
Abstract
Integrative approaches that harness large-scale molecular datasets can help develop mechanistic insight into findings from genome-wide association studies (GWAS). We have performed extensive analyses to uncover transcriptional and epigenetic processes which may play a role in complex trait variation. This was undertaken by applying Bayesian multiple-trait colocalization systematically across the genome to identify genetic variants responsible for influencing intermediate molecular phenotypes as well as complex traits. In this analysis, we leveraged high-dimensional quantitative trait loci data derived from the prefrontal cortex tissue (concerning gene expression, DNA methylation and histone acetylation) and GWAS findings for five complex traits (Neuroticism, Schizophrenia, Educational Attainment, Insomnia and Alzheimer’s disease). There was evidence of colocalization for 118 associations, suggesting that the same underlying genetic variant influenced both nearby gene expression as well as complex trait variation. Of these, 73 associations provided evidence that the genetic variant also influenced proximal DNA methylation and/or histone acetylation. These findings support previous evidence at loci where epigenetic mechanisms may putatively mediate effects of genetic variants on traits, such as KLC1 and schizophrenia. We also uncovered evidence implicating novel loci in disease susceptibility, including genes expressed predominantly in the brain tissue, such as MDGA1, KIRREL3 and SLC12A5. An inverse relationship between DNA methylation and gene expression was observed more than can be accounted for by chance, supporting previous findings implicating DNA methylation as a transcriptional repressor. Our study should prove valuable in helping future studies prioritize candidate genes and epigenetic mechanisms for in-depth functional follow-up analyses.
Similar content being viewed by others

Background
Genome-wide association studies (GWAS) have been integral in identifying thousands of genetic variants associated with complex traits and disease. The vast majority of genetic variants identified in these studies reside in intergenic or intronic regions of the genome and are therefore predicted to exert their effects on complex traits via changes in gene regulation1. Furthermore, there is evidence which suggests that GWAS hits are often located within regions of open chromatin and enhancers2. Typically, genetic variants associated with molecular phenotypes are enriched amongst single-nucleotide polymorphisms (SNPs) that are linked to traits and diseases3. Such variants are known as quantitative trait loci (QTL) and can affect molecular phenotypes such as: gene expression (eQTL), and epigenetic mechanisms including DNA methylation (mQTL) and histone acetylation (haQTL). DNA methylation and histone acetylation are alterations that affect gene expression without altering the DNA sequence. Several genetic variants have been identified that occur in the same genomic region and influence both gene expression and DNA methylation. In these cases, it is possible that the eQTL and mQTL share a common causal variant (CCV)4.
Several post-GWAS approaches exist to help functionally characterize non-coding variants5,6,7. In particular, there has been an emphasis on integrating eQTL and GWAS data together, which can be valuable in terms of identifying the underlying genes responsible for associations detected by GWAS. Recently, similar endeavours have extended the scope of their analysis to also evaluate additional molecular phenotypes (e.g. mQTL and haQTL) as well as gene expression8,9,10,11. A novel method in this paradigm involves calculating approximate Bayes factors12 to assess the likelihood that the genetic variant responsible for an association with a complex trait is also responsible for influencing intermediate molecular phenotypes (i.e. the likelihood they share a CCV). This multiple-trait colocalization (moloc) method has been shown to help characterize GWAS loci and develop mechanistic insight into the causal pathway from genetic variant to complex trait13. Furthermore, inclusion of an additional molecular trait into the analysis (e.g. complex trait, gene expression and DNA methylation vs. complex trait and gene expression alone) has been shown to increase power and assist in identifying novel disease susceptibility loci13.
The recent large influx of tissue-specific molecular data provides an unprecedented opportunity to assess the functional relevance of GWAS hits. Recently, a resource has become available that comprises QTL data derived from the dorsolateral prefrontal cortex in up to 494 subjects14. Brain xQTL Serve provides a list of SNPs associated with gene expression, DNA methylation and/or histone modifications specific to the same brain region14. Whilst progress has been made in terms of identifying genetic variants influencing neurological phenotypes and diseases, not enough is known about the biological effects of genetic risk factors. In this study, we have jointly analysed genetic variants identified across GWAS of five complex traits and diseases alongside the variants listed in the Brain xQTL Serve resource. In doing so, we aim to identify CCVs for complex traits and gene expression, and where possible, DNA methylation and histone acetylation. We selected a range of neurological, psychiatric, personality and behavioural traits (Neuroticism, Schizophrenia, Educational Attainment, Insomnia and Alzheimer’s disease) with publicly available large GWAS summary statistics, where we believed QTL data specific to the brain may be particularly biologically relevant. Uncovering evidence that epigenetic factors reside on the causal pathway along with gene expression can be extremely valuable for disease prevention due to early diagnosis. Additionally, the use of genetic evidence can be beneficial for identifying and selecting drug targets15.
