutat. 2017;38:180–92.
Schmidt M, Du Sart D. Functional disomies of the X chromosome influence the cell selection and hence the X inactivation pattern in females with balanced X-autosome translocations: a review of 122 cases. Am J Med Genet. 1992;42:161–9.
Moyses-Oliveira M, Guilherme RS, Meloni VA, Di Battista A, de Mello CB, Bragagnolo S, et al. X-linked intellectual disability related genes disrupted by balanced X-autosome translocations. American journal of medical genetics Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics. 2015;168:669–77.
Moyses-Oliveira M, Guilherme Rdos S, Dantas AG, Ueta R, Perez AB, Haidar M, et al. Genetic mechanisms leading to primary amenorrhea in balanced X-autosome translocations. Fertil Steril. 2015;103:1289–96 e2.
Moyses-Oliveira M, Giannuzzi G, Fish RJ, Rosenfeld JA, Petit F, Soares MF, et al. Inactivation of AMMECR1 is associated with growth, bone, and heart alterations. Hum Mutat. 2018;39:281–91.
Bragagnolo S, Colovati ME, Guilherme RS, Dantas AG, de Souza MZ, de Soares MF, et al. Wolf-Hirschhorn Syndrome with Epibulbar Dermoid: An Unusual Association in a Patient with 4p Deletion and Functional Xp Disomy. Cytogenet Genome Res. 2016;150:17–22.
Sisdelli L, Vidi AC, Moyses-Oliveira M, Di Battista A, Bortolai A, Moretti-Ferreira D, et al. Incorporation of 5-ethynyl-2'-deoxyuridine (EdU) as a novel strategy for identification of the skewed X inactivation pattern in balanced and unbalanced X-rearrangements. Hum Genet. 2016;135:185–92.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–81.
Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92.
Guilherme RS, Hermetz KE, Varela PT, Perez AB, Meloni VA, Rudd MK, et al. Terminal 18q deletions are stabilized by neotelomeres. Mol Cytogenet. 2015;8:32.
den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, et al. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat. 2016;37:564–9.
Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–6.
Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–80.
Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–7.
Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–30.
Gu W, Zhang F, Lupski JR. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1:1755–8417.
Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:30.
Bassani S, Cingolani LA, Valnegri P, Folci A, Zapata J, Gianfelice A, et al. The X-linked intellectual disability protein TSPAN7 regulates excitatory synapse development and AMPAR trafficking. Neuron. 2012;73:1143–58.
Lee SA, Suh Y, Lee S, Jeong J, Kim SJ, Park SK. Functional expression of dopamine D2 receptor is regulated by tetraspanin 7-mediated postendocytic trafficking. FASEB J. 2017;31:2301–13.
Tribioli C, Lufkin T. The murine Bapx1 homeobox gene plays a critical role in embryonic development of the axial skeleton and spleen. Development. 1999;126:5699–711.
Fischer S, Ludecke HJ, Wieczorek D, Bohringer S, Gillessen-Kaesbach G, Horsthemke B. Histone acetylation dependent allelic expression imbalance of BAPX1 in patients with the oculo-auriculo-vertebral spectrum. Hum Mol Genet. 2006;15:581–7.
Padovano V, Lucibello I, Alari V, Della Mina P, Crespi A, Ferrari I, et al. The POF1B candidate gene for premature ovarian failure regulates epithelial polarity. J Cell Sci. 2011;124:3356–68. (Pt 19)
Lacombe A, Lee H, Zahed L, Choucair M, Muller JM, Nelson SF, et al. Disruption of POF1B binding to nonmuscle actin filaments is associated with premature ovarian failure. Am J Hum Genet. 2006;79:113–9.
Du X, Hublitz P, Gunther T, Wilhelm D, Englert C, Schule R. The LIM-only coactivator FHL2 modulates WT1 transcriptional activity during gonadal differentiation. Biochim Biophys Acta. 2002;1577:93–101.
Dudding TE, Lawrence O, Winship I, Froyen G, Vandewalle J, Scott R, et al. Array comparative genomic hybridization for the detection of submicroscopic copy number variations of the X chromosome in women with premature ovarian failure. Hum Reprod. 2010;25:3159–60. author reply60-1.
