
Abstract
Attention-deficit/hyperactivity disorder (ADHD) is a neurodevelopmental disorder caused by an interplay of genetic and environmental factors. Epigenetics is crucial to lasting changes in gene expression in the brain. Recent studies suggest a role for DNA methylation in ADHD. We explored the contribution to ADHD of allele-specific methylation (ASM), an epigenetic mechanism that involves SNPs correlating with differential levels of DNA methylation at CpG sites. We selected 3896 tagSNPs reported to influence methylation in human brain regions and performed a case-control association study using the summary statistics from the largest GWAS meta-analysis of ADHD, comprising 20,183 cases and 35,191 controls. We observed that genetic risk variants for ADHD are enriched in ASM SNPs and identified associations with eight tagSNPs that were significant at a 5% false discovery rate (FDR). These SNPs correlated with methylation of CpG sites lying in the promoter regions of six genes. Since methylation may affect gene expression, we inspected these ASM SNPs together with 52 ASM SNPs in high LD with them for eQTLs in brain tissues and observed that the expression of three of those genes was affected by them. ADHD risk alleles correlated with increased expression (and decreased methylation) of ARTN and PIDD1 and with a decreased expression (and increased methylation) of C2orf82. Furthermore, these three genes were predicted to have altered expression in ADHD, and genetic variants in C2orf82 correlated with brain volumes. In summary, we followed a systematic approach to identify risk variants for ADHD that correlated with differential cis-methylation, identifying three novel genes contributing to the disorder.
Similar content being viewed by others
Introduction
Attention-deficit/hyperactivity disorder (ADHD) is a common neurodevelopmental disorder with a worldwide prevalence of around 5%1. Its main symptoms include inattention and/or hyperactivity-impulsivity (DSM-V)2. ADHD is among the most heritable psychiatric disorders, with about 76% of its etiology accounted by genetic risk factors3 and with single-nucleotide polymorphisms (SNPs) explaining around 22% of the phenotypic variance4. Furthermore, there is molecular evidence of shared genetic risk factors across many psychiatric disorders5. In ADHD, a recent genome-wide association study (GWAS) meta-analysis of 12 sample groups unraveled some of the specific genetic underpinnings of this polygenic disorder for the first time4. One of the challenges of GWAS is to establish the causal relationship between the associated genetic variants, especially those located outside genes, and the disorder. In this regard, the use of epigenetic information can improve the interpretation of functionality of non-coding genetic variation6. In addition, some studies have hypothesized the importance of sub-threshold variants derived from GWAS7,8, particularly those located in enhancer regions, with a potential impact on gene regulation9,10.
DNA methylation is one of the most stable epigenetic mechanisms, involving mainly cytosines of CpG dinucleotides. This mechanism plays an important role in the regulation of neurogenesis, differentiation, and brain development11. Furthermore, epigenetic alterations have been hypothesized to contribute to neurodevelopmental disorders12, including ADHD13, autism spectrum disorders (ASD)14,15, or borderline personality disorder16.
DNA methylation can be complementary if it involves both alleles, or non-complementary when it affects only one allele, as in chromosome X inactivation in females or allele-specific methylation (ASM)6. ASM is a common mechanism by which single nucleotide variants determine differential methylation levels of CpG sites. ASM can alter promoter activity, leading to allele-specific expression17 in combination with other still quite unknown factors, such as environmental effects6. It is quantitative and heterogeneous across tissues and individuals6. The environment affects DNA methylation leading to changes in gene regulation, although the underlying mechanism is still not well understood18. It has been suggested that, during embryonic development, ASM regions could be especially sensitive to environmental effects6. Investigating SNPs that display ASM could help to identify risk variants for common diseases, including neuropsychiatric disorders19, as shown by recent studies of bipolar disorder (BD) and schizophrenia10,20.
The present study investigated the possible contribution of ASM to ADHD using data from the largest GWAS meta-analysis performed to date in ADHD4. We also assessed its possible effect on gene expression and on brain volumes to identify new genes contributing to the disorder.
Materials and methods
Selection of ASM SNPs
SNP selection was made based on the results of two previous studies21,22, which identified ASM variants in multiple brain regions of post-mortem human samples. Gibbs et al.21, considered four brain regions (cerebellum, frontal cortex, caudal pons, and temporal cortex) of 150 subjects and Zhang et al.22, used only the cerebellum of 153 subjects. Gibbs et al.21, unlike Zhang et al.22, excluded those sequences of probes with significant correlation with methylation that contained polymorphisms. To discard possible artifacts in our results, we checked and confirmed that none of the probes used to detect the six highlighted CpG sites target genomic regions with SNP variants. The genotyping platforms used in the two studies were different (Gibbs et al.21 used Infinium HumanHap550 Beadchips and Zhang et al.22 used Affymetrix GeneChip Mapping 5.0K Array). Both studies evaluated DNA methylation using the HumanMethylation27 Beadchips, and performed linear regression analyses by PLINK23 to determine the correlation between each SNP and methylation of any CpG site21,22. Zhang et al.22, unlike Gibbs et al.21 applied quantile normalization to the residuals prior to the linear regression analyses.
