
Abstract
Identification of therapeutic targets from genome-wide association studies (GWAS) requires insights into downstream functional consequences. We harmonized 8,613 RNA-sequencing samples from 14 brain datasets to create the MetaBrain resource and performed cis- and trans-expression quantitative trait locus (eQTL) meta-analyses in multiple brain region- and ancestry-specific datasets (n ≤ 2,759). Many of the 16,169 cortex cis-eQTLs were tissue-dependent when compared with blood cis-eQTLs. We inferred brain cell types for 3,549 cis-eQTLs by interaction analysis. We prioritized 186 cis-eQTLs for 31 brain-related traits using Mendelian randomization and co-localization including 40 cis-eQTLs with an inferred cell type, such as a neuron-specific cis-eQTL (CYP24A1) for multiple sclerosis. We further describe 737 trans-eQTLs for 526 unique variants and 108 unique genes. We used brain-specific gene-co-regulation networks to link GWAS loci and prioritize additional genes for five central nervous system diseases. This study represents a valuable resource for post-GWAS research on central nervous system diseases.
Similar content being viewed by others


Main
Psychiatric and neurological diseases continue to be a massive global health burden: The World Health Organization estimated that in 2019, globally 280 million individuals were affected by depression, 39.5 million by bipolar disorder and 287.4 million by schizophrenia (SCZ)1 . Similarly, the number of people living with dementia is expected to rise from 50 million today to 152 million by 2050 (ref. 2), with similar trajectories for other neurodegenerative diseases. Although substantial progress has been made in uncovering the genetic basis of these diseases through genome-wide association studies (GWAS), much of how the identified genetic variants impact brain function remains unknown.
To translate from genetic signals to mechanisms, associations with gene expression levels or expression quantitative trait loci (eQTL) have shown great potential. Cis-eQTLs (nearby) and trans-eQTLs (distal) can aid the interpretation of GWAS loci in several ways. Cis-eQTLs provide direct links between genes and phenotypes through causal inference approaches such as Mendelian randomization (MR) and genetic co-localization analyses, whereas trans-eQTLs expose sets of downstream genes and pathways on which the effects of disease variants converge.
Expression quantitative trait loci are dynamic features and vary with tissue, cell type and additional factors such as response to stimulation. Therefore, eQTLs from disease-relevant tissues are desired for optimal interrogation of GWAS loci3. Previous brain eQTL meta-analyses by the PsychENCODE4 (n = 1,866) and AMP-AD5 (n = 1,433) consortia have been published to help interpret neurodegenerative and psychiatric disease GWAS loci. However, results from statistical approaches such as MR and co-localization are improved by robust effect-size estimates from even larger carefully curated eQTL datasets. In addition, large sample sizes are better suited to decompose eQTL effects to specific cell types.
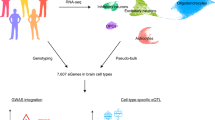
To maximize the potential of eQTL-based analyses of the brain, we combined and rigorously harmonized brain RNA-sequencing (RNA-seq) and genotype data from 14 different cohorts, including 8,613 RNA-seq samples from all major brain eQTL studies, and publicly available samples from the European Nucleotide Archive (ENA). We created a gene co-regulation network based on 8,544 RNA-seq samples covering different brain regions and performed cis- and trans-eQTL analyses of up to 2,683 individuals of European ancestry (EUR), with replication in up to 319 individuals of African ancestry (AFR). We made inferences on the brain cell types in which eQTLs operate and systematically conducted MR and co-localization analyses to find shared genetic effects between eQTLs and 31 brain-related GWAS traits. Our analyses prioritize probable causal genes and reveal cell type-dependent eQTLs that may be associated with disease risk (Fig. 1).

We downloaded publicly available RNA-seq and genotype data from 14 different datasets consisting of 8,613 RNA-seq measurements from seven main brain regions and 6,518 genotype samples. We created six eQTL meta-analysis datasets and performed cis-, trans- and interaction-eQTL analyses, built a brain-specific gene co-regulation network and prioritized genes using MR, co-localization and the co-regulation network. Image of sagittal cut of brain created with BioRender.com. This figure summarizes values from Supplementary Tables 1, 3, 8, 12 and 25–30.
To facilitate future studies, we have made all summary statistics and the co-expression network derived from our resource available at www.metabrain.nl.
Results
Harmonizing datasets for eQTL and co-regulation analysis
We combined 14 eQTL datasets into the ‘MetaBrain’ resource to maximize statistical power to detect eQTLs and create a brain-specific gene co-regulation network (Fig. 2, Supplementary Figs. 1–7 and Supplementary Table 1). Previous to quality control (QC), MetaBrain includes 7,604 RNA-seq samples and accompanying genotypes from the AMP-AD consortium (AMP-AD MAYO, ROSMAP and MSBB)6, Braineac7, the PsychENCODE consortium8 (Bipseq4, BrainGVEX4, CMC9, CMC_HBCC and UCLA_ASD4), BrainSeq10, NABEC11, TargetALS12 and GTEx3. In addition, we carefully selected 1,759 brain RNA-seq samples from the ENA13, which we subsequently genotyped and imputed (Fig. 2a, Supplementary Note and Supplementary Figs. 1–3). After realignment, removal of duplicate samples and stringent QC, 8,613 RNA-seq samples remained (Methods and Supplementary Figs. 4,5). Using slightly different QC thresholds, we created a gene network using 8,544 samples (Supplementary Note). For both datasets, we corrected the RNA-seq data for technical covariates and defined seven major tissue groups (amygdala, basal ganglia, cerebellum, cortex, hippocampus, hypothalamus and spinal cord): principal component analysis (PCA) on the RNA-seq data showed clear clustering by these major tissue groups, resembling brain physiology (Fig. 2b and Supplementary Fig. 6). The genotype data revealed individuals from different ancestries (Fig. 2c and Supplementary Fig. 2), including 5,138, 805 and 208 samples from EUR, AFR and East Asian (EAS) ancestries, respectively. After QC and deduplication, we created six cis-eQTL discovery datasets: Basal ganglia-EUR (n = 208), Cerebellum-EUR (n = 492), Cortex-EUR (n = 2,683), Cortex-AFR (n = 319), Hippocampus-EUR (n = 168) and Spinal cord-EUR (n = 108; Supplementary Table 1 and Fig. 2d). We used Cortex-EAS (n = 208) as a replication dataset. Cis-eQTLs were not calculated for the amygdala and hypothalamus tissue groups due to the small sample size (n < 100).

a, Number of samples per included cohort stratified according to the seven major brain regions. b, PCA dimensionality reduction plot of the normalized expression data after covariate correction. Each dot represents an RNA-seq sample and is colored according to the brain region. The figure shows that the samples cluster mainly on the brain region. c, Number of genotypes per cohort stratified according to ancestry. AMR, Admixed Americans; SAS, South Asian. d, Number of individuals per cohort, with each color representing an eQTL dataset. The number of individuals differ from the intersection between the number of RNA-seq samples and number of genotypes because not all samples with genotypes have RNA-seq samples and vice versa, and some individuals with genotypes have multiple RNA-seq measurements.
54% of cortex cis-eQTLs have independent associations
Within each discovery dataset, we performed a sample size-weighted cis-eQTL meta-analysis on common variants (minor allele frequency (MAF) > 1%), within 1 megabase (Mb) of the transcription start site (TSS) of a protein-coding gene. We identified 1,880 Basal ganglia-EUR, 10,577 Cerebellum-EUR, 4,797 Cortex-AFR, 16,169 Cortex-EUR, 1,265 Hippocampus-EUR and 998 Spinal cord-EUR cis-eQTL genes (q-value < 0.05; Fig. 3a and Supplementary Table 2). The observed eQTLs were consistent between datasets (Supplementary Fig. 8) but showed some sensitivity to RNA-seq alignment strategies (Supplementary Note and Supplementary Figs. 9,10). We next performed conditional analysis to identify independent associations in each cis-eQTL locus. In Cortex-EUR, 8,815 genes had a significant secondary cis-eQTL (54% of cis-eQTL genes identified in this dataset), 4,489 genes had tertiary and 2,065 had quaternary cis-eQTLs. We also identified secondary associations for the other discovery datasets, albeit to a lesser extent (Fig. 3a and Supplementary Tables 2,3). The properties of the Cortex-EUR cis-eQTLs conform to studies performed in blood14 and brain15: primary lead cis-eQTL single nucleotide polymorphisms (SNPs) were generally located close to the TSS (median distance, 33.6 kilobases (kb)) and cis-eQTL genes had a lower probability for loss-of-function intolerance (pLI > 0.9; χ2 P = 2.48 × 10−83). Genes with a cis-eQTL generally had a higher median expression (Wilcoxon P = 5.5 × 10−174; Fig. 3b); the other properties of cis-eQTLs were very comparable with earlier reports (Supplementary Note, Supplementary Fig. 11 and Supplementary Table 4).