Ayers KL, Lambeth LS, Davidson NM, Sinclair AH, Oshlack A, Smith CA. Identification of candidate gonadal sex differentiation genes in the chicken embryo using RNA-seq. BMC Genomics. 2015;16:704.
Sun YL, Zeng S, Ye K, Yang C, Li MH, Huang BF, et al. Involvement of FGF9/16/20 subfamily in female germ cell development of the Nile tilapia, Oreochromis niloticus. Fish Physiol Biochem. 2012;38:1427–39.
Zhao C, Zhang G, Yin S, Li Z, Wang Q, Chen S, et al. Integrated analysis of mRNA-seq and miRNA-seq reveals the potential roles of sex-biased miRNA-mRNA pairs in gonad tissue of dark sleeper (Odontobutis potamophila). BMC Genomics. 2017;18:613.
Basu M, Mukhopadhyay S, Chatterjee U, Roy SS. FGF16 promotes invasive behavior of SKOV-3 ovarian cancer cells through activation of mitogen-activated protein kinase (MAPK) signaling pathway. J Biol Chem. 2014;289:1415–28.
Hehir-Kwa JY, Marschall T, Kloosterman WP, Francioli LC, Baaijens JA, Dijkstra LJ, et al. A high-quality human reference panel reveals the complexity and distribution of genomic structural variants. Nat Commun. 2016;7:12989.
Chaisson MJ, Huddleston J, Dennis MY, Sudmant PH, Malig M, Hormozdiari F, et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature. 2015;517:608–11.
Redin C, Brand H, Collins RL, Kammin T, Mitchell E, Hodge JC, et al. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat Genet. 2017;49:36–45.
Chiang C, Jacobsen JC, Ernst C, Hanscom C, Heilbut A, Blumenthal I, et al. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat Genet. 2012;44:390–7. S1.
Rizzolio F, Pramparo T, Sala C, Zuffardi O, De Santis L, Rabellotti E, et al. Epigenetic analysis of the critical region I for premature ovarian failure: demonstration of a highly heterochromatic domain on the long arm of the mammalian X chromosome. J Med Genet. 2009;46:585–92.
Weckselblatt B, Rudd MK. Human Structural Variation: Mechanisms of Chromosome Rearrangements. Trends Genet. 2015;31:587–99.
Acknowledgements
We thank the patients and families, Danilo Moretti-Ferreira, Marco Antonio de Paula Ramos, Rosane Canonaco and Ana Beatriz Alvarez Perez, for the clinical data, and Lausanne Genomic Technologies Facility for technical help.
Funding
This work was supported by grants from Fundação de Amparo à Pesquisa do Estado de São Paulo [2014/11572-8 to MIM]; and the Swiss National Science Foundation [31003A_160203 to AR]. MMO, ADB and MZ are recipient of a FAPESP fellowship (Fundação de Amparo à Pesquisa do Estado de São Paulo). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Moysés-Oliveira, M., Di-Battista, A., Zamariolli, M. et al. Breakpoint mapping at nucleotide resolution in X-autosome balanced translocations associated with clinical phenotypes. Eur J Hum Genet 27, 760–771 (2019). https://doi.org/10.1038/s41431-019-0341-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0341-5
This article is cited by
-
Premature ovarian insufficiency is associated with global alterations in the regulatory landscape and gene expression in balanced X-autosome translocations
Epigenetics & Chromatin (2023)
-
Fold-back mechanism originating inv-dup-del rearrangements in chromosomes 13 and 15
Chromosome Research (2023)
-
Mechanisms of structural chromosomal rearrangement formation
Molecular Cytogenetics (2022)
-
Allelic and dosage effects of NHS in X-linked cataract and Nance–Horan syndrome: a family study and literature review
Molecular Cytogenetics (2021)
-
The genetic cause of intellectual deficiency and/or congenital malformations in two parental reciprocal translocation carriers and implications for assisted reproduction
Journal of Assisted Reproduction and Genetics (2021)