In the study by Zhang et al.22, a total of 12,117 SNP–CpG pairs associations were reported in cerebellum, and Gibbs et al.21 listed a total of 12,135 SNP–CpG pairs in frontal cortex, 11,374 in caudal pons, 16,734 in temporal cortex, and 12,102 in cerebellum (Fig. 1). We combined the information from both studies and obtained a total of 43,132 SNP–CpG pairs involving 33,944 different SNPs and 5306 CpG sites (Fig. 1). We considered all the ASM SNPs from all the tissues in the two studies, as there are multiple SNP–CpG pairs in common between them (Fig. S1).

SNPs tested in the ADHD GWAS meta-analysis and multiple testing correction. SNPs correlating with differential methylation of CpG sites and eQTLs in brain regions (only for genes in which the CpG site lies <5 kb from the transcription start site) are depicted
We subsequently applied different filters to generate a sub-list of 3896 SNPs (Figs. 1 and S2) out of these 33,944 variants to minimize redundancy: associations in cis between the SNP and the CpG site, correlation of the SNP with methylation levels of the CpG (R2) ≥ 0.2, as performed in Gibbs et al. (2010)21. We considered only autosomal SNPs and selected tagSNPs for each CpG site (r2 ≥ 0.85), by assessing linkage disequilibrium (LD) with Haploview software24 using the Central European (CEU) reference panel from 1000 Genomes Project Phase 325.
Case-control GWAS datasets
We explored the selected ASM SNPs in the summary statistics from a meta-analysis of 11 independent GWAS of ADHD conducted by the Psychiatric Genomics Consortium (PGC) and iPSYCH. This case-control study investigated 8,047,420 markers in 20,183 cases and 35,191 controls from Europe, USA, Canada, and China, with patients diagnosed according to the criteria detailed in Demontis et al. (2019)4.
Statistical analysis
To test whether risk variants for ADHD are enriched in ASM SNPs, we carried out an enrichment analysis using the Fisher’s exact test in R26 at p-value thresholds ranging from 5E−02 to 5E−08 considering the total number of ASM SNPs available from the ADHD GWAS meta-analysis4 (32,884 out of 33,944 SNPs).
From our selection of 3896 ASM tagSNPs, we could retrieve information on the association with ADHD of 3771 SNPs (96.8%) that were present in the summary statistics of the ADHD GWAS meta-analysis (Fig. 1)4. False discovery rate (FDR) was applied to correct for multiple testing. We used the q-value package for R27 and obtained a threshold p-value of 6.78E−05 corresponding to a 5% FDR. CpG sites highlighted by SNPs that were significant at this FDR threshold were followed-up in further analyses (Fig. 1). Additionally, we performed corrections for multiple testing, using Bonferroni and Genetic type 1 Error Calculator (GEC) methods (http://grass.cgs.hku.hk/gec/)28. The Bonferroni-corrected threshold was set at p ≤ 1.32E−05, which considered all the SNPs and tests to be independent (0.05/3771 SNPs). The GEC established the significance threshold at 1.98E−05, which addressed multiple testing for the set of 3771 dependent SNPs by estimating the independent number of tests. The LD between SNPs was calculated according to the 1000 Genomes EUR reference data25.
Finally, we considered and retrieved p-values of those tagged ASM SNPs in high LD (r2 ≥ 0.85) with the previous ones that also correlated in cis with the methylation levels of the same CpG sites (R2 ≥ 0.2) (Fig. 1).
Functional annotation of associated ASM SNPs
We applied four methods to obtain information about the possible functional impact of the ASM SNPs that were associated with ADHD. First, we evaluated the presence of possible enhancer or promoter regions using the Haploreg v4.1 tool29. To do this, we considered histone modifications related to enhancer regions (H3K4me1 and H3K27ac) and promoters (H3K4me3 and H3K9ac) of 10 different brain regions (hippocampus middle, substantia nigra, anterior caudate, cingulate gyrus, inferior temporal lobe, angular gyrus, dorsolateral prefrontal cortex, germinal matrix, and male and female fetal brain). Second, we evaluated the effect on gene expression through an eQTL analysis using GTEx data (Release V7)30. We considered eQTL information for all available brain tissues: amygdala, anterior cingulate cortex (BA24), caudate basal ganglia, cerebellar hemisphere, cerebellum, cortex, frontal cortex (BA9), hippocampus, hypothalamus, nucleus accumbens basal ganglia, putamen basal ganglia, spinal cord cervical c-1, and substantia nigra. Third, we considered all the SNPs, not only ASMs, located within ±1 Mb from the transcription start site (TSS) of each gene to infer if the genetically determined expressions of genes of interest correlated with ADHD. This analysis was carried out using MetaXcan31, the input being the summary statistics of the ADHD GWAS meta-analysis4 and prediction models trained with RNA-Seq data of 10 GTEx30 brain tissues and CommonMind32 dorsolateral prefrontal cortex. The SNP covariance matrices were generated using the 1000 Genomes Project Phase 325 EUR genotypes of the prediction model SNPs. Bonferroni correction for multiple testing was considered (p ≤ 2.27E−03; 0.05/22 tests). Finally, we examined the possible influence of the identified variants on subcortical brain structures. We obtained the summary statistics of a GWAS meta-analysis of eight MRI volumetric measures (nucleus accumbens, amygdala, caudate nucleus, hippocampus, pallidum, putamen, and thalamus) produced by the Enhancing Neuro Imaging Genetics through Meta-Analysis (ENIGMA) consortium33. This ENIGMA2 discovery sample included 13,171 subjects of European ancestry and contained association results between seven million markers and variance in the volumes of the mentioned structures33; we applied the Bonferroni correction (p ≤ 1E−03; 0.05/50 SNPs).