a, Number of conditional cis-eQTLs per eQTL dataset. b, Comparison of characteristics between primary and non-primary eQTLs. For the mean expression and pLI score values, each row compares the eQTL genes for that rank with eQTL genes from the previous rank (for example, for tertiary eQTLs, the non-significant (grey) distribution is from genes that have secondary but lack tertiary eQTLs). P values were calculated using a Wilcoxon test between significant and non-significant genes. Differences in mean gene expression levels (left), the distance between the most significant SNP–gene combination and the TSS (middle), and pLI scores (right) are shown. For primary, secondary and quaternary eQTLs, non-significant eQTLs have higher pLI scores. Vertical dotted lines indicate median of the distribution for the current rank (coloured) versus the previous rank (black). c,d, Number of overlapping eQTLs along with the Rb and standard error (s.e.) values of primary cis-eQTLs between the cortex eQTLs of different ancestries (c) and the different brain regions for the EUR datasets (d). n, sample size of the dataset. e, Correlation of effect sizes and standard error of primary cis-eQTLs of Cortex-EUR (discovery, excluding GTEx) in all of the GTEx tissues (replication). Each dot is a different GTEx tissue; the x axis indicates the number of eQTLs that are significant in both discovery and replication.
High eQTL agreement between ancestries and brain regions
We investigated ancestry, brain region, dataset and tissue-type differences in cis-eQTLs. Agreement between ancestries was high: allelic concordance (AC) and correlation of effect-size (Rb) estimates were high when different ancestries were compared (Rb > 0.78, AC > 92.95%; Fig. 3c, Supplementary Fig. 12 and Supplementary Table 5). The proportion of estimated true-positives (π1) and correlation of allelic fold change (caFC) estimates between ancestries were lower, potentially due to differences in sample size (for example, Cortex-EUR versus Cortex-EAS, caFC = 0.55 and π1 = 0.29; conversely, caFC = 0.85 and π1 = 0.95; Supplementary Fig. 12). Similarly, different brain regions showed high overall agreement (Rb > 0.76, caFC > 0.65, and AC > 91%), with π1 estimates dependent on the sample size (0.39–0.95). Cerebellum was an exception and showed lower agreement with the cerebral brain regions (Fig. 3d,e and Supplementary Fig. 12). Despite the limited sample size, we identified 477 cis-eQTL genes that are significant in Cerebellum-EUR but not in Cortex-EUR (Supplementary Fig. 13), perhaps due to low expression in the cortex or because they are regulated by transcription factors that are more active in the cerebellum (Supplementary Note and Supplementary Table 6). Next, we repeated Cortex-EUR eQTL discovery while excluding GTEx and compared the results with cis-eQTLs from different GTEx tissues (Fig. 3e, Supplementary Figs. 12,14 and Supplementary Table 5). There was high agreement between brain-related tissues (cerebral tissues, Rb > 0.8, caFC > 0.71 and AC > 96%; and cerebellar tissues, Rb > 0.76, caFC > 0.71, π1 > 0.55 and AC > 92%) compared with other tissue types. The lowest agreement was with tissues such as testis (Rb = 0.51, caFC = 0.48 and AC = 78%) and whole blood (Rb = 0.55, caFC = 0.53 and AC = 80%). The π1 scores were not higher for cerebral or cerebellar tissues compared with non-brain tissues. We also compared Cortex-EUR cis-eQTLs with eQTLGen14 (n = 31,684; blood-based, majority EUR ancestry), which supported the low agreement observed in GTEx blood. Of the overlapping eQTLs, 25% had an opposite allelic effect (AC = 75%, Rb = 0.52 and π1 = 0.83; Supplementary Fig. 15 and Supplementary Table 7) (ref. 16), which represents an increase over GTEx and suggests that many of the eQTLs are tissue-dependent. Combined, these results suggest that additional tissue- or ancestry-specific eQTLs can be identified when sample sizes increase. For instance, opposite effects may happen if two causal variants reside on the same haplotype but are specific for different tissues17, requiring large sample sizes for disentanglement. By revealing eQTLs with opposite allelic effects, our results highlight the relevance of tissue-dependent eQTL mapping to accurately assess the directionality of eQTLs17.
14% of cortex cis-eQTLs are dependent on the cell-type proportion
We evaluated the extent to which eQTLs are dependent on cell-type proportions by determining cell-type interaction eQTLs (ieQTLs)3,18,19. In the Cortex-EUR subset, we predicted cell-type proportions using single-cell RNA-seq-derived signature profiles4 (Supplementary Note and Supplementary Fig. 16). The cell-type proportions and reconstruction accuracy of our predictions (87%) were comparable to a previous study that used this reference profile on a subset of the Cortex-EUR samples4. We observed low-to-moderate correlations between predicted cell types (0.01 < Pearson’s correlation coefficient (r) < 0.55; Fig. 4a) and high positive correlations with immunohistochemistry (IHC) counts from the ROSMAP cohort20 (overall Pearson’s r = 0.89 and per cell-type Pearson’s r > 0.1; Fig. 4b). However, we note that the exact proportion for each cell type remains uncertain21,22.

a, Pearson’s correlations between the seven predicted cell-count proportions within cortex samples. b, Predicted cell-type proportions compared with cell-type proportions measured using IHC for 42 ROSMAP samples. Pearson’s correlation coefficients are provided. The cell-count predictions for most cell types closely approximate actual IHC cell counts. Shaded areas around regression lines indicate 95% confidence interval. c, Number of cell-type ieQTLs for Cortex-EUR deconvoluted cell types. The first 20 intersections with the highest overlap are shown. Oligodendrocytes have the most interactions, followed by astrocytes and other neurons. Notably, most interactions are unique for one cell type in 87.1% of the cases. d–f, Replication of cell-type ieQTLs for STMN4 (d), FAM221A (e) and CD38 (f), consisting of the scatterplot of the cell-type ieQTL in MetaBrain Cortex-EUR bulk RNA-seq (left) and a forest plot for the eQTL effect in the ROSMAP snRNA-seq data (right). Each dot in the scatterplots (left) represents a sample; colors indicate SNP genotype, with yellow being the minor allele; values under the genotypes are the Pearson’s correlation coefficients; interaction P values were determined using a one-sided F-test; eQTL P values were derived using the standard normal distribution from meta-analyzed z-scores. Forest plot (right): eQTL β values (dots) and standard error (error bars) with effect direction relative to the minor allele when replicating the eQTL effect in ROSMAP single-nucleus data (n = 38); each row denotes a cell type-specific dataset; cell types highlighted in bold reflect the equivalent to the cell type used in the ieQTL. Vertical dashed lines indicate an eQTL beta of 0. TMM, trimmed mean of M-values; AST, astrocytes; END, endothelial cells; EX, excitatory neurons; IN, inhibitory neurons; MIC, microglia; OPC, oligodendrocyte precursor cells; OLI, oligodendrocytes; NEU, other neuron; PER, pericytes.
We used Decon-QTL19 to identify ieQTLs for the 25,497 independent Cortex-EUR cis-eQTLs: 3,549 cis-eQTLs (13.9%) showed at least one significant ieQTL (4,095 ieQTLs; Benjamini–Hochberg false discovery rate (BH-FDR) < 0.05; Supplementary Table 8). The largest group of interactions were with excitatory, inhibitory and other neurons (1,627; 39.7%), probably because neurons are the most prevalent cell type. The majority of the ieQTLs (3,090; 75.5%) were uniquely mapped to one cell type (Fig. 4c), although we cannot exclude the possibility that these ieQTLs are also present in other cell types.
We replicated these findings in the Cortex-AFR dataset (n = 319) as well as in two independent single-nucleus RNA-seq (snRNA-seq) datasets from ROSMAP23 (n = 39) and Bryois et al.24 (n = 196; Supplementary Figs. 17–19 and Supplementary Table 9). Across all replication datasets, we observed moderate-to-high rates of agreement, depending on the cell-type frequency and sample size (Bryois et al.24: 0.78 <Rb < 0.86, median Rb = 0.84, 0.43 < π1 < 0.83, median π1 = 0.69, 81% <AC < 90%, median AC = 0.9; Supplementary Note). Examples of replicating ieQTLs include the oligodendrocyte ieQTL genes FAM221A, NKAIN1 and STMN4, which were previously identified as oligodendrocyte-specific25, and AMPD3 and CD82, of which the SNPs were previously associated with white-matter microstructure26, suggesting a role for oligodendrocytes (Fig. 4d,e and Supplementary Fig. 20a–e). The high replication rates indicate that our approach can accurately identify the cell type for a large number of eQTLs. We note that summary statistics were available for only 54% of ieQTLs in a well-powered replication dataset (Bryois et al.24), suggesting that our approach had the power to detect ieQTLs that are not yet identified in snRNA-seq datasets.
These ieQTLs can also provide valuable information about the cell types of interest for disease-associated SNPs. For example, the A allele of variant rs4698412, which is associated with increased risk for Parkinson’s disease (PD), also increased the expression of CD38, for which we identified a replicating astrocyte ieQTL (Fig. 4f and Supplementary Fig. 20f). This gene is an immunomodulatory agent and is mainly expressed in neurons, astrocytes and microglia27, and increased levels of CD38 are observed with neuroinflammation (Supplementary Note).
