
Abstract
Adamantinoma represents a distinct group of bone tumors showing both mesenchymal and epithelial differentiation most commonly involving the tibial diaphysis. Most adamantinomas contain a fibro-osseous component and an epithelial component consisting of squamous or basaloid cells. Adamantinomas are considered malignant neoplasms requiring en bloc excision that frequently recur locally and can rarely metastasize. Rare adamantinomas show an epithelial component consisting predominantly of monomorphic spindle cells, which, combined with an epithelial immunophenotype, can mimic monophasic synovial sarcoma. Synovial sarcoma is very rare in bone. It is considered a high-grade sarcoma that typically necessitates chemotherapy. However, the relationship between spindle cell adamantinoma and intraosseous synovial sarcoma has not been investigated. The current study was prompted by identification of a presumed spindle cell adamantinoma of the tibia with diffuse keratin expression that harbored a SS18 gene region rearrangement. FISH of eight additional bone tumors initially classified as spindle cell adamantinoma based on clinicoradiopathologic findings revealed one additional case with SS18 rearrangement. Histologically, both intraosseous synovial sarcoma and spindle cell adamantinoma demonstrated uniform fusiform nuclei with scant cytoplasm, short fascicles and low mitotic activity. The adamantinomas, but not the synovial sarcomas, were more likely to show overt epithelial differentiation in the form of pseudoglands or squamous nests. Immunohistochemistry of all cases, irrespective of SS18 status, showed diffuse keratin positivity in the spindle cell component, and less consistent EMA positivity. Clinical follow-up was available in both intraosseous synovial sarcomas, one of which recurred and the other metastasized. Two of the six spindle cell adamantinomas with follow-up metastasized. The above findings highlight the morphologic and immunophenotypic overlap between spindle cell adamantinoma and intraosseous synovial sarcoma of the tibia. Investigation of SS18 status to exclude synovial sarcoma is suggested prior to rendering a diagnosis of spindle cell adamantinoma.
Similar content being viewed by others
Introduction
Adamantinoma is a rare malignant biphasic tumor most commonly composed of an epithelial and fibro-osseous component chiefly involving the tibia and/or fibula in the third to fourth decade [1, 2]. A slight male preponderance is observed overall, but younger patients are more often female. Clinically, patients present with swelling, sometimes with pain, usually of years to decades duration [3]. On routine histology, adamantinoma is composed of fibro-osseous and epithelial components with the latter showing a basaloid, spindle cell, tubular or squamous pattern [4]. Adamantinoma is subclassified into “classic” and osteofibrous dysplasia-like variants. In “classic” adamantinoma, the lesion is dominated by the epithelial component with the fibro-osseous component usually restricted to the periphery [1]. It has also been recognized that recurrences and metastases more frequently show the spindle cell pattern though it is not clear whether it represents a more aggressive variant [5]. Recent evidence supports a pathogenetic relationship between osteofibrous dysplasia and adamantinoma but only the latter has malignant potential [6, 7]. On conventional radiographs, adamantinoma involves the tibia or fibula diaphysis and produces multiple lucencies with interspersed sclerosis of the cortex with an overall destructive appearance [8]. Medullary and soft tissue involvement is present in 60 and 30% of cases, respectively [8]. Few comprehensive genomic studies of adamantinoma exist in the literature. Aneuploidy has been identified in the epithelial component [9]. Recurrent numerical abnormalities of chromosomes 2, 7, 8, 10, 11, 12, 19 and/or 21 have been reported [1]. Isolated cases with translocations involving chromosomes 1, 8, 9, 16, 17, 19 and 21 have also been identified [10, 11]. To our knowledge, no adamantinoma with t(X;18) or molecular evidence of SS18 gene fusion has been reported. Furthermore, no genetic studies have focused specifically on the spindle cell subtype, which is most likely to mimic synovial sarcoma.
Adamantinoma is a locally aggressive tumor with potential for local recurrence that can rarely metastasize, even decades after diagnosis of the initial lesion [12,13,14]. Therapy consists of wide excision while the role of radiation and chemotherapy is not established [15].
Synovial sarcoma is a rare mesenchymal malignancy representing <10 percent of soft tissue sarcomas that shows a variable degree of epithelial differentiation. It shows a specific chromosomal translocation t(X;18)(p11.2;q11.2) resulting in fusion of the SS18 gene to one of the SSX genes [16]. It chiefly affects teens and young adults, though it has a wide age range. Most tumors arise in the deep soft tissues of the extremities but a wide anatomic distribution including mediastinum, sex organs and viscera is recognized [16].