Results
We investigated the possible association with ADHD of SNPs that show ASM in brain regions. Starting from two previous studies21,22 that describe ASM in brain tissues we obtained 43,132 SNP–CpG pairs involving 33,944 SNPs and 5306 CpG sites (Figs. 1 and S1). Genetic risk variants for ADHD are enriched in those ASM SNPs, as observed through enrichment analysis at different association p-value thresholds (Table S1).
We detected some overlaps and redundancies between studies and tissues (Fig. S1), so we performed a selection process ending up with a list of 3896 ASM tagSNPs (Figs. 1 and S2). Eight ASM tagSNPs were significantly associated with ADHD after correcting for multiple comparisons (5% FDR, p ≤ 6.78E−05) (Fig. 1 and Table S2). These eight SNPs correlated with differential methylation at six CpG sites in cis (three for cg20225915, two for both cg22930187 and cg06207804, and one for each of cg13047596, cg11554507, and cg04464446) in different brain areas (Figs. 2–4 and regional associational plots Figs. S3–S10, Table S2). Three of the eight ASM tagSNPs remained associated with ADHD after applying the Bonferroni and GEC corrections, all correlating with differential methylation at the cg20225915 site (Table S2).

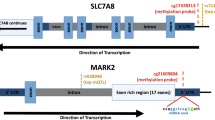
Genes are depicted in dark blue, showing the direction of transcription with an arrow; CpG sites inspected in the reference studies appear in brown; framed CpG sites indicate those sites showing differential levels of methylation for the associated ASM SNPs, and brown arrows indicate the effect on methylation of the ADHD risk variants, with indication of the brain regions where the ASMs were described. The tagSNPs are underscored. The colored rhombuses show the LD blocks present in each region. The colored dots for eQTLs indicate the effect on gene expression of the ADHD risk allele, according to the legend (red: over-expression, blue: under-expression). The number of enhancer (H3K4me1 and H3K27ac) and promoter (H3K4me3 and H3K9ac) histone marks found in the different brain areas are displayed for each SNP. ‘-' indicates no known enhancer or promoter histone marks

Genes are depicted in dark blue, showing the direction of transcription with an arrow; CpG sites inspected in the reference studies appear in brown; framed CpG sites indicate those sites showing differential levels of methylation for the associated ASM SNPs, and brown arrows indicate the effect on methylation of the ADHD risk variants, with indication of the brain regions where the ASMs were described. The tagSNPs are underscored. The colored rhombuses show the LD blocks present in each region. The colored dots for eQTLs indicate the effect on gene expression of the ADHD risk allele, according to the legend (red: over-expression, blue: under-expression). The number of enhancer (H3K4me1 and H3K27ac) and promoter (H3K4me3 and H3K9ac) histone marks found in the different brain areas are displayed for each SNP. ‘-' indicates no known enhancer or promoter histone marks

Genes are depicted in dark blue, showing the direction of transcription with an arrow; CpG sites inspected in the reference studies appear in brown; framed CpG sites indicate those sites showing differential levels of methylation for the associated ASM SNPs, and brown arrows indicate the effect on methylation of the ADHD risk variants, with indication of the brain regions where the ASMs were described. The tagSNPs are underscored. The colored rhombuses show the LD blocks present in each region. The colored dots for eQTLs indicate the effect on gene expression of the ADHD risk allele, according to the legend (red: over-expression, blue: under-expression). The number of enhancer (H3K4me1 and H3K27ac) and promoter (H3K4me3 and H3K9ac) histone marks found in the different brain areas are displayed for each SNP. ‘-' indicates no known enhancer or promoter histone marks
As considering only tagSNPs may overlook true causal variants, we retrieved association results from all the 52 ASM SNPs tagged by the previous ones (LD; r2 ≥ 0.85), ending up with 60 variants in eight LD blocks that show association with ADHD and correlate with methylation levels at six CpG sites (Figs. 2–4 and S11–S15 and Table S3). We also selected, for each LD block, the SNP showing the highest number of functional annotations (Table 1), as a putative causal SNP.
Consistently, the direction of the effect of the risk alleles on methylation levels is the same for all the SNPs correlating with the same CpG site. Thus, the risk alleles correlate with decreased methylation of cg22930187, cg06207804, cg11554507 and cg20225915 and with increased methylation of cg13047596 and cg0446444621,22 (Figs. 2–4 and Tables 1, S2, and S3).
All six CpG sites are located in possible promoter regions (<5000 bp upstream from a TSS) of six genes (Table 1), all of them expressed in brain: ARTN (cg22930187 and cg06207804), C2orf82 (cg13047596), NEUROD6 (cg11554507), PIDD1 (cg20225915), RPLP2 (cg20225915), and GAL (cg04464446) (Figs. 2–4). Furthermore, 85% of the 60 ASM SNPs are located within a region with enhancer or promoter histone marks in at least one brain area (Figs. 2–4 and Tables S4–S8). All putative causal SNPs selected from each LD block lie within a region with histone marks, ranging from 3 to 17 in enhancer regions and from 4 to 16 in promoter regions (Table 1).