Methods
Genome-wide association studies
We obtained summary statistics from five independent GWAS for the following complex traits: Neuroticism (n = 274,108)16, Schizophrenia (cases = 35,467, controls = 46,839)17, Educational Attainment (n = 293,723)18, Alzheimer’s disease (cases = 17,008, controls = 37,154)19 and Insomnia (n = 336,965)16. Information on all GWAS datasets can be found in Supplementary Table 1. Linkage disequilibrium (LD) clumping was undertaken using PLINK v1.920 with a reference panel consisting of European (CEU) individuals from phase 3 (version 5) of the 1000 genomes project21. This allowed us to identify the top independent loci for each set of results based on the conventional GWAS threshold (P < 5.0 × 10−08).
Brain tissue-derived QTL for three molecular phenotypes
All QTL data used in this study were obtained from the Brain xQTL Serve resource14. Genotype data in this resource was generated from 2093 individuals of European descent from the Religious Orders Study (ROS and Memory and Aging Project study cohorts (http://www.radc.rush.edu/). Gene expression (RNA-sequencing (RNA-seq); n = 494), DNA methylation (450 K Illumina array; n = 468) and histone modification (H3K9Ac ChIP-seq; n = 433) data were derived from the dorsolateral prefrontal cortex of post-mortem samples. eQTL were based on 13,484 expressed genes, mQTL on 420,103 methylation sites and haQTL on 26,384 acetylation domains. eQTL and haQTL results were available for variants within 1 Mb of their corresponding probes, whereas mQTL results were restricted to a 5 kb window14.
Gene-centric multiple-trait colocalization
We extracted effect estimates for all variants within 1 Mb of the lead SNP (i.e. P < 5.0 × 10–08) for each clumped region using results from each of the five GWAS. P values for molecular QTL were then extracted for the same set of SNPs using the Brain xQTL resource. Loci residing within the major histocompatibility complex region (chr6: 25−35 Mb) were removed due to extensive LD within this region, which may result in false-positive findings. The moloc method was then used to assess the likelihood that the variant at each region responsible for variation in complex traits was also responsible for influencing the expression of a nearby gene (i.e. within a 1 Mb distance of the lead GWAS SNP). As demonstrated previously13, we simultaneously investigated whether variants responsible for both gene expression and complex trait variation may also influence proximal epigenetic traits in a gene-centric manner. However, unlike previous work, which evaluated three traits at a time, we have investigated up to four traits in each analysis (i.e. complex trait, gene expression, DNA methylation and histone acetylation).
To achieve this, we used coordinates from Ensembl22 to map CpG sites and histone peaks to genes using a 50-kb window upstream and downstream of each gene. We then ran moloc to assess all gene–CpG–histone combinations within each region of interest. Summed posterior probabilities were computed for all scenarios where GWAS trait and gene expression colocalized. The reason for this is because if epigenetic mechanisms are responsible for mediating the effect of genetic variants on complex traits, then we would expect gene expression to also reside on this causal pathway. Therefore, 10 scenarios were considered of interest: GE, GE,M, GE,H, GE,M,H, GEM, GEM,H, GEH, GEH,M, GEMH, where evidence of a shared causal variants for GWAS complex traits is defined as ‘G', gene expression as ‘E’, DNA methylation as ‘M’ and histone acetylation as ‘H’. The ‘,’ denotes a scenario where there is a different causal variant for each molecular phenotype. For example, GE,M would represent a situation where the same causal variant is shared between the GWAS trait and gene expression, but a different causal variant for DNA methylation.
As recommended by the authors of moloc13, a summed posterior probability of association (PPA) ≥80% for these 10 scenarios was considered strong evidence that a genetic variant was responsible for changes in both molecular phenotype(s) and complex trait variation. Therefore, a GEMH scenario with a posterior probability ≥80% would represent a case where there is evidence that GWAS trait, gene expression, DNA methylation and histone acetylation colocalize and share a causal variant. When a gene–trait combination provided evidence of colocalization with multiple CpG sites or histone peaks, we only reported the association for the combination with the highest PPA. This was to reduce the number of findings detected due to co-methylation/probes within the same histone peak that were measuring the same epigenetic signatures.