Shared genetic effects between cis-eQTLs and central nervous system traits
We next linked Cortex-EUR cis-eQTLs to variants associated with brain-related traits and diseases. We determined the linkage-disequilibrium (LD) overlap between cis-eQTLs and GWAS SNPs, which indicated that primary eQTLs were 2.6-fold more likely to be in LD with a GWAS SNP compared with non-primary eQTLs (Fisher’s exact test, P = 7.4 × 10−125; Supplementary Note and Supplementary Table 10). To more formally test whether there was evidence for sharing the same genetic effect between cis-eQTLs and 31 neurological traits, we conducted MR using the Wald ratio method and co-localization analyses (Supplementary Table 11). Among the 359,763 Wald ratios tested across 11,270 genes, 1,531 Wald ratios for 1,088 genes passed a suggestive P-value threshold (P < 5 × 10−5; Supplementary Table 12). Of the cis-eQTL instruments from these findings, 294 were also cell-type ieQTLs. There were 549 significant Wald ratios that passed Bonferroni’s correction (P < 1.43 × 10−7), from which 186 also co-localized between the eQTL and GWAS traits when using coloc28 (posterior probability for co-localization of significant signals PP4 > 0.7; Fig. 5a and Supplementary Fig. 21), confirming that the two traits shared the same causal SNP. Of the prioritized findings, 282 (82 of which co-localized) were associated with the risk for 31 prioritized neurological and neuropsychiatric diseases (Table 1). We focus on multiple sclerosis (MS) and highlight two examples where MR and co-localization point to probable causal GWAS genes. For other traits, see Supplementary Note, Supplementary Fig. 22 and Supplementary Tables 11–16.

a, Number of significant Wald ratio effects (blue) and those with both Wald ratio and co-localization (coloc; red) evidence for 13 brain-related traits. b, Forest plots showing the SNP and effect allele (EA), eQTL β and GWAS odds ratio for 20 MS-associated genes that are both MR and co-localization significant as well as their Wald ratio P value. The dots indicate the eQTL β or odds ratio, the error bars indicate the 95% confidence interval. WR, Wald ratio; and OR, odds ratio. Grey dotted line indicates an OR of 1. c,d, Cell-type ieQTL for CYP24A1 (c) and CLECL1 (d) showing interactions with predicted excitatory neuron and microglia proportions, respectively. Each dot represents a sample. Colors indicate the SNP genotype, with yellow being the minor allele. Values under the alleles are the Pearson’s correlation coefficients. Interaction P values were derived using a one-sided F-test; eQTL P values were derived using the standard normal distribution from meta-analyzed z-scores. TMM, trimmed mean of M-values.
Tissue-specific genetic effects for MS
Using MR analysis for MS29, we identified 121 instruments (cis-eQTL SNPs) in 99 genes that passed the Bonferroni-adjusted P-value threshold of 1.43 × 10−7 (Supplementary Table 12); 25 of these instruments passed co-localization (Table 1 and Fig. 5b), of which 13 genes had a positive Wald ratio—indicating that increased gene expression increases disease risk—and the remainder a negative Wald ratio, indicating the opposite. A systematic comparison of the Wald ratio estimates on the 7,748 shared cis-eQTL genes between Cortex-EUR and eQTLGen14 (which instrumented the same genes but potentially with different SNPs) showed opposite effect directions for 3,173 (41.0%) genes (Supplementary Note, Supplementary Figs. 23,24 and Supplementary Table 14a). Although the agreement improved when the same SNP instrument was compared between studies, 2,671 (27.5%) of 9,728 MetaBrain Wald ratios still showed opposite directionality to eQTLGen (Supplementary Table 14b), underscoring the importance of tissue-specific differences when interpreting transcriptomics data.
Of the 172 genes with Wald ratio findings in Cortex-EUR, there were 47 without a significant eQTLGen instrument, including five (RGS1, SCO2, SLC12A5, CCDC155 and MYNN) that passed the MR and co-localization significance thresholds in MetaBrain (Supplementary Note and Supplementary Table 16). In comparisons of the blood and brain expression levels of these genes in GTEx30, SLC12A5 and CCDC155 had almost no expression in blood, whereas expression was comparable between tissues for RGS1, SCO2 and MYNN (Supplementary Note and Supplementary Fig. 25). The discrepancy in MR findings observed between Cortex-EUR and eQTLGen suggest the existence of tissue-dependent genetic effects for MS.
Cell type-specific cis-eQTLs linked to MS
Two MS-associated genes, CYP24A1 and CLECL1, showed cell type-specific cis-eQTLs (Fig. 5c,d). Another gene that was previously suggested to be neuron-specific31, SLC12A5, did not show a significant ieQTL in our data. In our analysis, we found that higher CYP24A1 expression is associated with increased risk for MS (Wald ratio = 0.13, P = 7.8 × 10−11) and that the eQTL and GWAS signals are co-localized (PP4 = 1.00). Furthermore, ieQTL analyses showed increasing expression of CYP24A1 with increasing excitatory neuron proportions for the risk allele rs2248137-C (interaction β = 1.92, interaction P = 1.98 × 10−11; Fig. 5c), similar to other neurons (Supplementary Table 12). CYP24A1 encodes for a protein that catalyzes the inactivation of 1,25-dihydroxyvitamin D3 (calcitriol), the active form of vitamin D32. Epidemiological studies have proposed vitamin D deficiency as a risk factor for MS33,34, which has recently been validated through MR35,36,37. Our findings are consistent with a previous report that indicates a shared signal for MS and CYP24A1 cis-eQTL in the frontal cortex38.
Decreased expression of CLECL1 was significantly associated with increased MS risk (Wald ratio = −0.16, P = 1.58 × 10−9) and showed clear co-localization (PP4 > 0.87). The ieQTL analysis indicated that rs7306304-A increased expression of CLECL1 with increased proportions of microglia (interaction β = −2.72, interaction P = 5.09 × 10−37; Fig. 5d), confirming a previous finding of a microglia cell type-specific cis-eQTL for CLECL1 at this MS risk locus29. This eQTL also replicates in the microglia single-cell analysis by Bryois et al.24 (eQTL β = −0.62, P = 3.2 × 10−20) with the same direction of effect. The rs7306304 SNP is in strong LD with the MS lead SNP rs7977720 (r2 = 0.84)29. CLECL1 encodes a C-type lectin-like transmembrane protein expressed at high levels in dendritic and B cells, which has been proposed to modulate immune response39. It has 20-fold higher expression in a purified microglia dataset29 than in cortical tissue, suggesting that decreased CLECL1 increases MS susceptibility through microglia-mediated immune processes in the brain.
MetaBrain allows for the identification of trans-eQTLs
Trans-eQTLs can identify the downstream consequences of disease-associated variants but their effects are usually small14. To maximize power, we combined the Cortex-EUR and -AFR datasets (n = 2,759, excluding the ENA). We reduced the multiple-testing burden by focusing on 228,819 unique genetic variants, including GWAS and cis-eQTL variants.
When correcting for an increasing number of principal components (PCs), we observed a decrease in the number of trans-eQTLs (Fig. 6a, Supplementary Note, Supplementary Fig. 7 and Supplementary Table 17) as well as in heterogeneity (Supplementary Fig. 26). The majority (85%) of the trans-eQTLs observed without PC correction were located in a 7p21.3 locus previously associated with frontotemporal lobar degeneration40, Alzheimer’s disease (AD)41 as well as changes in neuron proportions42 and gene expression levels43,44. We did not find evidence that these trans-eQTLs were dependent on AD status or neuron proportions and they were not significant when correcting for 100 PCs (Supplementary Note, Supplementary Figs. 26–29 and Supplementary Tables 17–24).

a, Location of the identified trans-eQTLs (SNP and gene positions) in the genome. The size of the dots indicates the P value of the trans-eQTL (larger is more significant). b, Two examples of convergent effects, where multiple independent SNPs affect the same genes in trans. Trans-eQTLs of rs1427407 and rs4895441 on HBG2 (top). Trans-eQTL of rs1150668 and rs106871 on ZNF31 and S100A5 (bottom). Both panels are derived from Supplementary Table 17.
We therefore concentrated on 737 trans-eQTLs, detected after correcting for 100 PCs, which reflect 526 unique SNPs and 108 unique genes; 127 SNPs had trans-eQTL effects on multiple genes and 461 (88%) of the trans-eQTL SNPs were associated with a significant cis-eQTL in Cortex-EUR. We observed that 150 (33%) of the 461 significant trans-eQTL SNPs overlapping a cis-eQTL SNP were also the cis-eQTL index SNP, which represents an enrichment (Fisher’s exact test, P = 1.2 × 10−28; Supplementary Note); 29 were also cis-eQTL SNPs in tissues other than the cortex (Supplementary Table 17). This indicates that cis-eQTL index SNPs yield trans-eQTL effects more often in the brain in comparison to other cis-eQTL variants.
Significant interactions with predicted cell-type proportions (BH-FDR < 0.05; Supplementary Table 22)—oligodendrocytes (n = 27), other neurons (n = 7), astrocytes (n = 7) and microglia (n = 2)—were also observed for 41 trans-eQTLs (5.9%). Four eQTLs—all influencing DTX4 and dependent on oligodendrocyte proportion—replicated significantly in the ROSMAP snRNA-seq dataset with the same direction of effect (Supplementary Fig. 30 and Supplementary Table 23).
