
Abstract
The category of “oncocytic renal tumors’’ includes well-recognized entities, such as renal oncocytoma (RO) and eosinophilic variant of chromophobe renal cell carcinoma (eo-ChRCC), as well as a group of “gray zone” oncocytic tumors, with overlapping features between RO and eo-ChRCC that create ongoing diagnostic and classification problems. These types of renal tumors were designated in the past as “hybrid oncocytoma-chromophobe tumors”. In a recent update, the Genitourinary Pathology Society (GUPS) proposed the term “oncocytic renal neoplasm of low malignant potential, not further classified”, for such solitary and sporadic, somewhat heterogeneous, but relatively indolent tumors, with equivocal RO/eo-ChRCC features. GUPS also proposed that the term “hybrid oncocytic tumor” be reserved for tumors found in a hereditary setting, typically arising as bilateral and multifocal ones (as in Birt–Hogg–Dubé syndrome). More recent developments in the “gray zone” of oncocytic renal tumors revealed that potentially distinct entities may have been “hidden” in this group. Recent studies distinguished two new entities: “Eosinophilic Vacuolated Tumor” (EVT) and “Low-grade Oncocytic Tumor” (LOT). The rapidly accumulated evidence on EVT and LOT has validated the initial findings and has expanded the knowledge on these entities. Both are uniformly benign and are typically found in a sporadic setting, but rarely can be found in patients with tuberous sclerosis complex. Both have readily distinguishable morphologic and immunohistochemical features that separate them from similar renal tumors, without a need for detailed molecular studies. These tumors very frequently harbor TSC/MTOR mutations that are however neither specific nor restricted to these two entities. In this review, we outline a proposal for a working framework on how to classify such low-grade oncocytic renal tumors. We believe that such framework will facilitate their handling in practice and will stimulate further discussions and studies to fully elucidate their spectrum.
Similar content being viewed by others
Why and when novel renal entities should be recognized?
What are the key principles of renal tumor classification?
Classification of renal neoplasia has evolved tremendously during the past half a century since its seminal division into “clear cell type” and “granular cell type”. In the contemporary practice, the key criteria for establishing any new entity include a stereotypical or recognizable morphology (or a set/constellation of morphologies), a reproducible (or specific) immunohistochemical profile, a consistent molecular/genetic profile, and an expected biologic behavior. Thus, the true question is not whether we “need” more entities, but whether a potential “new” entity fulfils the accepted recognition criteria, and whether its recognition results in a more accurate, improved diagnosis that leads to better patient treatment and prognosis. This process also helps to understand better the biologic complexity of renal neoplasia. In fact, pathologists are uniquely positioned to perform and lead this task, owing to their key diagnostic responsibilities.
Recognition of new entities reduces further the category of “unclassified renal tumors/carcinomas”
The need to recognize the evolving renal tumor landscape also stems from the fact that some new and currently emerging entities have been either “hidden” in the previously recognized and established ones, or have created perennial diagnostic and classification problems, because they did not fit into any of the already recognized categories. In practice, such diagnostic struggles translate into imprecise and descriptive diagnostic sign-outs, with a bottom line indicating “unclassified renal carcinoma (tumor), low grade or high-grade”. Thus, the recognition of novel entities reduces further the category of “unclassified renal carcinomas/tumors” and informs and improves the diagnostic practice. The process of entity “recognition” will ultimately be validated by the “test of time”, resulting in identification and recognition (or not) of the proposed new entities in practice.
Morphology and immunohistochemistry remain essential in identifying novel renal entities in the current era of “histo-molecular classification”
Currently, the morphology, complemented by a relatively limited immunohistochemistry panel, remains the cornerstones of the diagnosis of renal neoplasia, both old and new. In the last couple of decades, however, there was an evolution toward a blended “histo-molecular classification” of renal entities. Figuring out rare, new, and emerging renal tumors, by using novel techniques to study their underlying molecular pathogenesis and pathways involved, also helps us understand better the common tumors. In other words, understanding the molecular tumoral changes may clarify and help understand better the morphology and vice versa. Through this process of discovery, we may also identify clinical links and hereditary and syndromic associations of such novel and emerging tumors. It is therefore understandable why this topic has been a focus of ongoing research efforts, resulting in evaluations and reevaluations of the category of “unclassified” renal tumors1,2,3,4,5.
Novel, emerging and provisional renal entities—a practical approach
Several new and emerging renal tumors have been described in the literature since the publication of the World Health Organization (WHO) classification of genitourinary tumors in 2016. The growing evidence for the existence of such entities was also discussed at the International Society of Urologic Pathology (ISUP) consultation conference, and, in particular, the group of eosinophilic/oncocytic tumors was one of the subjects of interest6.
To further facilitate these efforts, the Genitourinary Pathology Society (GUPS) recently proposed criteria and a step-wise approach in recognizing new renal entities. GUPS introduced three categories: (i) “Novel entity”, validated by multiple independent studies; (ii) “Emerging entity”, with good compelling data available from at least two or more independent studies, but additional validation needed; and (iii) “Provisional entity”, with limited data available from one or two studies, requiring more work to validate them4.
Renal oncocytoma, chromophobe renal cell carcinoma, and cases “in-between”—why do we need a classification framework for oncocytic renal tumors?