Regions with fewer than 50 common SNPs (minor allele frequency ≤5%) were not considered in the moloc analysis in order to reduce the number of spurious findings. Prior probabilities of 1 × 10−04, 1 × 10−06, 1 × 10−07 and 1 × 10−08 were used in all analyses, which was also recommended by the authors of moloc. Furthermore, we used the option to adjust Bayes factors for overlapping samples as this was the case for the xQTL datasets. Manhattan plots to illustrate findings were subsequently generated using code adapted from the ‘qqman’ package23.
Identifying potentially novel loci in disease susceptibility
We also applied our analytical pipeline as described above to independent GWAS loci with P values between the conventional threshold (P < 5.0 × 10−08) and P ≤ 1.0 × 10−06. All parameters were the same as in the previous analysis. We hypothesized that incorporating additional evidence on molecular phenotypes could help to elucidate potentially novel loci, which are likely to be identified as sample sizes of future GWAS increase. Although the observed effects of these loci on traits alone do not meet the conventional GWAS threshold, we took evidence of colocalization (again defined as a combined PPA ≥80%) at these loci as novel evidence implicating them in disease, which can be used to prioritize them for future evaluation.
Functional informatics
Pathway analysis
For all scenarios where GWAS trait and gene expression colocalize based on a combined PPA of ≥80%, we compiled a list of associated genes for each trait. Where multiple genes at a region provided a PPA ≥80% for the same GWAS SNP, we took forward the gene with the highest PPA. Pathway analysis was then undertaken with a gene list for each complex trait using ConsensusPathDB24. This was to investigate whether multiple associated genes in our analysis reside along established biological pathways more than we would expect by chance.
Tissue-specific analysis
We also investigated whether any genes detected in our analysis were predominantly expressed in the brain tissue using three RNA-seq datasets: the Human Protein Atlas (HPA)25, the Genotype-Tissue Expression project (GTEx)26 and the Mouse ENCODE project27. We used the ‘TissueEnrich’ R Package to identify evidence of enrichment based on three definitions:25
-
Tissue enriched: Genes with an expression level ≥1 TPM (transcripts per million) or FPKM (fragments per kilobase of exon model per million reads mapped) plus at least 5-fold higher expression levels in a particular tissue when compared to all other tissues.
-
Group enriched: Genes with an expression level ≥1 TPM or FPKM plus at least 5-fold higher expression levels in a group of 2–7 tissues when compared to other tissues not considered to be ‘Tissue enriched’.
-
Tissue enhanced: Genes with an expression level ≥1 TPM or FPKM plus at least 5-fold higher expression levels in a particular tissue compared to the average levels in all other tissues, and not considered to be either ‘Tissue enriched’ or ‘Group enriched’.
Evidence of enrichment under the ‘Tissue enriched’ definition therefore suggests that a gene is specifically expressed in a single tissue type. This suggests that associations between this gene and a complex trait may be tissue specific (i.e. they would only be detected using data derived from one specific tissue). Enrichment based on the ‘Group enriched’ definition suggests that associations may be detected in a set of tissue types (i.e. 2–7 different types), which are expressed at least 5-fold higher than all other tissues considered. Evidence detected using the ‘Tissue enhanced’ definition suggests that this gene is more ubiquitously expressed (in more than seven different tissues), although expression in a specific tissue type is considerably higher than the others (5-fold higher compared to the average of all others). As such, genes detected using this definition may be strongly dependent on using data derived from one specific tissue, although associations may still be detected in other tissue types (i.e. they are not ‘tissue specific’).
For each dataset, we were only interested in genes predominantly expressed in the brain tissue, that is, ‘Cerebral Cortex’ in HPA, ‘Brain’ in GTEx and ‘Cerebellum’, ‘Cortex’ or ‘E14.5 Brain’ in the Mouse ENCODE project. Heatmaps to illustrate enrichment across all possible tissues from these datasets were generated using the ‘ggplot’ R package’28.