We subsequently assessed the possible effect of those 60 SNPs on gene expression and observed that 57 of them are eQTLs for different genes in brain regions (Table S3). Seven out of the eight putative causal SNPs are eQTLs in brain for at least one gene (Table 1). We focused on methylation in promoter regions, which is well established to inversely correlate with gene expression. The eQTLs for ARTN, C2orf82, and PIDD1 correlated with methylation of CpG sites lying on their possible promoter regions, showing opposite directions for methylation and gene expression levels (Figs. 2–4 and Tables 1 and S3). The ADHD risk alleles are associated with increased expression of ARTN (in cerebellum and a subcortical region) and PIDD1 (in cerebellum and cortex) and with decreased expression of C2orf82 (in cortical, subcortical, and cerebellar regions) (Figs. 2–4 and Tables 1 and S3).
Consistently, the predicted direction of the effect on gene expression for these three genes is the same when we consider all variants within ±1 MB from the TSS (and not only the ASM SNPs). We found significant associations of gene expression with ADHD for the same three genes in multiple brain tissues using MetaXcan: ARTN, PIDD1 showed increased expression (3.57 < Z-score <4.19 and 3.57 < Z-score < 5.37, respectively) and C2orf82 with a decreased expression (−3.64 < Z-score < −3.07) (Table S7), all of them surviving the Bonferroni correction.
We also evaluated the correlation of the 60 ADHD-associated SNPs with subcortical brain volume changes in ENIGMA2 data. SNPs correlating with methylation at cg13047596 and at cg04464446 correlate with nucleus accumbens and/or caudate nucleus volumes, while the only SNP correlating with cg11554507, which is present in ENIGMA2, correlates with thalamus volume (Table S10). Three of the putative causal SNPs showed correlation with brain volumes (Table 1).
Interestingly, the majority of ASM SNPs that correlate with methylation levels of cg13047596, located in the promoter region of C2orf82, are eQTLs in brain for this gene, lie in a region with histone marks and correlate with volume changes of nucleus accumbens and caudate nucleus (Figs. 2–4, Tables 1 and S3–S8 and S10). All this functional evidence highlights the C2orf82 gene as a good candidate for contributing to ADHD.
Discussion
This study is the first comprehensive assessment of the contribution to ADHD of genetic variants altering methylation in the brain. We identified a total of 60 variants from eight LD blocks associated with ADHD that correlate with differential levels of methylation at six different CpG sites21,22 (Tables 1 and S3). All the variants from six out of the eight LD blocks alter the methylation of CpG sites lying at potential promoter regions and are also eQTLs for one of the following three genes in multiple brain regions: ARTN, C2orf82, and PIDD1 (Figs. 2–4 and Tables 1 and S3). It is well known that DNA methylation in promoter regions inversely correlates with levels of gene expression18, and all these ASM variants associated with ADHD in our study are concordant with this statement.
The ARTN gene, highlighted by two tagSNPs, encodes Artemin, a ligand of the GDNF family (glial cell line-derived neurotrophic factor). Artemin supports the survival of sensory and sympathetic peripheral neurons in culture by interacting with GFRα3-RET and possibly also of dopaminergic neurons of the ventral mid-brain through activation of GFRα1-RET complex34. Gene Ontology (GO) pathways link it to key neurodevelopmental functions: axon guidance (GO:0007411), neuroblast proliferation (GO:0007405), and peripheral nervous system development (GO:0007422). Risk alleles for ADHD lead to an overexpression of ARTN. Previously, overexpression of ARTN has been studied in transgenic mice and been linked to an increase of neuron excitability that leads to hypersensitivity35,36. Another study in ARTN knockout mice reported aberrations in the sympathetic nervous system related to migration and axonal projection37. The C2orf82 gene (also known as SNORC) was highlighted by one tagSNP and it encodes a proteoglycan transmembrane protein that is expressed in brain more than in other tissues30. Little is known about its function. Finally, PIDD1 was highlighted by three tagSNPs. It is a cell life regulator gene and it has been linked to apoptotic and anti-apoptotic pathways. The PIDD protein initiates apoptosis as a component of the PIDDosome together with RAIDD (RIP-associated ICH-1/ECD3-homologous protein with a death domain) and procaspase-238 and it also activates an anti-apoptotic pathway involving the transcription factor NF-κB in response to genotoxic stress39.