Current criteria for the diagnosis of renal oncocytoma and chromophobe renal cell carcinoma
Traditionally, the group of oncocytic renal tumors included well recognized entities, such as renal oncocytoma (RO) and eosinophilic variant of chromophobe renal cell carcinoma (eo-ChRCC). With strict adherence to most typical features of RO and ChRCC on morphology and immunohistochemistry, an accurate and straightforward diagnosis can be established in majority of cases, according to the established criteria.7 Both RO and ChRCC workup often includes immunohistochemical evaluation for CD117 (KIT) and CK78. CD117 (KIT) is typically positive in both RO and eo-ChRCC, while diffuse reactivity for CK7 is generally considered to favor ChRCC, whereas CK7 labeling in oncocytoma is very focal, usually restricted to scattered cells; a threshold of 5% was suggested in a survey of genitourinary pathologists8. RO often exhibits a diploid karyotype, loss of chromosomes 1 or 14, or a few recurring rearrangements9. Conversely, ChRCC typically shows multiple chromosomal losses, including 1, 2, 6, 10, 13, 17, 21, and Y10. Numerous techniques for differentiating these two tumors have been evaluated in the past, including histochemical stains, immunohistochemistry, chromosomal changes, molecular assays, and electron microscopy8,11,12. More recent, comprehensive molecular studies have also focused on separating RO and ChRCC, using their complex molecular profile signatures13,14. In our view, however, HE staining and morphology remain the key for the routine diagnosis of oncocytoma. In limited specimens (for example, needle core biopsies) and when diagnostically necessary, an immunohistochemistry evaluation can support the diagnosis. Molecular genetic techniques may play a role in a research setting or to rule out a hereditary condition (for example, a Birt–Hogg–Dubé (BHD) syndrome and renal oncocytosis).
What about the cases “in between”—a historical perspective on an ongoing conundrum
However, the group of “oncocytic” tumors also contains tumors with “in-between” features, demonstrating overlapping and equivocal morphology between RO and eo-ChRCC, often with mixed, and sometimes confusing, immunohistochemical profiles. These were typically designated in the past as “hybrid oncocytoma-chromophobe tumors” (HOCT)15,16,17. When multiple and bilateral, such tumors with overlapping features between RO and eo-ChRCC, typically occur in a BHD syndrome and in renal oncocytosis18,19. Despite the debates and the discussions during the past two decades about the use and the exact meaning of the term “hybrid”, this term was invariably used both in practice and in published studies. In 2012, at the Vancouver ISUP Consensus Conference, HOCT was for the first time introduced into the official classification. In fact, HOCT was not separated as a specific entity, but it was proposed that HOCT should be provisionally included in the category of unusual ChRCC (with recommendation to include a detailed comment in the pathology report)20. The subsequent 2016 WHO classification of renal tumors essentially replicated this approach21. However, in daily clinical practice and in the literature, a broad spectrum of variable names were used to label such tumors, including “oncocytic RCC, NOS”, “oncocytic, low-grade RCC”, “oncocytic tumor, favor RO (or ChRCC)“, “unclassified oncocytic tumor”, “oncocytic tumor with uncertain/low malignant potential”, and “borderline tumor” (or with “borderline features”)7,8,17,22. Such terminological variability used for the tumors in the HOCT group reflected their heterogeneity, variable morphologies, immunohistochemical findings, and molecular-genetic features.
In the recent review update on existing renal tumors, GUPS proposed the term “oncocytic renal neoplasm of low malignant potential, not further classified”, to designate the tumors in this somewhat heterogeneous group of neoplasms, demonstrating borderline and equivocal features between RO and ChRCC7. In particular, it was suggested that this term should be restricted only to solitary and sporadic tumors with overlapping features7. For example, such category may include cases from a few recent studies labeled as “RO variants”23, “hybrid oncocytic/chromophobe renal tumors”24, or “oncocytic renal neoplasms with diffuse keratin 7 immunohistochemistry”25. GUPS also proposed that the term “hybrid oncocytic tumor” be reserved only for hereditary cases, that are typically bilateral and multifocal (as in BHD syndrome, illustrated in Fig. 1)7,8,26,27. A prospective standardized use of these two diagnostic categories—“oncocytic renal neoplasm of low malignant potential, not further classified”, for sporadic rumors, and “hybrid oncocytic tumor”, for hereditary ones, would create a working framework for reproducible classification of such cases7.

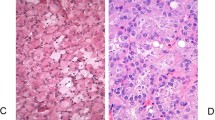
The patient had multifocal bilateral renal tumors, as well as multiple renal, liver, and lung cysts. A representative renal tumor is illustrated. A The tumor is non-encapsulated with solid growth; rare entrapped tubules are present at the periphery. Scattered cells with clear cytoplasm (“mosaic pattern”) can also be seen. B Higher magnification shows equivocal morphology between oncocytoma (mostly eosinophilic cells with round to oval nuclei) and chromophobe renal cell carcinoma (focal prominent cell membranes and nuclear halos). C Higher magnification of areas with “mosaic pattern” demonstrates scattered cells with clear cytoplasm, admixed with cells with eosinophilic cytoplasm. D Cytokeratin 7 is focally positive. E CD117 (KIT) is weakly positive only in focal areas. F Cathepsin K is diffusely positive.
Lumping different renal entities together is not the best solution—what are the implications?
Lastly, it should be mentioned that the tumors in this “oncocytic group” are generally expected to behave indolently, similar to RO and eo-ChRCC. Thus, it can be argued that lumping them in one group/category would make more sense, because separating them into more benign “new entities” does not really matter. When considering this question, in our view, it is important to avoid the pitfall of “lumping” cases in types/categories where they do not belong, which has been a revolving story in the classification of renal neoplasia. The “lumping” approach in the past often led to confusions, meandering, and delays in the formal recognition of some entities. For example, for many years, clear cell papillary RCC was (incorrectly) considered to be part of the low-end spectrum of the clear cell carcinoma, primarily based on morphology, despite its different immunoprofile with diffuse CK7 and consistent absence of VHL mutations/abnormalities. Moreover, the molecular profile of clear cell papillary RCC is “metabolically stable” and it is different from clear cell RCC; clear cell papillary RCC also has uniformly indolent behavior and it is now considered a separate entity7. The recognition of RCC with fibromyomatous stroma (FMS) has also been complicated by the use of different terminologies, lack of or incomplete molecular and genetic data in some studies, and difficulties in reproducing the findings between studies. Indeed, there are some clear cell RCCs that show a morphologic overlap with RCC FMS, and contain a significant component of smooth muscle stroma and variable CK7 expression. However, such clear cell RCCs typically have VHL mutations/abnormalities, and do not harbor TSC/MTOR mutations or TCEB1/ELOC mutations, as found in RCC FMS28,29. Therefore, some cases would require a molecular evaluation of VHL, TSC/MTOR, and TCEB1/ELOC genes that can be done in a reference lab, if not possible in-house, to establish the definitive diagnosis. On the other hand, it is also of paramount importance to fully establish the long-term indolent behavior of some of the recently proposed entities, for example eosinophilic vacuolated tumor (EVT) and low-grade oncocytic tumor (LOT), as the number of reported cases with long follow-up is still limited. Once the analytical part of the study of such potentially new entities is completed, a clinically relevant, evidence-based, and possibly simpler classification can be put forth. But we are not there yet.