Orienting directions of effect between molecular traits and regulatory region annotation
We oriented the direction of effect between transcriptional and epigenetic traits for detected associations: firstly, between gene expression and DNA methylation and then between gene expression and histone acetylation. For associations with evidence of colocalization between the two traits being assessed, we evaluated whether the lead SNP was correlated with molecular traits in the same direction using coefficients from the xQTL resource. We applied the hypergeometric test to investigate whether there was an enrichment of a particular direction of effect between molecular traits more than we would expect by chance. Background expectations were calibrated using randomly selected lead SNPs across the genome that were associated with both proximal gene expression and DNA methylation (P < 1.0 × 10−04). Permutation testing was applied for 10,000 iterations by sampling the same number of SNPs being evaluated.
Lastly, we obtained regulatory data from the Roadmap Epigenetics Project29 from 10 different types of the brain tissue. We used BEDtools30 to evaluate whether lead SNPs, CpG sites and histone peaks with evidence of colocalization from our study reside within promoters, enhancers and histone marks using these datasets. All analyses in this study were undertaken using R (version 3.3.1).
Results
Colocalization between gene expression, DNA methylation and histone acetylation at risk loci for five complex traits
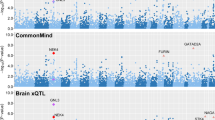
We applied the moloc method at loci with a trait-associated SNP (P < 5.0 × 10−08) using findings from five large-scale GWAS16,17,18,19 and molecular datasets (eQTL, mQTL and haQTL) derived from the brain tissue14. Across the five complex traits, we identified a total of 66 colocalization associations with GWAS loci and gene expression (Supplementary Tables 2–6). Of these, 40 provided evidence of colocalization with an epigenetic trait also. Altogether, four genetic loci colocalized with a complex trait and all three of the molecular phenotypes (gene expression, DNA methylation and histone acetylation). Figure 1 illustrates these associations for neuroticism and insomnia, whereas plots for the remaining traits can be located in Supplementary Figure 1.

Manhattan plots for a neuroticism and b insomnia. Shared causal variants with traits are represented for the following scenarios: gene expression (blue), gene expression and DNA methylation (red), gene expression and histone acetylation (green) and gene expression, DNA methylation and histone acetylation (yellow). For example, in a GNG12 is shown in yellow representing a case where there is a common causal variant (CCV) for neuroticism risk, expression of the gene GNG12, DNA methylation and histone acetylation at this locus. PAFAH1B1 is shown in red, indicating that there is a CCV for neuroticism risk, expression of the PAFAH1B1 gene and DNA methylation, but not for histone acetylation at this locus. The genome-wide significance threshold (P < 5 × 10−08) is illustrated in red
We identified evidence of colocalization between complex and molecular traits at loci previously reported as well as novel findings. For example, we were able to replicate findings reporting that the expression of KLC1 colocalizes with schizophrenia risk and DNA methylation13 (combined PPA = 97.9%). There were several other loci associated with schizophrenia that have been previously reported to colocalize with molecular traits (such as CNNM2 and PRMT713), as well as a several other genes where epigenetic mechanisms have not been previously detected to play a role in schizophrenia risk (such as TSNARE1 and ADOPT1) (Supplementary Table 3).
There were also novel associations with molecular phenotypes amongst the other complex traits. For instance, we uncovered evidence suggesting that neuroticism, gene expression and DNA methylation shared a CCV at the PAFAH1B1 locus (combined PPA = 89.9%). Figure 2a illustrates the overlapping distributions of effects on each of these traits for variants at this region. We also observed evidence of colocalization for several genes at the APOE locus that were associated with Alzheimer’s disease. This included TOMM40, where results suggested that there was also evidence of colocalization with DNA methylation (combined PPA = 99.3%). However, given the extensive LD at this region, findings should be interpreted with caution31.

Regional association plots illustrating colocalizations for the PAFAH1B1 gene (a) and MDGA1 gene (b) with neuroticism and insomnia, respectively. Effects for genetic variants on complex traits (plotted in grey) and gene expression (blue) were available within a 1 Mb distance of the lead variant at each locus, whereas effects on DNA methylation levels (red) were confined to a 5kb distance
Elucidating novel genes that may influence complex traits
We also applied our analytical pipeline to uncover potentially novel loci using a less stringent threshold (P ≤ 1.0 × 10−06). In this analysis, we identified 52 loci where complex traits and gene expression share a CCV, of which 33 provided evidence that these variants may also influence epigenetic traits (Fig. 1; Supplementary Figure 1). Our incentive for undertaking this analysis was that GWAS analyses may not identify evidence of association using observed effects on complex traits alone. However, by integrating evidence that SNPs at these loci also influence molecular traits derived from a relevant tissue type, we aimed to uncover novel loci in disease/trait variation. Table 1 provides an overview of the number of associations detected in our analysis.