Primary synovial sarcoma of bone is extremely rare, with fewer than 10 cases in the English literature, but these do include at least one in the tibia [17,18,19,20,21,22,23,24]. However, only four of these cases have documented t(X;18) or SS18 rearrangement [18,19,20, 25]. The few synovial sarcomas of bone so far described show similar histopathology to their soft tissue counterparts. Namely, either a monophasic pattern of uniform, tightly packed plump spindle cells with hyperchromasia and pale eosinophilic cytoplasm or a biphasic pattern that, in addition to the spindle cells, also includes variable amounts of epithelial differentiation. Although ossification can be seen as a secondary feature of synovial sarcoma of soft tissue [26], a distinct fibro-osseous component similar to adamantinoma has not been described. The epithelial component typically expresses epithelial membrane antigen (EMA) and keratins, but staining for these markers is usually weak and focal. Radiographically, soft tissue synovial sarcoma may erode bone in ~20% of cases [27]. Intraosseous tumors are lucent with either circumscribed or ill-defined margins and may show internal mineralization or soft tissue extension [18, 24]. Synovial sarcoma is an aggressive malignancy that requires wide excision, typically followed by radiation and/or chemotherapy [28, 29]. Five-year survival ranges from 60 to 80% [30].
The morphologic and immunophenotypic similarity between spindle cell adamantinoma and intraosseous synovial sarcoma is well recognized [31, 32]. The similarity raises the possibility that some tumors formerly classified as spindle cell adamantinoma, especially on small biopsies, actually represent intraosseous synovial sarcoma. In such cases, genetic and molecular techniques often allow more specific classification [33]. However, acid decalcification of bone tumors degrades nucleic acids [34], and can hinder nucleic acid-based testing.
We recently encountered a tibial synovial sarcoma initially classified as spindle cell adamantinoma based on clinicoradiopathologic features that exhibited SS18 rearrangement on reappraisal. Therefore, we sought to determine whether a subset of spindle cell adamantinomas harbor SS18 rearrangement and thus are misclassified intraosseous synovial sarcomas.
Materials and methods
Index case and case selection
This study was reviewed and approved by the Institutional IRBs. A 33-year-old previously healthy man presented to a referring institution with a swelling of the anterior mid-leg, which corresponded with a mixed lytic and sclerotic lesion of the tibia involving medulla and cortex. The initial diagnostic consideration was spindle cell adamantinoma or intraosseous synovial sarcoma. Fluorescence in situ hybridization (FISH) for SS18 gene region rearrangement was nondiagnostic after two attempts, presumably from decalcification required to process the tissue. However, FISH on a subsequent en bloc resection of a local recurrence 7 years after initial diagnosis, on undecalcified tissue, revealed a SS18 rearrangement. Based on the above finding, the pathology files of the authors’ institutions were searched for additional cases of spindle cell adamantinoma. These cases were screened with FISH for SS18 rearrangement and clinical, radiographic, histologic and immunophenotypic features were reviewed.
Fluorescence in situ hybridization
FISH was performed and evaluated according to previously published methods [35]. Briefly, 4 μm sections of formalin-fixed paraffin-embedded (FFPE) tissue were hybridized with fluorescent probes LSI SS18 mapped to 18q11.2 (Vysis) and counterstained with 4,6-diamidino-2-phenylindole. Two hundred consecutive nuclei showing complete (i.e., two green and two orange) signals were scored with the threshold of 20% break-apart signals set as a positive result. Nuclei with incomplete signals were omitted. If available, recurrent and metastatic tumors were evaluated in addition to the primary tumor. In total, 12 tumors were studied by FISH with 10 yielding informative results.