It is also important to acknowledge that when a diagnosis of “cancer” is established or mentioned in the diagnostic line (for example eo-ChRCC, or “unclassified RCC”), this matters to the patients and the clinicians. The word “cancer” carries a potential psychological burden for the patient and associated healthcare costs, as well as possible radiation exposure from surveillance and follow up imaging studies30. However, if a definitive benign diagnosis can be established with certainty, for example on needle-biopsy, such patients can be managed conservatively.
Chromophobe RCC, eosinophilic variant should be diagnosed using strict diagnostic criteria
A proposal for precise diagnostic criteria for chromophobe RCC, eosinophilic variant
A major conundrum in the diagnosis of renal oncocytic tumors stems from the lack of stringent criteria for the diagnosis of eo-ChRCC (Fig. 2). Based on the current 2016 WHO classification, the only requirement is that eo-ChRCC is “almost purely composed of eosinophilic cells”10. This is probably the main reason why some emerging oncocytic tumors with overlapping features have been included as “eo-ChRCC” in previous ChRCC studies, or as “RO” in the respective oncocytoma studies. For example, in 2011 Przybycin et al found in a large institutional cohort of 203 ChRCC that 16% of all ChRCC had a dominant population of >80% “oncocytoma-like eosinophilic cells” and “non-classic architecture”, which highlights the possibility of a morphologic overlap with similar entities. Importantly, in their study such cases lacked any adverse events on follow-up, in contrast to the typical/classic type ChRCC that showed potential for adverse behavior27. It was also proposed recently that “absence of pale cells could be used as the main histologic criterion to characterize eo-ChRCC”31, which may also be suboptimal, if used as the sole criterion for the diagnosis.

A The tumor shows broad trabecular and nested architecture and it is composed exclusively of eosinophilic cells. B At higher magnification, irregular (raisinoid) nuclei can be easily appreciated with extensive perinuclear halos (clearings). C CD117 (KIT) is diffusely positive. D Cytokeratin 7 is only focally positive, in contrast to diffuse reactivity typically seen in chromophobe renal cell carcinoma. However, such a result should be carefully correlated with the morphology, which in this case supports the diagnosis of chromophobe renal cell carcinoma (eosinophilic variant).
ChRCC typically has multiple losses of chromosomes 1, 2, 6, 10, 13, 17, 21, and Y10. In addition, some rare ChRCC variants, including ChRCC with pigmented microcystic/multi-cystic adenomatoid growth, ChRCC with neuroendocrine features, and ChRCC with papillary architecture, also show variable copy number abnormalities with frequent multiple chromosomal losses and, less often, gains32. However, in a large Japanese-Swiss cohort of ChRCC, Ohashi et al showed complete absence of any chromosomal losses in 10/24 (41.7%) cases diagnosed as “eo-ChRCC”, whereas only 6/69 (8.7%) of the “classic/typical ChRCC” showed absence of any chromosomal loss31.
To avoid further diagnostic confusion, and based on the previously published studies, we suggest that when diagnosing eo-ChRCC, more precise criteria should be used, primarily morphologic, and supported by immunohistochemistry, as well as, if necessary, by molecular/genetic studies:
-
1.
i) Presence of prominent, easily recognizable, nuclear irregularities (i.e. wrinkling or raisinoid appearances) (see the seminal paper by Thoenes et al., Fig. 1i)33, (ii) diffuse eosinophilic (oncocytic) appearance with absence of pale cells, and (iii) lack of any potentially adverse features, including microscopic necrosis, sarcomatoid differentiation, and vascular invasion, particular small vessel invasion26,27. ChRCC of large size (>7 cm) and higher stage (pT3a), should be particularly carefully and thoroughly sampled to ensure adequate diagnostic evaluation, because such ChRCCs are known to have potential for adverse behavior26,27.
-
2.
On immunohistochemistry, lack of diffuse CK7 reactivity and negative (or weak) CD117 (KIT), different from the CK7+/CD117+ profile typically seen in ChRCC, should be carefully correlated with the morphology, when considering a diagnosis eo-ChRCC.
-
3.
Lastly, in diagnostically equivocal cases, additional molecular genetic studies can aid in documenting major chromosomal losses (e.g. 1, 2, 6, 10, 13, 17, 21, and Y) or presence of p53 and PTEN mutations, as typically found in ChRCC vs presence of TSC/MTOR mutations, found in other similar oncocytic tumors, and typically not in eo-ChRCC.