As an example of this, there was evidence that insomnia risk and molecular traits share a CCV at the MDGA1 locus (combined PPA = 85.8%). However, the P value for the lead SNP at this region did not reach conventional GWAS thresholds (P = 7.7 × 10−07), suggesting that it would have been potentially overlooked based on GWAS evidence alone. As a validation of this finding, we found that a recent GWAS of insomnia with a larger sample size has found strong evidence of association at the MDGA1 locus, which survives conventional corrections (P = 4.0 × 10−12)32. Figure 2b illustrates the overlapping distribution of effects for genetic variants at MDGA1 on insomnia, gene expression and DNA methylation.
We identified several other instances from our analysis of loci with evidence of colocalization that have recently been detected by GWAS, suggesting that our analytical pipeline is valuable in terms of detecting novel findings. For example, we found that expression of the CD40 and SLC12A5 genes colocalize with risk of neuroticism. Both genes have subsequently been identified as associated with neuroticism at genome-wide significance in a GWAS meta-analysis33. Additionally, a recent large GWAS of educational attainment identified several genetic variants not previously found to reach genome-wide significance that we found to colocalize with molecular traits for the following genes: DNAJB4, RERE, Corf73, DHX30, CD164 and GLCC1134.
Pathway and tissue-specific enrichment analysis
Pathway analysis was conducted using ConsensusPathDB24 to investigate whether any sets of genes for each complex trait and disease reside along the same biological pathway (Supplementary Table 12). Amongst findings there was evidence that genes associated with neuroticism in our analysis (SLC12A5, GNAI2 and GNG12) reside on the GABAergic synapse pathway (enrichment P = 2.34 × 10−4).
Our tissue-specific analysis indicated that various genes with evidence of colocalization are predominantly expressed in the brain. SLC12A5, KLC1 and KIRREL3 are expressed specifically in the cerebral cortex using data from the Human Protein Atlas25, whereas MDGA1 was strongly expressed within the brain tissue using data from the GTEx26 project. RAP1GAP2 was predominantly expressed within the cortex tissue using findings from the Mouse ENCODE project, amongst other loci are enriched in the brain tissue based on this dataset (Supplementary Table 13, Supplementary Figure 2).
We observed enrichment of an inverse relationship between DNA methylation and gene expression across loci, which provided evidence of colocalization for these molecular traits (P = 1.98 × 10−03), supporting previous evidence implicating DNA methylation as a transcriptional repressor35 (Supplementary Table 14). This effect appeared to be driven by CpG sites located near the transcription start site of genes, as 11 of the 13 sites located at these regions were inversely correlated with gene expression (84.6%). Performing the same analysis except with gene expression and histone acetylation suggested that there was weak evidence of enrichment for a directional relationship (P = 0.37). The regulatory annotations within brain tissue datasets for lead SNPs and CpG sites can be found in Supplementary Tables 15–17.
Discussion
In this study, we have conducted an integrative analysis of GWAS and molecular QTL data to uncover mechanistic insight into the biological pathways underlying complex traits. We identified 118 colocalization associations between complex traits and gene expression, with 73 of these associations additionally colocalizing with proximal DNA methylation and/or histone acetylation in the brain tissue. Out of the 118 associations, 52 were potentially novel loci, which did not meet genome-wide significance corrections, but colocalized with molecular traits. Notably, several of these potentially novel loci have recently been validated by larger GWAS32,33,34, suggesting that other findings in our study are likely to be identified by GWAS as study sizes increase. Our findings should help future studies prioritize candidate genes and putative epigenetic mechanisms for functional follow-up analyses.
Validation of previous findings
Applying our analysis pipeline to GWAS loci associated with complex traits and disease (i.e. P < 5 × 10−08) replicated previous findings reported by functional studies. For instance, our findings are consistent with an in-depth evaluation of the KLC1 locus3. Variation at KLC1 provided strong evidence of colocalization in our study (combined PPA = 97.9%), where the highest individual posterior probability suggested that both gene expression and DNA methylation may be involved along the causal pathway to schizophrenia risk. This result also supports findings from an epigenome-wide association study implicating DNA methylation as potentially playing a role in schizophrenia risk at this locus36. Furthermore, Hi-C interactions have been identified at the promoter region of KLC1 within the brain tissue, which further helps validate the putative regulatory mechanism implicated by our results37.