We observed trans-eQTLs from multiple independent genomic loci for seven genes, suggesting convergent trans-eQTL effects (ARRDC4, HBG2, POP1, COX7A1, RFPL2, ZNF311 and ZNF404; Supplementary Table 17). This includes a convergent trans-eQTL on hemoglobin subunit ɣ-2 (HBG2; 11p15.4) that was previously identified in blood. HBG2 was affected in trans by two independent variants (rs1427407 on 2p16.1 and rs4895441 on 6q23.3; Fig. 6b), which have previously been associated with fetal hemoglobin levels45,46,47. We also found converging effects that were not identified in blood. For example, the ZNF311 gene (6p22.1) was affected by the rs1150668 variant in cis and the rs8106871 variant in trans (19q13.2), both of which have been previously associated with smoking48 and risk tolerance49. For both associations, the risk allele also increased ZNF311 expression. Furthermore, the risk allele rs1150668-G increased the expression of S100A5 in trans, and rs8106871-T decreased the expression of POU2F2 and increased expression of DEDD2 in cis (Fig. 6b). ZNF311 has been suggested to be a tumor-suppressor gene50 potentially involved in gliomas51, S100A5 is used as a biomarker for astrocytomas52 and POU2F2 has previously been associated with glioblastoma53. This example shows how multiple variants associated with smoking may alter multiple genes involved in cancer.
Brain co-regulation networks aid in GWAS interpretation
We generated brain region-specific co-regulation networks based on the RNA-seq data from 8,544 samples (Supplementary Note and Supplementary Figs. 31–33) using a similar approach to our previously developed multi-tissue GeneNetwork (n = 31,499)54,55. We applied Downstreamer56 to SCZ57, PD58, MS29, AD59 and amyotrophic lateral sclerosis (ALS) GWAS summary statistics60, using these networks to prioritize genes that are co-regulated with genes in their GWAS loci (Supplementary Note, Supplementary Fig. 34 and Supplementary Tables 25–30). For MS and AD, these were mostly immunity genes, whereas for PD, ALS and SCZ, these were genes that are specifically expressed in the brain (Supplementary Tables 25–30). For ALS and MS, we additionally created smaller networks for the cerebellum (n = 715) and cortex (n = 6,526) to identify brain region-specific effects.
For ALS, we applied Downstreamer to summary statistics from individuals with EUR ancestry (Supplementary Table 30) and a trans-ancestry meta-analysis including individuals with EUR and Asian ancestry60 (EUR + ASN; Supplementary Table 25). In contrast, whereas Downstreamer did not identify genes using GeneNetwork55 (n = 31,499), we identified a set of 27 unique co-regulated genes when applied to the smaller brain co-regulation networks (EUR + ASN summary statistics; Fig. 7a and Supplementary Table 25). Of the identified genes, HUWE1 was shared between the results from all brain regions and separate results from cortex, whereas UBR4 was shared between the cortex and cerebellum results. UBR4 encodes a ubiquitin ligase protein expressed throughout the body, which interacts with calmodulin, a protein regulating Ca2+—a process which has been linked to ALS disease-associated genes and motor-neuron vulnerability61. Furthermore, a previously discovered private mutation in UBR4 implicates its role in muscle coordination62. Many of the genes prioritized by Downstreamer are co-regulated with each other (Fig. 7b) and were enriched for genes implicated in causing gait disturbances (Fig. 7c). Our analysis identified genes that show strong co-regulation with positional candidate genes inside ALS-associated loci, suggesting that they must have a shared biological function.

a, Genes that are co-regulated with genes that are within ALS-associated loci. Co-regulation scores between genes were calculated using the MetaBrain cerebellum and MetaBrain cortex samples as well as the combined (all) MetaBrain samples. Except for URB4, cortex and cerebellum networks find different co-regulated genes for ALS. b, Co-regulation network using all MetaBrain samples for all genes prioritized for ALS by Downstreamer. c, Top five Human Phenotype Ontology (HPO) enrichments for the Downstreamer prioritized ALS-associated genes. d, Genes that are co-regulated with genes that are within MS-associated loci. Co-regulation scores between genes were calculated using a heterogeneous multi-tissue network, MetaBrain cerebellum samples and MetaBrain cortex samples. Most genes were found using a large heterogenous co-regulation network. e, Co-regulation network of all MetaBrain samples for 33 genes prioritized by Downstreamer in cortex. f, Top five Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichments for the Downstreamer prioritized MS genes in the cortex highlight neurotrophin signaling pathway enrichment (red). c,f, Enrichment P values were calculated using a two-sided χ2 test. Panels a and d are derived from Supplementary Tables 25, 29 and 30.
For MS, the GeneNetwork55 identified 257 unique genes that showed significant co-regulation with genes inside MS-associated loci (Fig. 7d and Supplementary Table 29), many of which were immunity genes, which is also expected for this disease. However, when we used the brain co-regulation networks, we identified a much smaller set of genes that showed strong enrichment for the neurotrophin signaling pathway (Fig. 7e,f). Neurotrophins are polypeptides secreted by immunological cell types and promote the survival and proliferation of neurons as well as synaptic transmission (Supplementary Note). The identified genes showed high expression in immunity-related tissues when using the GeneNetwork55 (Supplementary Fig. 31a) and high expression in the spinal cord but low expression in cortex samples (Supplementary Fig. 31b). This could implicate specific brain regions with MS development: for instance, the cerebellum is involved in muscle coordination and ataxia occurs in approximately 80% of patients with symptoms of MS63. But this could also implicate the ‘outside-in hypothesis’ that suggests the immune system may be a potential trigger for MS29,64.
Discussion
We describe an integrated analysis of the effects of genetic variation on gene expression levels in the brain with a sufficient sample size to identify robust cis-eQTLs and cell-type ieQTLs to compare cis-eQTLs between ancestries and identify brain trans-eQTLs that emanate from SNPs that have been previously linked to brain-related diseases. We have released harmonized results of the individual datasets to help others determine the robustness of eQTL effects.
We showed that eQTL-effect directions are generally shared between datasets, tissues and ancestries, but note that opposite allelic effects exist, which became apparent especially when we compared our results with a large blood eQTL dataset. We also identified non-primary cis-eQTLs, some of which reflect SNPs that are also the index variants for brain-related disorders, making them particularly interesting for subsequent follow-up.
We predicted cell-type proportions in the cortex and identified 3,549 cell-type ieQTLs. We compared the ieQTLs with snRNA-seq eQTLs and observed that the π1 estimates were low due to the low sample size or other limitations in the snRNA-seq datasets24,65,66. As we observed good Rb and AC values, we expect that the overlap and replication rates will improve once the sample sizes of snRNA-seq studies increase, highlighting the potential of ieQTL analysis in bulk RNA-seq.
This is a well-powered MR and co-localization analysis using brain cis-eQTLs as instruments for bipolar disease, epilepsy, frontotemporal dementia, MS, cognitive function and years of schooling GWAS outcomes. Interestingly, for SCZ, three signals for CILP2, MAU2 and TM6SF2 met our criteria that had not been reported in a recent psychiatric genomics consortium study67, further emphasizing the value of our large eQTL dataset (Supplementary Note). Our results also identify increased CYP24A1 expression to be associated with MS risk and propose excitatory neurons as the cell type most susceptible to CYP24A1 expression changes and probably active vitamin D levels. The potentially novel role of CYP24A1 in the brain could play an important role in MS etiology, as may lowered expression of CLECL1 in microglia.
The analyses identified trans-eQTLs in the brain cortex for many variants, some of which are brain-specific. Similar to blood, the trans-eQTL-effect sizes in the brain were usually small, emphasizing the importance of increasing the sample size of brain eQTL studies. The identified trans-eQTLs allowed us to gain insights into the functional effect of several disease-associated variants. We observed that trans-eQTL SNPs were enriched for cis-eQTL index SNPs, indicating consequences beyond the gene regulated in cis. Our trans-eQTL analysis focused on a single brain region and was limited to trait-associated and cis-eQTL SNPs. We expect that a genome-wide approach will identify many more trans-eQTLs. Furthermore, we note that true separation of cis- and trans-eQTL effects would require investigation of the allelic function of the associated SNPs (Supplementary Note).
We used brain-specific co-regulation networks to study several brain-related GWAS studies and prioritized genes that show significantly enriched co-regulation with genes inside GWAS loci. For ALS, this revealed a limited but significant set of genes located outside of currently known ALS loci, which might lead to a better understanding of the poorly understood pathways that cause ALS.