A proposal for a working classification of low-grade oncocytic tumors with inclusion of EVT and LOT
More recent work in the “gray zone” of oncocytic renal tumors distinguished two additional entities: “Eosinophilic Vacuolated Tumor” (EVT) and “Low-grade Oncocytic Tumor” (LOT). EVT was initially described as “high-grade oncocytic tumor” and as “sporadic RCC with eosinophilic and vacuolated cytoplasm”34,35. In the recent GUPS update, a unifying name EVT was accepted for this tumor; to date, more than 40 such cases have been reported22,23,34,35,36,37. The second tumor that emerged in this group, was proposed in 2019 as “Low-grade Oncocytic Tumor” (LOT)38. Since the initial description, more than 100 patients with such tumors were documented in the literature from multiple groups23,38,39,40,41,42,43,44 (see also recent review by Mansoor et al.45). Despite their recent descriptions, both EVT and LOT have received immediate attention, resulting in subsequent studies that have confirmed and further expanded the knowledge on these tumors, supporting the notion that both likely represent separate entities23,37,38,39,40,41,42. The findings from the published studies on EVT and LOT are discussed in more detail below and are summarized in Tables 1, 2.
In summary, we would like to propose the following categories for the classification framework of oncocytic renal neoplasms:
-
1.
Renal oncocytoma
-
2.
Eosinophilic chromophobe renal cell carcinoma
-
3.
Oncocytic renal neoplasm of low malignant potential, not further classified
-
4.
Hybrid oncocytic tumor
-
5.
Eosinophilic vacuolated tumor
-
6.
Low-grade oncocytic tumor
The key morphologic and immunohistochemical features helpful to distinguish entities in this group of oncocytic renal tumors are summarized in Table 3. Since RO, eo-ChRCC, “oncocytic renal neoplasm of low malignant potential, not further classified”, and “hybrid oncocytic tumor” have been covered in the recent review update by GUPS7, in this review, we would like to provide an update on the most recent developments in EVT and LOT that have occurred after the publication of the GUPS papers.
EVT and LOT are different from other oncocytic tumors and can be diagnosed primarily by morphology and immunohistochemistry - summary of the evidence
The accumulated evidence on EVT and LOT has validated the initial findings and has further expanded the knowledge on these entities, as well as on the whole group of oncocytic renal tumors that pose diagnostic and classification challenges. Both entities appear to have readily distinguishable morphologic, immunohistochemical, genetic, and molecular features that separate them from other similar renal oncocytic tumors, supporting the conclusion that they are distinct entities. Importantly, they can be distinguished from RO and eo-ChRCC primarily by morphology and immunohistochemistry, and without detailed molecular studies. So far, it appears that both behave uniformly in a benign fashion. Moreover, in our experience, and based on some published studies, both entities are not that rare in practice, but are still labeled as RO, eo-ChRCC, or descriptively, as “in-between” cases, or as “unclassified oncocytic tumors”. For example, two recent retrospective institutional studies found that LOT represented 4% of the cases initially diagnosed as either ChRCC46 or RO39. In another recent single-institution study, LOT was found to represent 6.7% of “unclassified RCC or low-grade oncocytic/eosinophilic renal neoplasms”41.
Eosinophilic vacuolated tumor (EVT)
EVT is an oncocytic tumor characterized by the finding of large intracytoplasmic vacuoles. This tumor type was initially described by He et al as “high-grade oncocytic tumor” (abbreviated as HOT) and by Chen et al as “sporadic RCC with eosinophilic and vacuolated cytoplasm34,35. EVT was considered to be sporadic tumor in both studies; however, it was later also identified in rare patients with tuberous sclerosis complex (TSC)22,23,36,37.
Clinical features and behavior
EVT is typically detected incidentally and occurs more frequently in women (M:F = 1:2.5). It is found in patients of broad age range from 25 to 73 years (mean 50.9, median 54 years)4,34,35,47.
All reported EVT cases to date were found to have indolent behavior, without any evidence of local recurrence or metastatic disease4,48. The follow-up period was relatively long, mean 56.3 months and median 41.5 months (range 12–198 months), in the largest study of 19 cases47.
Gross description
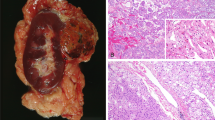
In the great majority of cases, EVT is solitary and sporadic tumor5,34,35,36,47, with only rare examples found in TSC patients, often associated with other tumors typically seen in that setting22,23,37. Based on the largest reported cohort, the mean tumor size was 4.3 cm (median 3.8 cm; range 1.5–11.5 cm)47. EVT is typically solid, gray, or tan to brown tumor (Fig. 3A) that usually lacks a well-formed capsule. Necrosis or extensive hemorrhage have not been documented grossly in the reported cases4,34,35,36,47.

A Grossly, EVT is solid, mahogany brown, and well-circumscribed tumor. B At low magnification, large vessels are invariably present at the periphery (lower left), with adjacent entrapped tubules, while a well-formed capsule is absent. C At low to medium power, EVT may superficially resemble chromophobe renal cell carcinoma. It has mostly solid architecture, often admixed with nested and tubulocystic areas (inset). D At high magnification, neoplastic cells have large intracytoplasmic vacuoles and round to oval nuclei with prominent nucleoli. E CD117 (KIT) is uniformly positive and only rare CK7 positive cells are present (inset), mimicking the immunoprofile of oncocytoma. F Cathepsin K and antimitochondrial antigen antibody (inset) are also positive in EVT.
Microscopic features
EVT typically has solid microscopic architecture, in some cases focally admixed with nested and tubulocystic areas. Thick-walled vessels are virtually always found at the periphery and entrapped tubules are also common, particularly at the border with the normal renal parenchyma (Fig. 3B, C). Although the overall appearance at low magnification resembles ChRCC or RO, the cells have often quite large intracytoplasmic vacuoles (Fig. 3D). The nuclei are round to oval, with enlarged nucleoli (corresponding to WHO/ISUP grade 3); in some cases the nucleoli can be focally quite prominent, resembling viral inclusions34,35,48.
Immunohistochemistry
EVT is positive for CD117 (KIT), CD10, antimitochondrial antigen antibody, and cathepsin K in great majority of cases, albeit in some cases focally. EVT is also uniformly positive for PAX8 and AE1/AE3, but it is completely negative for vimentin. CK7 expression is typically restricted only to scattered cells, usually not exceeding 5–10%34,36,47. Fumarate hydratase and SDHB were retained in all evaluated cases. The key immunohistochemistry findings are illustrated in Fig. 3E, F.