Alterations in the expression of these three genes (upregulation of ARTN and PIDD1 and downregulation of C2orf82) in different brain regions seem to be related to ADHD. Interestingly, most of these regions are relevant for this disorder. Neuroimaging studies have implicated the cerebellum, subcortical and prefrontal regions in ADHD, suggesting a link to problems in the processing of temporal information40. Structural anomalies in the cerebellum have been reported in ADHD individuals through neuroimaging studies41,42,43. Cerebellar developmental trajectories and hippocampal volumes are linked to the severity of ADHD symptoms44,45,46. Structural and functional abnormalities in cerebellum and basal ganglia have been associated with motor impairments47, which are frequent in nearly half of ADHD cases48. Subcortical regions identified through our expression analyses have also been related to ADHD, for instance: (i) remarkably different shapes of caudate-putamen basal ganglia and smaller volumes have been reported in ADHD boys49,50,51,52; (ii) in adult males with ADHD, right caudate volume correlates with poor accuracy on sensory selection tasks53 and also with hyperactivity/impulsivity54,55; (iii) nucleus accumbens, caudate nucleus, putamen, amygdala, and hippocampus are structurally altered in the brains of ADHD patients56. Remarkably, all the ASM SNPs in the LD block for C2orf82 with available information nominally correlate with increased volumes of nucleus accumbens and caudate nucleus subcortical regions. Also, the eQTL effect sizes of these SNPs are the largest for caudate basal ganglia, which volume correlates with the SNP genotype variation. There is evidence about the role in ADHD of cortical thickness, cortical volume and functional connectivity in the anterior cingulate cortex, a region involved in cognitive control, attention, affect and drive57,58,59,60,61,62,63. Furthermore, delayed cortical development, e.g. in prefrontal regions has been reported in ADHD patients64,65 and this appears to be stronger in ADHD children with below median intelligence quotient66. All the above mentioned fronto-subcortical structures and pathways are rich in catecholamines, the molecular targets in pharmacological treatments for ADHD48,52,64,67.
Interestingly, the methylation of cg20225915 has also been associated with PIDD1 expression in peripheral blood68, turning it into a good candidate as a biomarker. The expression of ARTN was found to be altered in blood of major depressive disorder (MDD) patients69 and the C2orf82 gene has been associated to schizophrenia70,71. Furthermore, C2orf82 was highlighted in a cross-disorder GWAS of eight psychiatric conditions, including ADHD and schizophrenia72, with the rs778353 lead SNP, located 47 kb downstream from the gene, showing a genome-wide significant association with the phenotype. All three genes overlap with several CNVs that contribute to autism, intellectual disability or aggressive behavior, conditions often comorbid with ADHD (Table S11). It is noteworthy that some of the CNVs reported in ARTN, C2orf82, and PIDD1 are related to brain-specific and overall developmental delay at both fetal and postnatal stages. Thus, it is reasonable to assume that altered expression of these genes might affect brain volumes and cognition. Overall, the fact that these genes have previously been related to neuropsychiatric disorders that are often comorbid with ADHD73 make them appealing candidates to be pursued.
ARTN is the only gene highlighted in our study that is present in one of the top regions reported in the ADHD GWAS meta-analysis4, although it did not contain SNPs surviving genome-wide significance. The GWAS findings in the region could be accounted for by one of several genes: ST3GAL3, PTPRF, KDM4A, RP11-184I16.4, XR_246316.1, KDM4A-AS1, and SLC6A9. ST3GAL3 had the most signals. Although two of the reported ASM variants associated with ADHD are intronic to ST3GAL3, this gene was not highlighted in our study as none of the associated variants correlated with differential methylation of CpG sites near the ST3GAL3 TSS (distance from the nearest CpG site: 197 kb) or were eQTLs for the gene in brain tissues. Instead, these SNPs correlated with a nearby gene, ARTN, both in terms of methylation and gene expression. This suggests the importance of finding functional connections between disease-associated SNPs and genes, besides considering the genes in the physical vicinity of variants. Furthermore, another of the highlighted genes, PIDD1, although not being among the top findings in the ADHD GWAS meta-analysis4, it is pointed out by the gene-based association analysis performed in the same study.
Genetic variants surpassing genome-wide significance in GWAS explain only a small part of the SNP-based heritability and associations not reaching the significance threshold also contribute to disease susceptibility4,9. An omnigenic model has been recently proposed suggesting that the sub-threshold variants could point at regulatory elements of core genes7,8. Indeed, a previous study on a cardiovascular cardiac phenotype reported that nominally significant associations are enriched in enhancer regions9, consistent with our findings. Therefore, although none of the variants that we identified in our study display genome-wide significant association with ADHD, they may contribute to the susceptibility to ADHD, as they do have a functional impact (methylation, expression, and in some cases brain structure) via genes that are expressed in brain.
Brain-specific ASM information has also been utilized to detect key genes and pathways in BD20. Also, a higher enrichment of brain ASM was observed in a schizophrenia GWAS in comparison to non-psychiatric GWAS10. This, together with the enrichment of ASM in ADHD-associated variants found in the present study, reinforces the rationale of utilizing ASM SNPs to highlight genes that are relevant to psychiatric disorders from GWAS data.
There are some strengths and limitations in our study that should be discussed. Strengths: (i) We used the largest GWAS meta-analysis of ADHD performed so far, including around 20,000 cases and 35,000 controls. (ii) The genetic variants identified as associated with ADHD have a functional impact on epigenetic regulation, expression or brain volumes. (iii) Two of the highlighted genes in this study, ARTN and C2orf82, have previously been associated with other psychiatric disorders. (iv) For two of the genes there is more than one LD block showing the same effect on CpG site methylation. (v) Our results are concordant with eQTL information that had been assessed in an independent sample, with all the SNPs showing the opposite effect on methylation of the promoter region and on the expression of a given gene in brain (more promoter methylation and less gene expression or vice versa), even for the different LD blocks from each region. Limitations: (i) We did not perform a follow-up study to replicate the association findings in an independent sample. (ii) The previous studies that we used for the selection of ASM SNPs were performed on different genotyping platforms that do not include all the existing SNPs in the genome, and therefore we could not test all possible ASMs. (iii) We only considered cis-associated ASM variants, which are the vast majority, although non-cis ASM also occurs. (iv) There is an overrepresentation of ASM SNPs from cerebellum compared to the other studied tissues.