Amongst other established GWAS loci, there was evidence suggesting that expression of the TOMM40 gene and DNA methylation may play a role in Alzheimer’s disease. An exploratory analysis has found that regulatory element methylation levels in the TOMM40-APOE-APOC2 gene region correlate with Alzheimer’s disease38. However, there is also evidence that, although SNPs at this region are known to influence Alzheimer’s disease, gene expression, and DNA methylation, they may be attributed to different causal variants39. Moreover, there is a complex LD structure at this region31, suggesting that further analysis is required to fully understand the mechanisms underlying this association.
In cases where gene expression was found to colocalize with DNA methylation, we observed evidence of enrichment for an inverse relationship between these molecular phenotypes. Such inverse correlations support established biology that DNA methylation plays a role in silencing gene transcription40. However, recently there has been conflicting reports concerning whether DNA methylation on its own is sufficient to lead to transcriptional repression of promoters35,41. Further analysis investigating the epigenetic mechanisms identified by our study should prove valuable in fully understanding the role of DNA methylation in gene regulation.
Novel leads
We were also able to identify evidence of colocalization at GWAS loci that have not been linked previously by functional analyses or integrative studies harnessing molecular traits. For instance, the underlying biology explaining a GWAS association with neuroticism on chromosome 17 (lead SNP = rs12938775) has yet to be thoroughly evaluated. Our findings suggest that PAFAH1B1 may be the likely causal gene at this locus, as well as implicating the involvement of DNA methylation along the causal pathway to neuroticism susceptibility as well (combined PPA = 90.0%). PAFAH1B1 (also known as LIS1) is involved in neuronal migration, the process by which different classes of neurons are brought together so that they can interact appropriately42. Functional evaluations of how changes in DNA methylation may influence neurological function at loci such as this may prove valuable in understanding epigenetic contributions to disease susceptibility. Moreover, doing so will help improve the accuracy of early disease prognosis.
As well as helping characterize associations detected by GWAS studies, we have also uncovered evidence for many novel genes, which may influence complex trait variation and therefore represent promising candidates for future endeavours. The association with insomnia risk at the MDGA1 locus is an example of this, particularly given that it has recently been validated by a large-scale GWAS32. Furthermore, our results may provide functional insight into this association, by suggesting that MDGA1 may be the responsible causal gene and that DNA methylation may also play a role in disease risk at this locus (combined PPA = 85.8%). Similar to PAFAH1B1, MDGA1 has also been report to play a role in neuronal migration43 and based on our tissue-specific analysis is predominantly expressed in the brain tissue.
SLC12A5, associated with neuroticism in our analysis (combined PPA = 99.0%), was amongst other promising candidates which has yet to be discovered by GWAS. This gene encodes the neuronal KCC2 channel, which plays a crucial role in fast synaptic inhibition44. SLC12A5 was also amongst the genes associated with neuroticism in our analysis that resides along the GABAergic synapse pathway (along with GNAI2 and GNG12). A recent study has suggested that GABAegic neurons are causally associated with risk of bipolar disorder45, a condition previously linked with higher global measures of neuroticism46. The association between KIRREL3 and neuroticism (combined PPA = 92.2%) is another finding that has yet to be identified by GWAS, which warrants in-depth functional evaluation. KIRREL3 regulates target-specific synapse formation and has been previously linked with neurodevelopmental disorders47. Our tissue-specific analysis suggests that both SLC12A5 and KIRREL3 are predominantly expressed in the brain tissue.
Limitations of this study
In terms of limitations of this study, we recognize that integration of GWAS results with QTL data is limited by the sample sizes used to derive summary statistics, which is particularly noteworthy for QTL data available in the brain. It may be the case that replication in blood can provide greater power due to the larger sample sizes available. It has been shown that top cis-eQTL and mQTL are highly correlated between blood and brain tissues48. Future work could take advantage of this correlation and the higher power in these blood datasets.
There is also evidence that the expression of certain genes is both highly tissue and disease specific45. Recently, it has been shown that both tissue-specific and tissue-shared eQTL provide a substantial polygenic contribution to various complex traits27. Further investigation into the tissue specificity of our results could be interesting since the ROSMAP/Brain xQTL14 dataset comes specifically from the dorsolateral prefrontal cortex region of the brain. Analysis of effects in other regions of the brain may be interesting to potentially identify disease-relevant regions. We were also limited as the mQTL data was confined to 5 kb windows affecting the coverage we could get within a genomic region. Whilst the nature of this mQTL dataset means we may have missed some true effects, it also means we are unlikely to have identified false positives. It is also worth noting that as the number of molecular studies increases, so too does the likelihood of detecting incidental QTL-GWAS overlaps3. Hence, developments concerning robust methods in colocalization should prove to be extremely valuable and important for future research.