Molecular genetic features
The copy number variation (CNV) pattern found in EVT is somewhat variable, but loss of chromosome 1 was commonly reported, as well as losses of chromosome 19p or 19q, and loss of heterozygosity at 16p11 and 7q3134. Complete losses or gains of other chromosomes have not been found. Loss of chromosome 1, along with concurrent TSC/MTOR mutations, seem to be the key molecular genetic findings in EVT35,36. In a recent study, Farcas et al. demonstrated non-overlapping mutations in MTOR, TSC2, and TSC1 in all 19 evaluated cases, associated invariably with low mutational rates. However, in one case in their study, MTOR mutation coexisted with a RICTOR missense mutation that is also part of the MTOR pathway47. Based on the current knowledge, EVT is a renal tumor associated with either germline or somatic mutations leading to mTORC1 activation36. In our view, however, molecular-genetic testing is not necessary to diagnose great majority of EVTs, as they can be distinguished from their mimickers primarily based on their characteristic morphologic and immunohistochemical features. In cases with overlapping morphology or in cases where morphology is not convincing, analysis of MTOR pathway genes would be useful to establish the diagnosis.
Low-grade oncocytic tumor (LOT)
Clinical features and behavior
Great majority of LOT cases documented in the literature were sporadic tumors, typically detected incidentally. Rare examples have also been found in patients with TSC23,42. Although LOT is typically found as a single tumor, multiple LOT tumors, measuring from few millimeters up to 14.2 cm, have also been rarely reported, either in patients with end-stage kidney disease39, or in patients with TSC42. Lerma et al recently reported 4 patients, in whom LOT was associated with other tumors typically seen in TSC patients, including eosinophilic solid and cystic renal cell carcinoma (ESC RCC), EVT, RCC FMS, AML, and papillary adenoma23.
Overall, there was a slight female predominance (1:1.3) although the reported gender distribution has been somewhat variable in individual studies. LOT was usually found in older patients, but it was identified in patients of broad age range from 10 to 87 years. To date, all reported LOTs with available follow-up have behaved indolently, without evidence of disease progression, including metastatic disease. The mean follow-up was 42.5 months (range 0 to 344 months)23,38,39,40,41,42,46.
Gross description
LOT is typically a small tumor with median size between 3 and 4 cm, as reported in the largest studies38,39,41; in great majority of cases the stage was pT1a or pT1b. Rare large tumors have also been documented, exceeding 10 cm38,39,41.
On gross section, the tumors were solid and compact, without grossly visible necrosis or cysts. The cut surface was typically tan-yellow or brown, similar to RO. However, hemorrhagic areas may also be found, usually in the more central parts of the tumor.
Microscopic features
LOT has a solid, compact nested, or focally tubular, tubuloreticular, or trabecular growth. The tumors are sharply demarcated from the renal parenchyma, but without a capsule, and rare entrapped renal tubules may be seen at the periphery4,5,38 (Fig. 4A). The neoplastic cells are uniformly eosinophilic with finely granular cytoplasm. The nuclei are round to oval, without significant irregularities (i.e. lack “raisinoid shapes”), and may focally show delicate perinuclear halos or clearings, which can be more prominent in some cases (Fig. 4B). The nucleoli are typically inconspicuous (usually equivalent to WHO/ISUP grade 2).

A LOT shows “onocytoma-like” appearance at low power; it has typically a sold growth and capsule is absent. B At higher magnification, LOT is composed of eosinophilic cells with round to oval, “low-grade” nuclei, focally showing perinuclear halos. C, D Frequent finding is a sharp transition from solid areas into loose stromal areas that may show hemorrhage and contain scattered, often elongated and irregularly distributed, individual cells (“boats in a bay” arrangement). E CK7 is diffusely positive. F CD117 (KIT) is typically negative or it is very focally and weakly positive in rare cases. Note rare normal mast cells that mark for CD117.
A fairly characteristic finding is the presence of edematous stromal areas, sharply delineated from the solid tumoral areas (Fig. 4C, D). They are hypocellular and contain scattered individual cells, elongated cells, and/or cord-like cell formations, resembling a picture of individual “boats in a bay”. The stromal areas often contain fresh hemorrhage. Small lymphocytic aggregates can also be found in the solid areas4. Worrisome morphologic features, such as coagulative necrosis, nuclear pleomorphism, significant cell atypia, multinucleation, and mitotic activity are typically absent.
Muller-Mowry colloidal iron special stain is typically negative in LOT, at most restricted to only limited, apical/luminal reactivity38. On electron microscopy, LOT contains abundant, closely packed cytoplasmic mitochondria, similar to RO42,48. Both of these techniques, however, are rarely used in the contemporary practice as they are of limited diagnostic value.
Immunohistochemistry
When found with the appropriate morphology, the immunohistochemical profile of LOT strongly supports the diagnosis. Virtually all tumors were diffusely positive for CK7 and negative for CD117 (KIT), as illustrated in Fig. 4E, F. However, rare cases had very weak and focal CD117 (KIT) reactivity. Such CK7+/CD117- profile would be unusual for RO that typically exhibits only rare or patchy CK7 positive cells and diffuse CD117 reactivity, or for a typical ChRCC that shows diffuse staining for both. LOT is also positive for AE1/AE3, PAX8, e-cadherin, BerEP4, and MOC31. Negative stains include CAIX, CK20, CK5/6, p63, CD15, HMB45, Melan A, and vimentin. CD10 and AMACR can be either negative or focally positive. Fumarate hydratase and SDHB were retained in all examined cases. Ki67 showed reactivity in less than 5% of the cells38,40,41. We have also recently found consistent expression of GATA3 in LOT (unpublished observations and personal communication with Dr. Omar Hameed). In our experience, GATA3 is negative in RO, but GATA3 reactivity has been reported in about half of the ChRCC cases in one study49. These recent observations should be considered preliminary and require further confirmation. It has also been recently found that LOT consistently and, at least focally, expresses p-S6 and p-4EBP1, both markers associated with MTOR pathway activation42,46. Absent or very low immunohistochemical expression of another novel marker FOXI1 has also been recently found in LOT; of note, FOXI1 marks the intercalated cells in the normal kidney and it is typically expressed in both RO and ChRCC13,46,50.