This study has several limitations. First, our eQTL analyses were limited to single tissues and excluded replicate RNA-seq measurements. A joint analysis with random-effects models68,69 could increase the effective sample size, which would be especially useful for trans-eQTL identification. Second, our GWAS overlap analysis may have failed to identify previously identified genes due to differences in sample size, effect size, variant density, LD structure and imputation quality. For example, our results did not include the MAPT gene for AD because the H1/H2 haplotype separating SNP rs8070723 had an eQTL P value of 1.8 × 10−5 due to our alignment strategy (Supplementary Note). This might have been an issue for other genes as well. Graph-based alignment tools or long-read sequencing methods are required to ultimately determine the true effects on such genes. Third, the GWAS overlap methods we used have known limitations (for example, Supplementary Note). For the MR analysis, we opted to perform single-SNP MR instead of multi-SNP MR (such as inverse-variance weighted70), which requires multiple independent associations per gene. As this was the case for only a limited proportion of the tested cis-eQTLs and there were no genes with more than five independent eQTLs, we reasoned this would not provide for reliable inverse-variance-weighted estimation. Inverse-variance-weighted estimation could potentially be applied on a genome-wide trans-eQTL analysis, resulting in many more independent instruments per gene. However, such an approach would be more susceptible to confounding because of horizontal pleiotropy71, where a gene is affected by multiple indirect effects. Finally, our co-localization approach was based on the single-causal-variant assumption, which is not applicable to cis-eQTL genes with multiple independent associations (for example, TREM2; Supplementary Note), and therefore we may have failed to detect co-localizing signals in such loci. Recently published co-localization methods72 do not have this assumption, which may improve future co-localization results. Finally, it is possible that bulk RNA-seq eQTL studies generally capture eQTL effects for genes that are not dosage sensitive and do not cause disruptive downstream consequences73. Furthermore, many eQTLs can only be detected in certain contexts74 for which single-cell experiments are best suited.
We expect that this resource will prove valuable for post-GWAS brain research. Our dataset can be utilized to disambiguate GWAS loci, point to causal pathways and prioritize targets for drug discovery. We expect that through future integration with single-cell eQTL studies that have higher resolution but still lower power, our results will help to pinpoint transcriptional effects in specific brain cell types for many disease-associated genetic variants.
Methods
Dataset collection and description
We collected published bulk brain RNA-seq samples from the AMP-AD consortium (AMP-AD MAYO, ROSMAP and MSBB)6, Braineac7 and the PsychENCODE consortium8 (Bipseq4, BrainGVEX4, CMC9, CMC_HBCC and UCLA_ASD4) from Synapse.org using the Python 2.7.5 package synapseclient v1.9.2 (ref. 75). The NABEC and GTEx datasets were retrieved from NCBI dbGaP and the TargetALS data were provided directly by the investigators. In addition, we collected brain bulk RNA-seq samples from the ENA and performed rigorous QC, resulting in 1,759 samples (Supplementary Note, Supplementary Fig. 1 and Supplementary Table 28). Combined with the other datasets, this yielded a total of 9,363 samples (Supplementary Table 1).
RNA-seq alignment and QC
RNA-seq data were processed using a pipeline built with molgenis-compute76. FASTQ files were aligned against the GRCh38.p13 primary assembly using the GENCODE77 v32 annotation with STAR78 (v2.6.1c), while excluding patch sequences (Supplementary Note) with the following parameter settings: outFilterMultimapNmax = 1, twopassMode Basic and outFilterMismatchNmax = 8 for paired-end sequences; and outFilterMismatchNmax = 4 for single-end sequences. Gene quantification was performed using STAR, similar to gene quantification using HTSeq79, with default settings. The gene counts were then TMM-normalized80 per cohort using edgeR81 (v3.20.9) with R82 (v3.5.1). Quantification for the GeneNetwork was done using Kallisto83 v0.43.1 (Supplementary Note).
To measure the FASTQ and alignment quality we used FastQC84 (v0.11.3), STAR metrics and Picard Tools85 metrics (v2.18.26; MultipleMetrics and RNAseqMetrics). Samples were filtered out if aligned reads had <10% coding bases (Supplementary Fig. 4a), <60% reads aligned (Supplementary Fig. 4b) or <60% unique mapping. Among the RNA-seq samples, 117 did not pass this filter, mostly from GTEx81. The other quality measurements were visually inspected but contained no outliers. To identify outliers that had not been captured by these statistics, we performed a PCA-based filtering approach, after which 8,868 samples remained (Supplementary Note and Supplementary Fig. 5a–c). To adjust for between-dataset differences observed in the data (Supplementary Fig. 6a), we correlated the RNA-seq data with 77 covariates from the different QC tools and regressed-out the top-20 correlated covariates using ordinary least squares (OLS; Supplementary Note), after which clustering of datasets in PC1 and PC2 were no longer present (Supplementary Fig. 6b).
Our collection of RNA-seq samples consisted of 36 different tissue labels, many of which were represented by only a few samples. Therefore, we next defined major brain regions present in our dataset, including samples from the amygdala, basal ganglia, cerebellum, cortex, hippocampus and spinal cord. We noted that some samples (especially from the ENA) were not annotated with a specific major brain region. To resolve this, we performed PCA over the sample correlation matrix and then performed k-nearest neighbors on the first two PCs (k = 7) to classify samples to the major brain regions. Using this approach, we defined a set of 86 amygdala, 574 basal ganglia, 723 cerebellum, 6,601 cortex, 206 hippocampus, 252 hypothalamus and 285 spinal cord samples (Fig. 2a and Supplementary Table 1).
Genotype QC and definition of eQTL datasets
The genotype data for the included datasets were generated using different platforms, including genotypes called from whole-genome sequencing (AMP-AD, TargetALS12 and GTEx3), genotyping arrays (NABEC11 and Braineac7) and haplotype reference consortium86-imputed genotypes (PsychENCODE datasets), or were called from RNA-seq directly (ENA dataset; Supplementary Note). A total of 22 different genotyping datasets were available, reflecting 6,658 genotype samples (Supplementary Table 1). We performed QC on each dataset separately, using slightly different approaches per platform (Supplementary Note) and used a PCA-based approach to assign ancestries to each individual sample. Most of the included samples were of EUR ancestry: 5,138 samples had an EUR assignment, 805 samples had an AFR assignment and 573 samples were assigned to the other ancestries (Fig. 2b and Supplementary Table 1). We next assessed links between RNA-seq samples and genotyped individuals, and were able to identify 7,644 links (Supplementary Table 1). For eQTL discovery, we grouped these links based on brain region and ancestry, in which we required at least 30 samples spanning at least two cohorts. We next removed sample mix-ups and duplicate samples (Supplementary Note), resulting in the following final eQTL discovery datasets: Basal ganglia-EUR (n = 208), Cerebellum-EUR (n = 492), Cortex-EUR (n = 2,683), Cortex-AFR (n = 319), Hippocampus-EUR (n = 208) and Spinal cord-EUR (n = 108; Fig. 2c and Supplementary Table 1).
eQTL analysis
We performed cis-eQTL analysis in each of the eQTL discovery datasets by calculating Spearman correlations within each cohort, followed by a sample size-weighted z-score meta-analysis approach, as described previously14. We opted for this approach to minimize the influence of potential heterogeneity between cohorts and showed that it performs comparably to FastQTL/QTLTools87 (Supplementary Note and Supplementary Fig. 35). To correct for multiple testing, we used an approach similar to FastQTL/QTLTools87, where we used 1,000 permutations of the sample labels to fit a β-distribution per gene and, after adjustment using this distribution, calculated the q-values88 over the top association per gene to determine significance (Supplementary Note). Genes with q-value < 0.05 were deemed significant. We limited these analyses to 19,373 protein-coding genes and to SNPs located within 1 Mb of the TSS, with MAF > 1% and Hardy–Weinberg P > 0.0001. The RNA-seq data were corrected for up to 20 technical covariates, dataset indicator variables and four multidimensional scaling components derived from the genotype data using OLS. In addition, we evaluated the impact of regressing out increasing numbers of PCs and defined the optimal numbers of PCs to remove for each eQTL discovery dataset (Supplementary Note and Supplementary Fig. 7). To identify secondary, tertiary, quaternary and other non-primary cis-eQTLs, we repeated the procedure in an iterative conditional approach, where in each subsequent iteration, we regressed out the cis-eQTL effect of the previous iterations using OLS and identified cis-eQTLs using the residuals, followed by an LD pruning step to circumvent SNP missingness between included cohorts (Supplementary Note).