To conclude, the present study points to the ARTN, C2orf82, and PIDD1 genes as potential contributors to ADHD susceptibility. The identified risk variants have an impact on the methylation levels of different CpG sites located in promoter regions and they inversely correlate with expression of the corresponding genes in brain. This finding is supported by a prediction of increased expression of ARTN and PIDD1, and a decreased expression of C2orf82 in ADHD. Moreover, variants correlating with methylation at cg13047596 (near C2orf82) influence the volumes of nucleus accumbens and/or caudate nucleus. Further studies are required to elucidate the mechanisms by which these genes contribute to ADHD.
References
Polanczyk, G., De Lima, M. S., Horta, B. L., Biederman, J. & Rohde, L. A. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am. J. Psychiatry 164, 942–948 (2007).
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Arlington, VA, US: American Psychiatric Publishing, Inc. (2013).
Faraone, S. V. et al. Molecular genetics of attention-deficit/hyperactivity disorder. Biol. Psychiatry 57, 1313–1323 (2005).
Demontis, D. et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 51, 63–75 (2019).
Anttila, V. et al. Analysis of shared heritability in common disorders of the brain. Science 360, eaap8757 (2018).
Meaburn, E. L., Schalkwyk, L. C. & Mill, J. Allele-specific methylation in the human genome: implications for genetic studies of complex disease. Epigenetics 5, 578–582 (2010).
Boyle, E. A., Li, Y. I. & Pritchard, J. K. An expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177–1186 (2017).
Liu, X., Li, Y. I. & Pritchard, J. K. Trans effects on gene expression can drive omnigenic inheritance. Cell 177, 1022–1034 (2019).
Wang, X. et al. Discovery and validation of sub-threshold genome-wide association study loci using epigenomic signatures. Elife 5, e10557 (2016).
Gagliano, S. A. et al. Allele-skewed DNA modification in the brain: relevance to a Schizophrenia GWAS. Am. J. Hum. Genet. 98, 956–962 (2016).
Ladd-Acosta, C. et al. DNA methylation signatures within the human brain. Am. J. Hum. Genet. 81, 1304–1315 (2007).
Jang, H. S., Shin, W. J., Lee, J. E. & Do, J. T. CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes 8, E148 (2017).
Xu, Y. et al. Multiple epigenetic factors predict the attention deficit/hyperactivity disorder among the Chinese Han children. J. Psychiatr. Res. 64, 40–50 (2015).
Schanen, N. C. Epigenetics of autism spectrum disorders. Hum. Mol. Genet. 15, R138–R150 (2006).
Grayson, D. R. & Guidotti, A. Merging data from genetic and epigenetic approaches to better understand autistic spectrum disorder. Epigenomics 8, 85–104 (2016).
Dammann, G. et al. Increased DNA methylation of neuropsychiatric genes occurs in borderline personality disorder. Epigenetics 6, 1454–1462 (2011).
Gaur, U., Li, K., Mei, S. & Liu, G. Research progress in allele-specific expression and its regulatory mechanisms. J. Appl. Genet. 54, 271–283 (2013).
Li, E. & Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 6, a019133 (2014).
Do, C. et al. Genetic-epigenetic interactions in cis: a major focus in the post-GWAS era. Genome Biol. 18, 120 (2017).
Chuang, L.-C., Kao, C.-F., Shih, W.-L. & Kuo, P.-H. Pathway analysis using information from allele-specific gene methylation in genome-wide association studies for bipolar disorder. PLoS ONE 8, e53092 (2013).
Gibbs, J. R. et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 6, e1000952 (2010).
Zhang, D. et al. Genetic control of individual differences in gene-specific methylation in human brain. Am. J. Hum. Genet. 86, 411–419 (2010).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Barrett, J. C. Haploview: visualization and analysis of snp genotype. Data. Cold Spring Harb. Protoc. 4, 1–5 (2009).
The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015).
R Core Team. R: A Language and Environment for Statistical Computing. https://www.r-project.org/ (2014).
Storey, John D. with contributions from Bass, Andrew J. AD and, Robinson D. qvalue: Q-value estimation for false discovery rate control. R Package Version 2.6.0. http://github.com/jdstorey/qvalue (2015).
Li, M.-X., Yeung, J. M. Y., Cherny, S. S. & Sham, P. C. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet. 131, 747–756 (2012).
Ward, L. D. & Kellis, M. HaploRegv4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 44, D877–D881 (2016).
GTEx Consortium et al. The Genotype-Tissue Expression (GTEx) project. Nat. Publ. Gr. 45, 580–585 (2013).
Barbeira, A. N. et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 9, 1825 (2018).
Fromer, M. et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 19, 1442–1453 (2016).
Hibar, D. P. et al. Common genetic variants influence human subcortical brain structures. Nature 520, 224–229 (2015).