Molecular genetic features
Regarding the CNV in LOT, Trpkov et al. found frequent deletions at 19p13 (7/9), 1p36 (5/9), and 19q13 (4/9), and a disomic chromosomal status in 2/9 cases38. As loss of 1p36 and diploid pattern are commonly found in RO, it has been confirmed that CCND1 rearrangements are not found in LOT (unlike in oncocytoma in which they are frequent)39. No other whole-length chromosomal gains or losses were identified in LOT.
Recent studies strongly point toward involvement of the MTOR pathway genes in LOT. Morini et al. recently identified variations in MTOR pathway related genes in 80% (8/10) of evaluated LOT cases, including MTOR (7/8) and TSC1 (1/8)46. Similarly, Kapur et al. found somatic, likely activating, mutations in MTOR (4/6) and RHEB (1/6) in 6 evaluable LOTs; additionally, one patient with multiple bilateral LOTs had a pathogenic germline mutation in TSC1 (1/6)42. Lerma et al. also found TSC1 germline mutations in two TSC patients who had multiple LOTs23. As in EVT, molecular-genetic analysis should be reserved for diagnostically uncertain cases, but should not be considered a primary diagnostic tool.
TSC/MTOR mutations are not specific or exclusive for any renal entity
An intriguing finding on molecular-genetic level has been the frequent presence of TSC/MTOR mutations in EVT and LOT, but also in some other novel and emerging entities. These mutations generally indicate activation of the MTOR pathway. While such mutations support a specific diagnosis in a set of typical morphology or constellation of morphologies, and compatible immunoprofiles, they are not specific for any individual entity. Indeed, TSC/MTOR mutations are found in TSC patients that exhibit heterogeneous spectrum of renal neoplasia51, which includes not only EVT and LOT, but also ESC RCC and RCC FMS, as well as more common ones, such as AML (or PEComa). All these entities, essentially with identical morphology, can be found much more commonly in a sporadic setting, along with TSC/MTOR mutations. However, these MTOR pathway abnormalities are nor exclusive for this group of tumors, and have been found in metastatic clear cell RCC, papillary RCC, chromophobe RCC, acquired cystic disease associated (ACKD) RCC, and also in some unclassified aggressive RCCs2,52,53,54,55,56. Therefore, the spectrum of renal tumors that have TSC/MTOR mutations is quite heterogeneous, and they also exhibit different biologic behaviors. This argues strongly against lumping all these entities in a family (or a “superfamily“) of “TSC/MTOR pathway associated renal neoplasia“, as these molecular alterations are neither specific, nor pathognomonic for any individual entity. Additional work is also needed to clarify if these mutations are driver or passenger ones, and if they are somatic or germline ones in the individual tumor types4.
Unifying terminology should be used for unclassifiable low-grade oncocytic tumors—sporadic and syndromic (hereditary) ones
We would also recommend use of uniform terminology for other tumors in this oncocytic group, as already outlined: “oncocytic renal neoplasm of low malignant potential, not further classified”, for sporadic ones that do not fit any of the categories, and “hybrid oncocytic tumor”, for hereditary ones (such as BHD syndrome)7. This framework, including the use of more precise criteria for eo-ChRCC, would result in a more reproducible categorization of such tumors, and would allow better comparison between studies7,15,16,17.
Open questions remain regarding the classification of low-grade oncocytic tumors, but constructive and evidence-based debates help move the field forward
Open questions still remain about the broader spectrum of the “hybrid oncocytic tumors” that occur in a hereditary setting, such as BHD. In our practice, these tumors have a wider morphologic spectrum than reflected in the literature, and not all of them appear prima facie to belong to the group of “oncocytic tumors”, resembling either RO, or ChRCC, or showing a typical “mosaic pattern”. For example, we have identified rare examples of such sporadic tumors with FLCN mutations, mimicking EVT on morphology47. It is also challenging to study these tumors, as they are rare and larger studies can be performed only through multi-institutional collaborations or through national registries/institutions.
We expect that this review will stimulate further discussions, and will also initiate additional studies to fully elucidate the spectrum of oncocytic renal tumors, resulting in new knowledge for their better understanding and classification.
References
Perrino, C. M. et al. Morphological spectrum of renal cell carcinoma, unclassified: An analysis of 136 cases. Histopathology 72, 305–319 (2018).
Chen, Y. B. et al. Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat. Commun. 7, 13131 (2016).
Andeen, N. K. et al. Clinical utility of chromosome genomic array testing for unclassified and advanced-stage renal cell carcinomas. Arch. Pathol. Lab. Med. 143, 494–504 (2019).
Trpkov, K. et al. Novel, emerging and provisional renal entities: The Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod. Pathol. 34, 1392–1424 (2021).
Trpkov, K. & Hes, O. New and emerging renal entities: A perspective post-WHO 2016 classification. Histopathology 74, 31–59 (2019).
Williamson, S. R. et al. Report from the International Society of Urological Pathology (ISUP) Consultation Conference on Molecular Pathology of Urogenital Cancers: III: Molecular Pathology of Kidney Cancer. Am. J. Surg. Pathol. 44, e47–e65 (2020).
Trpkov, K. et al. New developments in existing WHO entities and evolving molecular concepts: The Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod .Pathol. 34, 1392–1424 (2021).