Conclusions
By integrating GWAS findings with data concerning brain cortex-derived molecular phenotypes, we have helped uncover putative epigenetic and transcriptomic drivers of complex traits and disease. The polygenic nature of many psychiatric disorders has led to many challenges in terms of developing novel therapeutics49. Our work has focused on the prioritization of GWAS hits to uncover loci, which may be potential targets for therapeutic intervention. Genetically informed targets have been shown to have a higher success rate in clinical development, with such targets being more likely to progress to phase III trials15.
Animal studies are one possible approach to validate and help translate the findings of our study. Knockout studies, for example, could be used to investigate the impact of whole gene loss-of-function on a particular phenotype50. Furthermore, the loci we have uncovered associated with changes in DNA methylation could be validated by studies interested in using this epigenetic marker to help predict later life disease events. Such endeavours may help elucidate CpG sites that may be valuable for disease prediction and patient stratification.
References
Edwards, S. L., Beesley, J., French, J. D. & Dunning, A. M. Beyond GWASs: illuminating the dark road from association to function. Am. J. Hum. Genet. 93, 779–797 (2013).
Hannon, E., Weedon, M., Bray, N., O’Donovan, M. & Mill, J. Pleiotropic effects of trait-associated genetic variation on DNA methylation: utility for refining GWAS loci. Am. J. Hum. Genet. 100, 954–959 (2017).
Gusev, A. et al. Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat. Genet. 50, 538–+ (2018).
Pierce, B. L. et al. Co-occurring expression and methylation QTLs allow detection of common causal variants and shared biological mechanisms. Nat. Commun. 9, 12 (2018).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Hormozdiari, F. et al. Colocalization of GWAS and eQTL signals detects target genes. Am. J. Hum. Genet. 99, 1245–1260 (2016).
Wu, Y. et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun. 9, 918 (2018).
Gutierrez-Arcelus, M. et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. eLife 2, 18 (2013).
Pai, A. A., Pritchard, J. K. & Gilad, Y. The genetic and mechanistic basis for variation in gene regulation. PLoS Genet. 11, 8 (2015).
Richardson, T. G. et al. Systematic Mendelian randomization framework elucidates hundreds of CpG sites which may mediate the influence of genetic variants on disease. Hum. Mol. Genet. 27, 3293–3304 (2018).
Wakefield, J. Bayes factors for genome-wide association studies: comparison with P-values. Genet. Epidemiol. 33, 79–86 (2009).
Giambartolomei, C. et al. A Bayesian framework for multiple trait colocalization from summary association statistics. Bioinformatics bty147–bty147 34, 2538–2545 (2018).
Ng, B. et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat. Neurosci. 20, 1418–+ (2017).
Nelson, M. R. et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856 (2015).
Bycroft, C. et al. Genome-wide genetic data on ~500,000 UK Biobank participants. bioRxiv 166298 (2017).
Ripke, S. et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–+ (2014).
Okbay, A. et al. Genome-wide association study identifies 74 loci associated with educational attainment. Nature 533, 539–+ (2016).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–U1206 (2013).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
1000 Genomes Project, Abecasis, G. R. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Zerbino, D. R. et al. Ensembl 2018. Nucleic Acids Res. 46, D754–D761 (2018).
Turner, S. D. qqman: an R package for visualizing GWAS results using Q–Q and Manhattan plots. Journal of Open Source Software 3, 731 (2018). https://doi.org/10.21105/joss.00731.
Kamburov, A., Wierling, C., Lehrach, H. & Herwig, R. ConsensusPathDB-a database for integrating human functional interaction networks. Nucleic Acids Res. 37, D623–D628 (2009).
Uhlen, M. et al. Tissue-based map of the human proteome. Science 347, 10 (2015).
Ardlie, K. G. et al. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
Gamazon, E. R. et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat. Genet. 50, 956–967 (2018).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, New York, 2016).
Kundaje, A. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Quinlan, A. R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 47, 11 12 11–34 (2014).
Yu, C. E. et al. Comprehensive analysis of APOE and selected proximate markers for late-onset Alzheimer’s disease: Patterns of linkage disequilibrium and disease/marker association. Genomics 89, 655–665 (2007).