Baloh, R. H. et al. Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRα3-RET receptor complex. Neuron 21, 1291–1302 (1998).
Elitt, C. M. et al. Artemin overexpression in skin enhances expression of TRPV1 and TRPA1 in cutaneous sensory neurons and leads to behavioral sensitivity to heat and cold. J. Neurosci. 26, 8578–8587 (2006).
Elitt, C. M., Malin, S. A., Koerber, H. R., Davis, B. M. & Albers, K. M. Overexpression of artemin in the tongue increases expression of TRPV1 and TRPA1 in trigeminal afferents and causes oral sensitivity to capsaicin and mustard oil. Brain Res. 1230, 80–90 (2008).
Honma, Y. et al. Artemin is a vascular-derived neurotropic factor for developing sympathetic neurons. Neuron 35, 267–282 (2002).
Tinel, A. & Tschopp, J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 304, 843–846 (2004).
Janssens, S., Tinel, A., Lippens, S. & Tschopp, J. PIDD mediates NF-kappaB activation in response to DNA damage. Cell 123, 1079–1092 (2005).
Toplak, M. E., Dockstader, C. & Tannock, R. Temporal information processing in ADHD: findings to date and new methods. J. Neurosci. Methods 151, 15–29 (2006).
Berquin, P. C. et al. Cerebellum in attention-deficit hyperactivity disorder: a morphometric MRI study. Neurology 50, 1087–1093 (1998).
Castellanos, F. X. et al. Quantitative brain magnetic resonance imaging in attention-deficit hyperactivity disorder. Arch. Gen. Psychiatry 53, 607–616 (1996).
Castellanos, F. X. et al. Quantitative brain magnetic resonance imaging in girls with attention-deficit/hyperactivity disorder. Arch. Gen. Psychiatry 58, 289–295 (2001).
Mackie, S. et al. Cerebellar development and clinical outcome in attention deficit hyperactivity disorder. Am. J. Psychiatry 164, 647–655 (2007).
Plessen, K. J. et al. Hippocampus and amygdala morphology in attention-deficit/hyperactivity disorder. Arch. Gen. Psychiatry 63, 795–807 (2006).
Friedman, L. A. & Rapoport, J. L. Brain development in ADHD. Curr. Opin. Neurobiol. 30, 106–111 (2015).
Pasini, A. & D’agati, E. Pathophysiology of NSS in ADHD. World J. Biol. Psychiatry 10, 495–502 (2009).
Curatolo, P., D’Agati, E. & Moavero, R. The neurobiological basis of ADHD. Ital. J. Pediatr. 36, 79 (2010).
Qiu, A. et al. Basal ganglia volume and shape in children with attention deficit hyperactivity disorder. Am. J. Psychiatry 166, 74–82 (2009).
Greven, C. U. et al. Developmentally stable whole-brain volume reductions and developmentally sensitive caudate and putamen volume alterations in those with attention-deficit/hyperactivity disorder and their unaffected siblings. JAMA Psychiatry 72, 490–499 (2015).
Sobel, L. J. et al. Basal ganglia surface morphology and the effects of stimulant medications in youth with attention deficit hyperactivity disorder. Am. J. Psychiatry 167, 977–986 (2010).
Rubia, K., Alegría, A. A. & Brinson, H. Brain abnormalities in attention-deficit hyperactivity disorder: a review. Rev. Neurol. 58(Suppl. 1), S3–S16 (2014).
Casey, B. J. et al. Implication of right frontostriatal circuitry in response inhibition and attention-deficit/hyperactivity disorder. J. Am. Acad. Child Adolesc. Psychiatry 36, 374–383 (1997).
Singh, A., Yeh, C. J., Verma, N. & Das, A. K. Overview of attention deficit hyperactivity disorder in young children. Heal Psychol. Res. 3, 23–35 (2015).
Onnink, A. M. H. et al. Brain alterations in adult ADHD: Effects of gender, treatment and comorbid depression. Eur. Neuropsychopharmacol. 24, 397–409 (2014).
Hoogman, M. et al. Subcortical brain volume differences in participants with attention deficit hyperactivity disorder in children and adults: a cross-sectional mega-analysis. Lancet Psychiatry 4, 310–319 (2017).
Shaw, P. et al. Longitudinal mapping of cortical thickness and clinical outcome in children and adolescents with attention-deficit/hyperactivity disorder. Arch. Gen. Psychiatry 63, 540–549 (2006).
Ambrosino, S., de Zeeuw, P., Wierenga, L. M., van Dijk, S. & Durston, S. What can cortical development in attention-deficit/hyperactivity disorder teach us about the early developmental mechanisms involved? Cereb. Cortex 27, 4624–4634 (2017).
Castellanos, F. X. & Aoki, Y. Intrinsic functional connectivity in attention-deficit/hyperactivity disorder: a science in development. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 1, 253–261 (2016).
Makris, N. et al. Cortical thinning of the attention and executive function networks in adults with attention-deficit/hyperactivity disorder. Cereb. Cortex 17, 1364–1375 (2007).
Makris, N. et al. Anterior cingulate volumetric alterations in treatment-naïve adults with ADHD: a pilot study. J. Atten. Disord. 13, 407–413 (2010).