Williamson, S. R. et al. Diagnostic criteria for oncocytic renal neoplasms: A survey of urologic pathologists. Hum. Pathol. 63, 149–156 (2017).
Hes, O. et al. Oncocytoma. In WHO Classification of Tumours of the Urinary System and Male Genital Organs. 4. (eds. Moch, H. et al.) 43–44 (Lyon, IARC, 2016).
Paner, G. et al. Chromophobe renal cell carcinoma. WHO Classification of Tumours of the Urinary System and Male Genital Organs. (eds Moch, H. et al.) 27–28 (Lyon, IARC, 2016).
Ng, K. L. et al. Differentiation of oncocytoma from chromophobe renal cell carcinoma (RCC): Can novel molecular biomarkers help solve an old problem? J. Clin. Pathol. 67, 97–104 (2014).
Ng, K. L. et al. A systematic review and meta-analysis of immunohistochemical biomarkers that differentiate chromophobe renal cell carcinoma from renal oncocytoma. J. Clin. Pathol. 69, 661–671 (2016).
Skala, S. L. et al. Next-generation RNA sequencing-based biomarker characterization of chromophobe renal cell carcinoma and related oncocytic neoplasms. Eur. Urol. 78, 63–74 (2020).
McGillivray, P. D. et al. Distinguishing benign renal tumors with an Oncocytic Gene Expression (ONEX) Classifier. Eur. Urol. 79, 107–111 (2021).
Mai, K. T., Dhamanaskar, P., Belanger, E. & Stinson, W. A. Hybrid chromophobe renal cell neoplasm. Pathol. Res. Pract. 201, 385–389 (2005).
Petersson, F. et al. Sporadic hybrid oncocytic/chromophobe tumor of the kidney: A clinicopathologic, histomorphologic, immunohistochemical, ultrastructural, and molecular cytogenetic study of 14 cases. Virchows Arch. 456, 355–365 (2010).
Hes, O., Petersson, F., Kuroda, N., Hora, M. & Michal, M. Renal hybrid oncocytic/chromophobe tumors—a review. Histol. Histopathol. 28, 1257–1264 (2013).
Pavlovich, C. P. et al. Renal tumors in the Birt-Hogg-Dube syndrome. Am. J. Surg. Pathol. 26, 1542–1552 (2002).
Gobbo, S. et al. Renal cell neoplasms of oncocytosis have distinct morphologic, immunohistochemical, and cytogenetic profiles. Am. J. Surg. Pathol. 34, 620–626 (2010).
Srigley, J. R. et al. The International Society of Urological Pathology (ISUP) Vancouver Classification of Renal Neoplasia. Am. J. Surg. Pathol. 37, 1469–1489 (2013).
Moch, H., Humphrey, P. A., Ulbright, T. M. & Reuter, V. E., eds. WHO Classification of Tumours of the Urinary System and Male Genital Organs. (Lyon, IARC, 2016).
Trpkov, K. et al. High-grade oncocytic tumour (HOT) of kidney in a patient with tuberous sclerosis complex. Histopathology 75, 440–442 (2019).
Lerma, L. A., Schade, G. R. & Tretiakova, M. S. Co-existence of ESC-RCC, EVT, and LOT as synchronous and metachronous tumors in six patients with multifocal neoplasia but without clinical features of tuberous sclerosis complex. Hum. Pathol. 116, 1–11 (2021).
Ruiz-Cordero, R. et al. Hybrid oncocytic/chromophobe renal tumors are molecularly distinct from oncocytoma and chromophobe renal cell carcinoma. Mod. Pathol. 32, 1698–1707 (2019).
Mohanty, S. K. et al. Oncocytic renal neoplasms with diffuse keratin 7 immunohistochemistry harbor frequent alterations in the mammalian target of rapamycin pathway. Mod. Pathol. 35, 361–375 (2022).
Amin, M. B. et al. Chromophobe renal cell carcinoma: Histomorphologic characteristics and evaluation of conventional pathologic prognostic parameters in 145 cases. Am. J. Surg. Pathol. 32, 1822–1834 (2008).
Przybycin, C. G. et al. Chromophobe renal cell carcinoma: A clinicopathologic study of 203 tumors in 200 patients with primary resection at a single institution. Am. J. Surg. Pathol. 35, 962–970 (2011).
Hakimi, A. A. et al. TCEB1-mutated renal cell carcinoma: A distinct genomic and morphological subtype. Mod. Pathol. 28, 845–853 (2015).
Shah, R. B. et al. “Renal Cell Carcinoma With Leiomyomatous Stroma” harbor somatic mutations of TSC1, TSC2, MTOR, and/or ELOC (TCEB1): clinicopathologic and molecular characterization of 18 sporadic tumors supports a distinct entity. Am. J. Surg. Pathol. 44, 571–581 (2020).
Stewart-Merrill, S. B. et al. Oncologic surveillance after surgical resection for renal cell carcinoma: a novel risk-based approach. J. Clin. Oncol. 33, 4151–4157 (2015).
Ohashi, R. et al. Loss of CDKN1A mRNA and protein expression are independent predictors of poor outcome in chromophobe renal cell carcinoma patients. Cancers 12, 465 (2020).
Alaghehbandan, R. et al. Comprehensive review of numerical chromosomal aberrations in chromophobe renal cell carcinoma including its variant morphologies. Adv Anat Pathol 28, 8–20 (2021).
Thoenes, W. et al. Chromophobe cell renal carcinoma and its variants—a report on 32 cases. J Pathol 155, 277–287 (1988).
He, H. et al. “High-grade oncocytic renal tumor”: Morphologic, immunohistochemical, and molecular genetic study of 14 cases. Virchow Arch. 473, 725–738 (2018).