Jansen, P. R. et al. Genome-wide analysis of insomnia (N = 1,331,010) identifies novel loci and functional pathways. bioRxiv 214973 (2018).
Nagel, V. et al. Meta-analysis of genome-wide association studies for neuroticism in 449, 484 individuals identifies novel genetic loci and pathways. Nat. Genet. 50, 920–+ (2018).
Lee, J. J. et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 50, 1112–1121 (2018).
Ford, E. E. et al. Frequent lack of repressive capacity of promoter DNA methylation identified through genome-wide epigenomic manipulation. bioRxiv (2017).
Pidsley, R. et al. Methylomic profiling of human brain tissue supports a neurodevelopmental origin for schizophrenia. Genome Biol. 15, 483 (2014).
Won, H. J. et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–+ (2016).
Shao, Y. et al. DNA methylation of TOMM40-APOE-APOC2 in Alzheimer’s disease. J. Hum. Genet. 63, 459–471 (2018).
Marioni, R. E. et al. GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 8, 7 (2018).
Moore, L. D., Le, T. & Fan, G. P. DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38 (2013).
Korthauer, K. & Irizarry, R. A. Genome-wide repressive capacity of promoter DNA methylation is revealed through epigenomic manipulation. bioRxiv 381145 (2018).
Tokuoka, S. M. et al. Involvement of platelet-activating factor and LIS1 in neuronal migration. Eur. J. Neurosci. 18, 563–570 (2003).
Takeuchi, A. & O’Leary, D. D. M. Radial migration of superficial layer cortical neurons controlled by novel Ig cell adhesion molecule MDGA1. J. Neurosci. 26, 4460–4464 (2006).
Stodberg, T. et al. Mutations in SLC12A5 in epilepsy of infancy with migrating focal seizures. Nat. Commun. 6, 9 (2015).
Finucane, H. K. et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 50, 621–+ (2018).
Sparding, T., Palsson, E., Joas, E., Hansen, S. & Landen, M. Personality traits in bipolar disorder and influence on outcome. BMC Psychiatry 17, 10 (2017).
Martin, E. A. et al. The intellectual disability gene Kirrel3 regulates target-specific mossy fiber synapse development in the hippocampus. eLife 4, 14 (2015).
Qi, T. et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat. Commun. 9, 12 (2018).
Breen, G. et al. Translating genome-wide association findings into new therapeutics for psychiatry. Nat. Neurosci. 19, 1392 (2016).
Boerwinkle, E. & Heckbert, S. R. Following-up genome-wide association study signals: lessons learned from Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium Targeted Sequencing Study. Circ. Cardiovasc. Genet. 7, 332–334 (2014).
Acknowledgements
We are grateful to the developers of the Brain xQTL resource (http://mostafavilab.stat.ubc.ca/xqtl/) and authors of the GWAS studies cited in this study for making their summary statistics publicly available. We would also like to thank Claudia Giambartolomei, the lead developer of ‘moloc’, for all her time and help with questions regarding our analysis plan. This publication is the work of the authors and T.G.R. will serve as guarantor for the contents of this paper. This study and C.H. were supported by a 4-year studentship fund from the Wellcome Trust Molecular, Genetic and Lifecourse Epidemiology Ph.D. programme at the University of Bristol (108902/B/15/Z). The MRC Integrative Epidemiology Unit receives funding from the UK Medical Research Council and the University of Bristol (MC_UU_00011/4 and MC_UU_00011/5). T.G.R. is a UKRI Innovation Research Fellow (MR/S003886/1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hatcher, C., Relton, C.L., Gaunt, T.R. et al. Leveraging brain cortex-derived molecular data to elucidate epigenetic and transcriptomic drivers of complex traits and disease. Transl Psychiatry 9, 105 (2019). https://doi.org/10.1038/s41398-019-0437-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-019-0437-2
This article is cited by
-
meQTL mapping in the GENOA study reveals genetic determinants of DNA methylation in African Americans
Nature Communications (2023)
-
A systematic review of neuroimaging epigenetic research: calling for an increased focus on development
Molecular Psychiatry (2023)
-
Genetic impacts on DNA methylation: research findings and future perspectives
Genome Biology (2021)
-
Profiling haplotype specific CpG and CpH methylation within a schizophrenia GWAS locus on chromosome 14 in schizophrenia and healthy subjects
Scientific Reports (2020)