Yang, X.-R., Carrey, N., Bernier, D. & MacMaster, F. P. Cortical thickness in young treatment-naive children with ADHD. J. Atten. Disord. 19, 925–930 (2015).
Qiu, M. et al. Changes of brain structure and function in ADHD children. Brain Topogr. 24, 243–252 (2011).
Cortese, S. The neurobiology and genetics of Attention-Deficit/Hyperactivity Disorder (ADHD): what every clinician should know. Eur. J. Paediatr. Neurol. 16, 422–433 (2012).
Shaw, P. et al. Attention-deficit/hyperactivity disorder is characterized by a delay in cortical maturation. Proc. Natl Acad. Sci. USA 104, 19649–19654 (2007).
de Zeeuw, P. et al. Differential brain development with low and high IQ in attention-deficit/hyperactivity disorder. PLoS ONE 7, e35770 (2012).
Pliszka, S. R., Lancaster, J., Liotti, M. & Semrud-Clikeman, M. Volumetric MRI differences in treatment-naive vs chronically treated children with ADHD. Neurology 67, 1023–1027 (2006).
Wu, Y. et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun. 9, 918 (2018).
Otsuki, K. et al. Altered expression of neurotrophic factors in patients with major depression. J. Psychiatr. Res. 42, 1145–1153 (2008).
Schizophrenia Working Group of the Psychiatric Genomics. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Ripke, S. et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 45, 1150–1159 (2013).
Lee, P. H. et al. Genome wide meta-analysis identifies genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. bioRxiv. 528117 (2019).
Brown T. E. ADHD Comorbidities: Handbook for ADHD Complications in Children and Adults. Washington, DC, US: American Psychiatric Press (2009).
Acknowledgements
Major financial support for this research was received by B.C. from the Spanish ‘Ministerio de Economía y Competitividad’ (SAF2015-68341-R, RTI2018-100968-B-I00) and AGAUR, ‘Generalitat de Catalunya’ (2017-SGR-738). The research leading to these results has also received funding from the European Union Seventh Framework Program [FP7/2007–2013] under grant agreement no. 602805 and from the European Union H2020 Program [H2020/2014-2020] under grant agreements nos. 667302 and 643051, the latter supporting the contract of A.S. L.P.-C. and J.C.-D. were supported by ‘Generalitat de Catalunya’ (2016 FI_B 00728 and 2015 FI_B 00448, respectively). L.P.-C. was also supported by ‘Ministerio de Educación, Cultura y Deporte’ (FPU15/03867). N.F.-C. was supported by contracts of the ‘Centro de Investigación Biomédica en Red de Enfermedades Raras’ (CIBERER). V.R. was supported by the Graduate School of Health from the University of Aarhus. The iPSYCH team acknowledges support from the Lundbeck Foundation. Finally, S.F. was supported by the European Union’s Seventh Framework Program for research, technological development and demonstration under grant agreement no. 602805, the European Union’s Horizon 2020 research and innovation program under grant agreements nos. 667302 and 728018 and NIMH grants 5R01MH101519 and U01 MH109536-01. We are thankful to Roser Corominas (Universitat de Barcelona) for helpful advice. We are also grateful to the ADHD Working Group of the Psychiatric Genomics Consortium (PGC) and the iPSYCH team for distributing the summary statistics of the ADHD GWAS meta-analysis. This study is part of the International Multicentre persistent ADHD Collaboration (IMpACT); www.impactadhdgenomics.com). IMpACT unites major research centres working on the genetics of ADHD persistence across the lifespan and has participants in The Netherlands, Germany, Spain, Norway, the United Kingdom, the United States, Brazil and Sweden. Principal investigators of IMpACT are: Barbara Franke (chair), Andreas Reif (co-chair), Stephen V. Faraone, Jan Haavik, Bru Cormand, J. Antoni Ramos-Quiroga, Marta Ribasés, Philip Asherson, Klaus-Peter Lesch, Jonna Kuntsi, Claiton H.D. Bau, Jan Buitelaar, Alejandro Arias-Vásquez, Tetyana Zayats, Henrik Larsson, Alysa Doyle, and Eugenio H. Grevet. Access to the PGC ADHD data was obtained through dbGaP project number 10608 that includes the following datasets: phs000016.v2.p2, phs000407.v1.p1, phs000358.v1.p1, and phs000490.v1.p1. We thank the ENIGMA consortium for sharing the summary statistics of genome-wide association meta-analyses of MRI phenotypes.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pineda-Cirera, L., Shivalikanjli, A., Cabana-Domínguez, J. et al. Exploring genetic variation that influences brain methylation in attention-deficit/hyperactivity disorder. Transl Psychiatry 9, 242 (2019). https://doi.org/10.1038/s41398-019-0574-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-019-0574-7
This article is cited by
-
DNA methylation signatures in autism spectrum disorders
The Nucleus (2021)
-
Biallelic mutations in the death domain of PIDD1 impair caspase-2 activation and are associated with intellectual disability
Translational Psychiatry (2021)
-
Biochemical characteristics of the chondrocyte-enriched SNORC protein and its transcriptional regulation by SOX9
Scientific Reports (2020)
-
Prenatal Risk Factors for Tourette Syndrome: a Systematic Review Update
Current Developmental Disorders Reports (2020)