Chen, Y. B. et al. Somatic mutations of TSC2 or MTOR characterize a morphologically distinct subset of sporadic renal cell carcinoma with eosinophilic and vacuolated cytoplasm. Am. J. Surg. Pathol. 43, 121–131 (2019).
Kapur, P. et al. Eosinophilic vacuolated tumor of the kidney: A review of evolving concepts in this novel subtype with additional insights from a case with MTOR mutation and concomitant chromosome 1 loss. Adv. Anat. Pathol. 28, 251–257 (2021).
Gupta, S. et al. Renal Neoplasia in Tuberous Sclerosis: A study of 41 patients. Mayo Clin. Proc. 96, 1470–1489 (2021).
Trpkov, K. et al. Low-grade oncocytic tumour of kidney (CD117-negative, cytokeratin 7-positive): A distinct entity? Histopathology 75, 174–184 (2019).
Kravtsov, O. et al. Low grade oncocytic tumor of kidney (CK7-positive, CD117-negative). A single institution experience with incidence and clinicopathologic characteristics. Hum. Pathol. (2021). Online ahead of print.
Guo, Q. et al. Characterization of a distinct low-grade oncocytic renal tumor (CD117-negative and cytokeratin 7-positive) based on a tertiary oncology center experience: The new evidence from China. Virchows Arch. 478, 449–458 (2021).
Akgul, M., Al-Obaidy, K. I., Cheng, L. & Idrees, M. T. Low-grade oncocytic tumour expands the spectrum of renal oncocytic tumours and deserves separate classification: A review of 23 cases from a single tertiary institute. J. Clin. Pathol. (2021). Online ahead of print.
Kapur, P. et al. Germline and sporadic mTOR pathway mutations in low-grade oncocytic tumor of the kidney. Mod. Pathol. 35, 333–343 (2022).
Ishikawa, N. et al. A case of low-grade oncocytic tumor/chromophobe renal cell carcinoma (oncocytic variant) of the kidney. Case Rep. Pathol. 2021, 6684777 (2021).
Sharma, D., Pai, T., Prakash, G., Desai, S. & Menon, S. Low-grade oncocytic tumor: Report of two cases of an emerging entity in the spectrum of oncocytic renal neoplasms. Turk. Pathol. Derg. (2021). Online ahead of print.
Mansoor, M., Siadat, F. & Trpkov, K. Low-grade oncocytic tumor (LOT) – a new renal entity ready for a prime time: An updated review. Histol. Histopathol. https://doi.org/10.14670/HH-18-435. (2022). Online ahead of print.
Morini, A. et al. Low-grade oncocytic renal tumor (LOT): Mutations in mTOR pathway genes and low expression of FOXI1. Mod. Pathol. 35, 352–360 (2022).
Farcas, M. et al. Eosinophilic vacuolated tumor (EVT) of kidney demonstrates sporadic TSC/MTOR mutations: Next-generation sequencing multi-institutional study of 19 cases. Mod Pathol. 35, 344–351 (2022). (2021).
Siadat, F. & Trpkov, K. ESC, ALK, HOT and LOT: Three letter acronyms of emerging renal entities knocking on the door of the who classification. Cancers 12, 168 (2021).
Miettinen, M. et al. GATA3: A multispecific but potentially useful marker in surgical pathology: A systematic analysis of 2500 epithelial and nonepithelial tumors. Am. J. Surg. Pathol. 38, 13–22 (2014).
Tong, K. & Hu, Z. FOXI1 expression in chromophobe renal cell carcinoma and renal oncocytoma: A study of The Cancer Genome Atlas transcriptome-based outlier mining and immunohistochemistry. Virchows Arch. 478, 647–658 (2021).
Guo, J. et al. Tuberous sclerosis-associated renal cell carcinoma: A clinicopathologic study of 57 separate carcinomas in 18 patients. Am. J. Surg. Pathol. 38, 1457–1467 (2014).
Schultz, L. et al. Immunoexpression status and prognostic value of mTOR and hypoxia-induced pathway members in primary and metastatic clear cell renal cell carcinomas. Am. J. Surg. Pathol. 35, 1549–1556 (2011).
Roldan-Romero, J. M. et al. Molecular characterization of chromophobe renal cell carcinoma reveals mTOR pathway alterations in patients with poor outcome. Mod. Pathol. 33, 2580–2590 (2020).
Shah, A. et al. Acquired Cystic Kidney Disease-associated Renal Cell Carcinoma (ACKD-RCC) harbor recurrent mutations in KMT2C and TSC2 Genes. Am. J. Surg. Pathol. 44, 1479–1486 (2020).
Chaux, A. et al. Dysregulation of the mammalian target of rapamycin pathway in chromophobe renal cell carcinomas. Hum. Pathol. 44, 2323–2330 (2013).
Kwiatkowski, D. J. et al. Mutations in TSC1, TSC2, and MTOR are associated with response to rapalogs in patients with metastatic renal cell carcinoma. Clin. Cancer Res. 22, 2445–2452 (2016).
Acknowledgements
Authors wish to thank Dr. Farshid Siadat for his technical help in preparation of the figures.
Funding
This study was supported by the Charles University Research Fund (project number Q39) and by the grant of Ministry of Health of the Czech republic-Conceptual Development of Research Organization (Faculty Hospital in Plzen-FNPl 00669806).
Author information
Authors and Affiliations
Contributions
O.H. and K.T. contributed equally to the conception, design, drafting, and editing of the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study was performed in accordance with the Declaration of Helsinki. Ethics committee approval was not required by Charles University and University Hospital Plzen and Cumming School of Medicine, University of Calgary.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hes, O., Trpkov, K. Do we need an updated classification of oncocytic renal tumors?. Mod Pathol 35, 1140–1150 (2022). https://doi.org/10.1038/s41379-022-01057-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01057-z
This article is cited by
-
A pilot radiometabolomics integration study for the characterization of renal oncocytic neoplasia
Scientific Reports (2023)