To identify trans-eQTLs, we performed a limited analysis to reduce the multiple-testing burden by focusing on 228,819 variants with a known interpretation. This set constituted variants that were either previously associated with traits, having a GWAS P < 5 × 10−8 in the IEU OpenGWAS database89 or EBI GWAS catalog90 on 3 May 2020, and additional neurological traits (Supplementary Table 17) or that showed an association with q-value < 0.05 in any of our discovery cis-eQTL analyses (including non-primary associations identified in the iterative conditional analysis). To maximize power, we combined the Cortex-EUR and Cortex-AFR datasets but excluded the ENA cohort due to the potential for genotypes of poorer quality (n = 2,759; Supplementary Note). For this dataset, we also repeated the cis-eQTL analysis (Supplementary Table 2) and normalization approach, including the selection of optimal number of 100 PCs to regress out (Supplementary Fig. 7). We assessed those combinations of SNPs and genes where the SNP–TSS distance was >5 Mb or where the gene and SNP were on different chromosomes as trans-eQTLs. To determine significance, we employed a previously used FDR estimation method91 using ten genome-wide permutations (Supplementary Note) and deemed trans-eQTLs with an FDR < 0.05 significant. We finally used an alignment-based approach to detect potential crossmapping artifacts, after which the FDR estimates were recalculated (Supplementary Note).
eQTL agreement
We used four different measurements of agreement of eQTL effects when comparing different brain regions or tissues: AC, π1, Rb and caFC92. Each of these measures evaluates different aspects of replication: AC is an indication of the proportion of effects that have a shared direction of effect within the set of eQTLs that is significant in both the discovery and replication datasets and is expected to be 50% for random eQTL effects, π193 estimates the proportion of eQTL effects that are true positive in the replication cohort but does not take into account the effect direction and can be dependent on the replication dataset sample size, Rb94 effectively estimates the correlation between the eQTL-effect slopes (for example, β values from linear regression) while controlling for potential covariance in standard errors of those slopes and caFC measures the correlation between estimates of the fold change in expression values between alleles92. We note that whereas AC, π1 and Rb can be calculated from summary statistics, caFC requires access to genotype and expression data. We therefore limited the caFC analysis to comparisons within the MetaBrain datasets and comparisons with the GTEx tissues. Details on how we calculated these measures of agreement are in the Supplementary Note. For AC, π1 and Rb comparisons with GTEx, we used the summary statistics for GTEx-v8 that were downloaded from the GTEx portal website. Calculations of the aFC per eQTL were performed with the original aFC script by Mohammadi and colleagues92 using the settings –log_xform 1 and –log_base 2, after which Pearson correlation was used to calculate caFC across shared eQTLs. For all comparisons with GTEx tissues, we performed discovery in Cortex-EUR while excluding the GTEx cohort.
Identification of cell type-dependent eQTL effects in bulk RNA-seq
We predicted cell-type proportions of the MetaBrain Cortex-EUR and -AFR datasets using the method and single-cell profiles previously published by the PsychENCODE consortium4 (Supplementary Note). We decided to discard the developmental cell types as we expected that these cell types are very rare or not present in adult human brain and because their signatures were obtained from fetal cells. The remaining cell types included all major cell types in the brain: neurons (excitatory, inhibitory and other), oligodendrocytes, astrocytes, microglia and endothelial cells. We then predicted the cell-type proportions as previously described4 (Supplementary Note). However, to enable the joint analysis of samples, we chose to correct the log2-transformed transcript-per-million gene counts for 20 RNA-seq quality metrics using OLS as we observed that this removed dataset biases in the predictions. To maintain the information captured by relative expression differences between genes required for deconvolution, we rescaled the residuals to the original log2-transformed mean and standard deviation, and replaced negative values with zero. For the deconvolution step, we used the non-negative least squares95 implementation in SciPy (v1.4.1)96. Given that the average proportions of cell subtypes were often very low (that is, <1%; Supplementary Fig. 16), we opted to sum all the subtypes of cells for excitatory neurons, inhibitory neurons and oligodendrocytes (oligodendrocyte precursor cells and oligodendrocytes). We observed a high correlation between the predicted cell proportions and PCs, indicating that cell-proportion differences contribute to a substantial variance in bulk gene expression levels (Supplementary Fig. 36).
Using the predicted cell-type proportions, we aimed to identify cell type-dependent eQTLs. To increase the robustness of our results, we excluded 50 samples with a cell-proportion z-score > 4 on one or more cell type and limited the analysis to eQTLs with <95% missingness per dataset, a joint MAF > 5% and a joint Hardy–Weinberg P < 0.0001. With the remaining 25,497 eQTLs and 2,633 samples, we used Decon-QTL19, which employs a non-negative least-squares model to identify cell-type interaction effects. For this analysis, we used the steps as described in the Decon-QTL manuscript19. For the pre-processing of the TMM expression counts, we corrected for dataset indicator variables, 20 RNA-seq alignment metrics and four genotype multidimensional scaling components using OLS. As an additional step, we forced the data to the normal distribution per gene to reduce outliers. Finally, we evaluated whether the multiple-testing correction applied by Decon-QTL properly reflects the null distribution by comparing a permutation-based method to the default BH-FDR multiple-testing correction. We found that the vast majority (87.76%) of FDR significant interactions were also significant using permutations (Supplementary Note and Supplementary Figs. 37,38).
To confirm cell type-specific eQTL effects identified in Cortex-EUR, we used three replication datasets: Cortex-AFR and the snRNA-seq datasets Bryois et al.24 and ROSMAP4. For the Cortex-AFR replication, we applied the same cell-type prediction and Decon-QTL interaction analysis as for Cortex-EUR. Over the ieQTLs significant in Cortex-EUR, we calculated BH-FDR estimates and deemed ieQTLs with a BH-FDR < 0.05 as significant. Given that Decon-QTL does not return any standard errors, we predicted β and standard errors using the sample size, MAF, interaction β and interaction P value97 to calculate the Rb metrics. For the ROSMAP dataset, encompassing 80,660 single-nucleus transcriptomes from the prefrontal cortex of 48 individuals with varying degrees of AD pathology23, we re-processed the expression matrix to create a pseudo-bulk expression matrix for each broad cell type and subsequently mapped cis-eQTLs using the same procedure as the trans-eQTL analysis in bulk data (Supplementary Note and Supplementary Figs. 39,40). To correct for multiple testing, we confined the analysis to only test for primary cis- or trans-eQTLs that had a significant interaction with one or more cell types in MetaBrain Cortex-EUR (BH-FDR < 0.05), while also permuting the sample labels 100 times. Finally, we attempted replication of our findings using the snRNA-seq eQTL summary statistics of the recent preprint by Bryois and colleagues24. We overlapped their summary statistics with the Cortex-EUR cis-eQTLs and found (depending on the cell type) that between 9,402 and 13,764 overlapped. We calculated a BH-FDR on the P values of the ieQTLs that were significant in Cortex-EUR in the respective cell type. Given that the summary statistics did not include standard errors or MAF values, we predicted β and standard errors using the MetaBrain Cortex-EUR MAF together with the eQTL sample size, β and P value97 from Bryois et al. to calculate the Rb metrics.
Single-SNP MR analysis
We conducted MR between the Cortex-EUR eQTLs and 31 neurological traits (21 neurological disease outcomes, two quantitative traits and eight brain-volume outcomes; Supplementary Table 11). For this purpose, we used the Wald ratio method, which computes the change in disease risk per s.d. change in gene expression, explained through the cis-eQTL instrument(s) for that gene. To obtain our instruments, Cortex-EUR eQTLs at a genome-wide significant P-value threshold (P < 5 × 10−8) were selected and then LD-clumped using the ld_clump() function in the ieugwasr package v0.1.4 (ref. 98) with the default settings (10,000 kb clumping window with r2 cut-off of 0.001 using the 1000 Genomes EUR reference panel). SNP associations for each of the eQTL instruments were then looked up in the outcome GWAS. If the SNP could not be found in the outcome GWAS using the dbSNP rsid, then a proxy search was performed to extract the next closest SNP available in terms of pairwise LD, providing a minimum r2 threshold of 0.8 with the eQTL. These steps were performed using the associations() function in the ieugwasr package. To ensure correct orientation of effect alleles between the eQTL and outcome associations, the SNP effects were harmonized using the harmonise_data() function in TwoSampleMR70 selecting Action 2, which assumes that the alleles are forward-stranded in the GWASs (so no filtering or re-orientation of alleles according to frequency was conducted on the palindromic SNPs). Single-SNP MR was then performed using the mr_singlesnp() function in TwoSampleMR. We reported all of the MR findings that passed a P-value threshold of 5 × 10−5 but note that the Bonferroni-corrected P = 0.05 threshold for multiple-testing correction is P = 1.43 × 10−7.
Co-localization
Following the MR analysis, co-localization analysis was performed on the MR findings that passed the suggestive threshold to determine whether the eQTL and trait shared the same underlying signal. We ran co-localization28 using both the default parameters (p1 = p2 = 10−4 and p12 = 10−5) and parameters based on the number of SNPs in the region (p1 = p2 = 1/(number of SNPs in the region) and p12 = p1 / 10). We considered the traits to co-localize if either of the parameter runs yielded PP4 > 0.7. In addition, a systematic co-localization analysis was performed to compare findings between Cortex-EUR eQTLs and other existing cortex eQTL datasets (Supplementary Note).
Ethical compliance
All cohorts included in this study enrolled participants with informed consent and collected and analyzed data in accordance with ethical and institutional regulations. Information about individual institutional review board approvals is available in the original publications for each cohort. Where applicable, data access agreements were signed by the investigators previous to acquisition of the data, either to the UMCG (AMP-AD, CMC, GTEx, CMC_HBCC, BrainSeq, UCLA_ASD, BrainGVEX, BipSeq and NABEC) or Biogen (TargetALS and Braineac), which state the data usage terms. To protect the privacy of the participants, data access was restricted to the investigators of this study, as defined in those data access agreements. Per data use agreements, only summary level data is made publicly available and strictly mentioned in the disclaimer that they cannot be used to re-identify study participants.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Our study is comprised of previously published human brain eQTL datasets. The majority of these datasets are available on request or through online repositories after signing data access agreements. Summary statistics for the performed cis-eQTL analyses are available through the MetaBrain website (https://www.metabrain.nl), after registering name, institute and e-mail address. The mode of access for each of the included datasets was as follows. TargetALS12 data were pushed directly from the New York Genome center (https://www.targetals.org/) to our SFTP server. CMC99 (accession code: syn2759792), CMC_HBCC (accession code: syn10623034), AMP-AD5 (accession code: syn2580853; the snRNA-seq data were collected using the Synapse accession code syn18485175 and the IHC data were from https://github.com/ellispatrick/CortexCellDeconv/tree/master/CellTypeDeconvAnalysis/Data), BrainSeq (accession code: syn12299750), UCLA_ASD data (accession code: syn4587609), BrainGVEx (accession code: syn4590909) and BipSeq (accession code: syn5844980) data were downloaded from Synapse (https://www.synapse.org/) using synapseclient (https://python-docs.synapse.org/build/html/index.html). GTEx69 was downloaded from the Sequence Read Archive (SRA) using fastq-dump of the SRA toolkit (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?cmd=show&f=software&m=software&s=software). Access was requested and granted through dbGaP (accession code: phs000424.v7.p2). Braineac7 data were pushed to our SFTP server by Biogen. The identifiers of the 76 included studies and 2,021 brain samples downloaded from the ENA13 are listed in Supplementary Table 31. NABEC data (accession code: phs001301.v1.p1) were downloaded from dbgap. The Bryois et al. eQTL summary statistics were downloaded from Zenodo in January 2022 from https://doi.org/10.5281/zenodo.5543734. Other databases or datasets that we have used: SkyMap100. Source data are provided with this paper.
Code availability
Code is available at Zenodo (https://doi.org/10.5281/zenodo.7376855)101. The eQTL mapping software (cis) is available at https://github.com/molgenis/systemsgenetics/tree/master/mbQTL. The eQTL mapping software (trans) is available at https://github.com/molgenis/systemsgenetics/tree/master/eqtl-mapping-pipeline/. Other custom scripts used in this manuscript are available at https://github.com/molgenis/metabrain. The allelic fold change code is available at https://github.com/secastel/aFC.
References
Vos, T. et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 396, 1204–1222 (2020).
Patterson, C. World Alzheimer Report 2018. The state of the art of dementia research: New frontiers. (Alzheimer’s Disease International, 2018). https://www.alzint.org/u/WorldAlzheimerReport2018.pdf
Donovan, M. K. R., D’Antonio-Chronowska, A., D’Antonio, M. & Frazer, K. A. Cellular deconvolution of GTEx tissues powers discovery of disease and cell-type associated regulatory variants. Nat. Commun. 11, 955 (2020).
Wang, D. et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 362, eaat8464 (2018).
Raj, T. et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat. Genet. 50, 1584–1592 (2018).
Hodes, R. J. & Buckholtz, N. Accelerating medicines partnership: Alzheimeras disease (AMP-AD) knowledge portal aids Alzheimeras drug discovery through open data sharing. Expert Opin. Therapeutic Targets 20, 389–391 (2016).
Ramasamy, A. et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci. 17, 1418–1428 (2014).
The PsychENCODE Consortium. Revealing the brain’s molecular architecture. Science 362, 1262–1263 (2018).
Fromer, M. et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 19, 1442–1453 (2016).
BrainSeq Consortium. BrainSeq: neurogenomics to drive novel target discovery for neuropsychiatric disorders. Neuron 88, 1078–1083 (2015).
Gibbs, J. R. et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 6, e1000952 (2010).
Prudencio, M. et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 18, 1175–1182 (2015).
Leinonen, R. et al. The European Nucleotide Archive. Nucleic Acids Res. 39, D28–D31 (2011).
Võsa, U. et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53, 1300–1310 (2021).
Dobbyn, A. et al. Landscape of conditional eQTL in dorsolateral prefrontal cortex and co-localization with schizophrenia GWAS. Am. J. Hum. Genet 102, 1169–1184 (2018).
Foley, C. N. et al. A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat. Commun. 12, 764 (2021).
Fu, J. et al. Unraveling the regulatory mechanisms underlying tissue-dependent genetic variation of gene expression. PLoS Genet. 8, e1002431 (2012).
Glastonbury, C. A., Couto Alves, A., El-Sayed Moustafa, J. S. & Small, K. S. Cell-type heterogeneity in adipose tissue is associated with complex traits and reveals disease-relevant cell-specific eQTLs. Am. J. Hum. Genet. 104, 1013–1024 (2019).
Aguirre-Gamboa, R. et al. Deconvolution of bulk blood eQTL effects into immune cell subpopulations. BMC Bioinform 21, 243 (2020).
Patrick, E. et al. Deconvolving the contributions of cell-type heterogeneity on cortical gene expression. PLoS Comput. Biol. 16, e1008120 (2020).
Herculano-Houzel, S. The human brain in numbers: a linearly scaled-up primate brain. Front. Hum. Neurosci 3, 31 (2009).
von Bartheld, C. S., Bahney, J. & Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: a review of 150 years of cell counting. J. Comp. Neurol. 524, 3865–3895 (2016).
Mathys, H. et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019).
Bryois, J. et al. Cell-type-specific cis-eQTLs in eight human brain cell types identify novel risk genes for psychiatric and neurological disorders. Nat. Neurosci. 25, 1104–1112 (2022).
Ng, B. et al. Using transcriptomic hidden variables to infer context-specific genotype effects in the brain. Am. J. Hum. Genet. 105, 562–572 (2019).
Zhao, B. et al. Large-scale GWAS reveals genetic architecture of brain white matter microstructure and genetic overlap with cognitive and mental health traits (n = 17,706). Mol. Psychiatry 26, 3943–3955 (2021).
Guerreiro, S., Privat, A.-L., Bressac, L. & Toulorge, D. CD38 in neurodegeneration and neuroinflammation. Cells 9, 471 (2020).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 365, eaav7188 (2019).
GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
International Multiple Sclerosis Genetics Consortium & The Wellcome Trust Case Control Consortium 2 Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219 (2011)..
Jones, G., Prosser, D. E. & Kaufmann, M. 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): its important role in the degradation of vitamin D. Arch. Biochem. Biophys. 523, 9–18 (2012).
Agnello, L. et al. CYP27A1, CYP24A1, and RXR-α polymorphisms, vitamin D, and multiple sclerosis: a pilot study. J. Mol. Neurosci. 66, 77–84 (2018).
Pierrot-Deseilligny, C. & Souberbielle, J. C. Is hypovitaminosis D one of the environmental risk factors for multiple sclerosis? Brain 133, 1869–1888 (2010).
Rhead, B. et al. Mendelian randomization shows a causal effect of low vitamin D on multiple sclerosis risk. Neurol. Genet. 2, e97 (2016).
Jacobs, B. M., Noyce, A. J., Giovannoni, G. & Dobson, R. BMI and low vitamin D are causal factors for multiple sclerosis: a Mendelian randomization study. Neurol. Neuroimmunol. Neuroinflamm 7, e662 (2020).
Jiang, X., Ge, T. & Chen, C.-Y. The causal role of circulating vitamin D concentrations in human complex traits and diseases: a large-scale Mendelian randomization study. Sci. Rep. 11, 184 (2021).
Ramasamy, A. et al. Genetic evidence for a pathogenic role for the vitamin D3 metabolizing enzyme CYP24A1 in multiple sclerosis. Mult. Scler. Relat. Disord. 3, 211–219 (2014).
van Luijn, M. M. et al. Multiple sclerosis-associated CLEC16A controls HLA class II expression via late endosome biogenesis. Brain 138, 1531–1547 (2015).
Li, Z. et al. Genetic variants associated with Alzheimer’s disease confer different cerebral cortex cell-type population structure. Genome Med. 10, 43 (2018).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022).
Li, Z. et al. The TMEM106B FTLD-protective variant, rs1990621, is also associated with increased neuronal proportion. Acta Neuropathol. 139, 45–61 (2020).
Ren, Y. et al. TMEM106B haplotypes have distinct gene expression patterns in aged brain.Mol. Neurodegeneration 13, 35 (2018).
Yang, C. et al. Rewiring neuronal glycerolipid metabolism determines the extent of axon regeneration. Neuron 105, 276–292 (2020).
Sankaran, V. G. et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 322, 1839–1842 (2008).
Jiang, J. et al. cMYB is involved in the regulation of fetal hemoglobin production in adults. Blood 108, 1077–1083 (2006).
Métais, J.-Y. et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv 3, 3379–3392 (2019).
Liu, M. et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet. 51, 237–244 (2019).
Karlsson Linnér, R. et al. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat. Genet. 51, 245–257 (2019).
Yu, J. et al. Zinc-finger protein 331, a novel putative tumor suppressor, suppresses growth and invasiveness of gastric cancer. Oncogene 32, 307–317 (2013).
Ge, X. et al. Prognostic biomarker ZNF311 and its correlation with tumor progression and immune infiltrates in Glioma. World Neurosurg. 151, e37–e46 (2021).
Camby, I. et al. Differential expression of S100 calcium-binding proteins characterizes distinct clinical entities in both WHO grade II and III astrocytic tumours. Neuropathol. Appl Neurobiol. 26, 76–90 (2000).
Yang, R. et al. POU2F2 regulates glycolytic reprogramming and glioblastoma progression via PDPK1-dependent activation of PI3K/AKT/mTOR pathway. Cell Death Dis 12, 433 (2021).
Pers, T. H. et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat. Commun. 6, 5890 (2015).
Deelen, P. et al. Improving the diagnostic yield of exome- sequencing by predicting gene–phenotype associations using large-scale gene expression analysis. Nat. Commun. 10, 2837 (2019).
Bakker, O. B. et al. Linking common and rare disease genetics through gene regulatory networks. Preprint at medRxiv https://doi.org/10.1101/2021.10.21.21265342 (2021).
Ripke, S. et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019).
van Rheenen, W. et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 53, 1636–1648 (2021).
Leal, S. S. & Gomes, C. M. Calcium dysregulation links ALS defective proteins and motor neuron selective vulnerability. Front. Cell Neurosci. 9, 225 (2015).
Conroy, J. et al. A novel locus for episodic ataxia: UBR4 the likely candidate. Eur. J. Hum. Genet. 22, 505–510 (2014).
Redondo, J. et al. Purkinje cell pathology and loss in multiple sclerosis cerebellum. Brain Pathol. 25, 692–700 (2015).
Baecher-Allan, C., Kaskow, B. J. & Weiner, H. L. Multiple sclerosis: mechanisms and immunotherapy. Neuron 97, 742–768 (2018).
Wijst, M. G. Pvander et al. Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat. Genet. 50, 493–497 (2018).
Jerber, J. et al. Population-scale single-cell RNA-seq profiling across dopaminergic neuron differentiation. Nat. Genet. 53, 304–312 (2021).
Trubetskoy, V. et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604, 502–508 (2022).
Davenport, E. E. et al. Discovering in vivo cytokine-eQTL interactions from a lupus clinical trial. Genome Biol. 19, 168 (2018).
GTEx Consortium. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 7, e34408 (2018).
Hemani, G., Bowden, J. & Davey Smith, G. Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum. Mol. Genet. 27, R195–R208 (2018).
Wallace, C. Eliciting priors and relaxing the single causal variant assumption in colocalisation analyses. PLoS Genet. 16, e1008720 (2020).
Wang, X. & Goldstein, D. B. Enhancer domains predict gene pathogenicity and inform gene discovery in complex disease. Am. J. Hum. Genet. 106, 215–233 (2020).
Umans, B. D., Battle, A. & Gilad, Y. Where are the disease-associated eQTLs? Trends Genet. 37, 109–124 (2021).
The Synapse Engineering Team. synapseclient: A client for Synapse, a collaborative compute space that allows scientists to share and analyze data together. https://pypi.org/project/synapseclient/
Swertz, M. A. et al. The MOLGENIS toolkit: rapid prototyping of biosoftware at the push of a button. BMC Bioinform 11, S12 (2010).
Frankish, A. et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ (2021)
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Babraham Bioinformatics. FastQC A Quality Control tool for High Throughput Sequence Data (2019) https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Broad Institute. Picard Toolkit. https://broadinstitute.github.io/picard/ (2019)
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Delaneau, O. et al. A complete tool set for molecular QTL discovery and analysis. Nat. Commun. 8, 15452 (2017).
Storey, J. D. A direct approach to false discovery rates. J. R. Stat. Soc. B 64, 479–498 (2002).
Lyon, M. et al. The variant call format provides efficient and robust storage of GWAS summary statistics. Genome Biol. 22, 32 (2021).
Buniello, A. et al. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019).
Westra, H.-J. et al. Systematic identification of trans-eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243 (2013).
Mohammadi, P., Castel, S. E., Brown, A. A. & Lappalainen, T. Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change. Genome Res. 27, 1872–1884 (2017).
Holland, D. et al. Estimating effect sizes and expected replication probabilities from GWAS summary statistics. Front. Genet. 7, 15 (2016).
Qi, T. et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat. Commun. 9, 2282 (2018).
Lawson, C. L. & Hanson, R. J. Solving Least Squares Problems (Society for Industrial and Applied Mathematics, 1995).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Elsworth, B. et al. The MRC IEU OpenGWAS data infrastructure. Preprint at bioRxiv https://doi.org/10.1101/2020.08.10.244293 (2020).
Ng, B. et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat. Neurosci. 20, 1418–1426 (2017).
Tsui, B., Dow, M., Skola, D. & Carter, H. Extracting allelic read counts from 250,000 human sequencing runs in Sequence Read Archive. In Biocomputing 2019. 196–207 (2019)
de Klein, N. et al. Software for the manuscript ‘Brain expression quantitative trait locus and network analysis reveals downstream effects and putative drivers for brain-related diseases’. Zenodo https://doi.org/10.5281/zenodo.7376855 (2022).
Acknowledgements
We thank the donors of the brain tissues underlying the RNA-seq data used for this study and their families for their willingness to donate samples for research. We also thank all researchers involved with the included cohorts for making their data available for use. We have acknowledged each of the included cohorts and datasets in the Supplementary Note. We thank the Center for Information Technology of the University of Groningen for their support and for providing access to the Peregrine high-performance computing cluster, as well as the UMCG Genomics Coordination Center, the UG Center for Information Technology and their sponsors BBMRI-NL and TarGet for storage and computing infrastructure. D.B. and T.R.G. are supported by funding from the UK Medical Research Council (MRC Integrative Epidemiology Unit at the University of Bristol, grant no. MC_UU_00011/4) and a sponsored research collaboration with Biogen. L.F. is supported by a grant from the Dutch Research Council (grant no. ZonMW-VICI 09150182010019), an ERC Starting Grant (grant agreement 637640 (ImmRisk)), an Oncode Senior Investigator grant and a sponsored research collaboration with Biogen. This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 772376–EScORIAL to J.V.). The authors thank the Biogen CellMap team (Z. Ouyang, N. Bourgeois, E. Lyashenko, P. Cundiff, K. Li, X. Zhang, F. Casey, S. Engle, R. Kleiman, B. Zhang and M. Zavodszky) for the advice they provided towards deriving the cell type-specific expression profiles.
Author information
Authors and Affiliations
Contributions
N.d.K., O.E.G. and H.-J.W. processed the RNA-seq and genotype data. N.d.K. and H.-J.W. were responsible for data management as well as for the cis-eQTL analysis. H.-J.W. was responsible for the trans-eQTL analysis. M.V. was responsible for, with assistance from Z.O. and M.I.Z., the cell-type proportion prediction. M.V. and R.O. were responsible for the ROSMAP single-nucleus data processing. N.d.K. and M.V. were responsible for the cell-type interaction analysis. S.v.D. was responsible for the selection of brain samples from the ENA. D.B., Y.H., C.-Y.C., E.E.M., T.R.G. and E.A.T. were responsible for the MR and co-localization analysis and interpretation. J.V., M.K.B. and W.v.R. supplied ALS GWAS summary statistics and aided in the interpretation of ALS-related results. P.D., O.B.B. and L.F. were responsible for the Downstreamer analysis. L.F., E.A.T. and H.R. acquired funding and supervised the study. N.d.K., E.A.T., M.V., D.B., Y.H., C.-Y.C., O.B.B., H.R., L.F. and H.-J.W. drafted the manuscript. All authors have proof-read the manuscript.
Corresponding authors
Ethics declarations
Competing interests
E.A.T., D.B., Y.H., C.-Y.C., Z.O., E.E.M., M.I.Z. and H.R. are full-time employees and hold stock options at Biogen. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Jason Stein and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Note and Supplementary Figs. 1–40.
Supplementary Table
Supplementary Tables 1–31.
Source data
Source Data Fig. 2
Source data for Fig. 2.
Source Data Fig. 3
Source data for Fig. 3.
Source Data Fig. 4
Source data for Fig. 4.
Source Data Fig. 5
Source data for Fig. 5.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
de Klein, N., Tsai, E.A., Vochteloo, M. et al. Brain expression quantitative trait locus and network analyses reveal downstream effects and putative drivers for brain-related diseases. Nat Genet 55, 377–388 (2023). https://doi.org/10.1038/s41588-023-01300-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-023-01300-6
This article is cited by
-
PICALO: principal interaction component analysis for the identification of discrete technical, cell-type, and environmental factors that mediate eQTLs
Genome Biology (2024)
-
Brain tissue- and cell type-specific eQTL Mendelian randomization reveals efficacy of FADS1 and FADS2 on cognitive function
Translational Psychiatry (2024)
-
Identification of 27 allele-specific regulatory variants in Parkinson’s disease using a massively parallel reporter assay
npj Parkinson's Disease (2024)
-
Genomic insights into the comorbidity between type 2 diabetes and schizophrenia
Schizophrenia (2024)
-
Proteo-genomics of soluble TREM2 in cerebrospinal fluid provides novel insights and identifies novel modulators for Alzheimer’s disease
Molecular Neurodegeneration (2024)