Reverse transcriptase-polymerase chain reaction analysis
Total RNA was isolated from FFPE tumor tissue sections using the Ambion RecoverAll Total Nucleic Isolation kit (Thermo Fisher Scientific), following the manufacturer’s instructions. The RNA was eluted in Ambion Elution Solution and stored at −80˚C. The quantity and quality of the isolated RNA were determined in 2 µL of the sample using a NanoDrop 2000 UV-Vis. Approximately 300 ng of the extracted total RNA was reverse transcribed into complementary DNA (cDNA), using random primers (2.6 ng/µL) and SuperScript III Reverse Transcriptase (5.3 U/µL; Thermo Fisher Scientific) (6 min at 95 ˚C, 1 h at 37 ˚C and 5 min at 95 ˚C). Polymerase chain reaction (PCR) amplification of the resulting cDNA was performed using the following M13-tagged primers: SYT-SSX_FFPE_M13-F (5ʹ-CAAGGTCAGCAGTATGGAGGAT-3ʹ) and SYT-SSX_FFPE_M13-R (5ʹ-TCACGCAGTCTGTGGGTC-3ʹ). The reaction products were electrophoresed in 2% agarose gels and visualized by ethidium bromide staining. The size of the PCR products was ~300 bp for the SS18-SSX1 and SS18–SSX2 fusion gene transcripts, and ~230 bp for β-actin, which was used as RNA control. The reverse transcriptase-PCR procedure was performed in duplicate. A cloned, known positive synovial sarcoma specimen, was used as a positive control. The adequacy of the extracted RNA was assessed by reverse transcriptase-PCR using primers for β-actin. A negative control RNA isolated from a normal control tissue and a reaction control without RNA were also used. Positive results were confirmed by Sanger sequencing using BigDye Terminator v1.1 (Thermo Fisher Scientific) and Agencourt CleanSEQ system (Beckman Coulter) with a 3130 Genetic Analyzer (Applied Biosystems, Thermo Fisher Scientific). Sequences were viewed with FinchTV software v1.4.0 and queried against NCBI Reference RNA database (refseq_rna) for humans (taxid:9605), excluding Models (XM/XP), using NCBI BLASTN 2.8.0 + optimized for highly similar sequences (megablast). The breakpoints in the fusion transcripts were determined using BLASTN alignments with SS18 transcript NM_005637.3, SSX1 transcript NM_001278691.1 and SSX2 transcript NM_175698.2.
Results
Including the index case, we identified nine primary bone tumors initially diagnosed as spindle cell adamantinoma for which paraffin tissue, imaging and clinical information was available. In addition to the primary tumor, recurrent and/or metastatic tumor was available for histologic and genetic evaluation in three cases. No cases had pre-existing malignancy elsewhere to suggest that the bone lesions represented metastasis.
Clinicopathologic and genetic findings of intraosseous synovial sarcoma
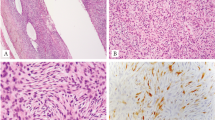
Two cases initially classified as spindle cell adamantinoma (Table 1, cases 1 and 2) showed SS18 rearrangement by FISH (Figs. 1a, b). RT-PCR followed by Sanger sequencing demonstrated SS18–SSX2 and SS18–SSX1 fusions in cases 1 and 2, respectively (Figs. 1c, d). Based on the genetic results, these two tumors were reclassified as intraosseous synovial sarcoma. The tumors were 4 and 5 cm, respectively, arose in the diaphysis of the tibia, in men in their 30s. Neither case had a significant soft tissue component at initial presentation. Both tumors recurred, case 1 after curettage and intramedullary nail, and case 2 after en bloc resection. Case 1 also metastasized to an ipsilateral groin node and received adjuvant chemotherapy. Both patients were alive at last follow-up (84 and 96 months, respectively).

SS18–SSX fusions were identified in two tumors supporting the diagnosis of synovial sarcoma. Fluorescence in situ hybridization showing SS18 break-apart signals (arrows) (green, centromeric; orange, telomeric) (a, case 1; b, case 2) corresponding to SS18–SSX2 (c, case 1) and SS18-SSX1 (d, case 2) fusions. In contrast, remaining cases showed normal fused SS18 signal (e, case 6, f, case 9)
Radiographically, both tumors showed destructive, mottled areas of sclerosis in the diaphyses involving cortex and medulla (Figs. 2g, h and 3h). In case 1, the earliest imaging available was from recurrence after initial fracture repair and curettage. The findings for both cases were compatible with adamantinoma, but other aggressive tumors and infection could not be excluded.

Pathologic and radiographic findings of intraosseous synovial sarcoma (case 1). The tumor displayed ramifying sheets of monomorphic spindle cells in short fascicles amid a cellular fibrous stroma (a, b) invading articular cartilage (c) with areas of woven bone showing conspicuous osteoblastic and osteoclastic activity (d). Case 1 was diffusely and strongly positive for keratin (e) and focally for EMA (f). Conventional AP radiograph (g) and lateral (h) radiographs of recurrence of case 1 showed a mixed lytic and sclerotic lesion, with an intramedullary rod and screw from the initial curettage

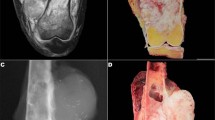
Pathologic and radiographic findings of intraosseous synovial sarcoma (case 2). This tumor displayed highly cellular monomorphic spindle cells with more intervening pericellular collagen (a, b). In some areas, more conspicuous fibrous stroma contained vague gland-like spaces formed by tumor cells (c). The tumor also entrapped native lamellar bone (d). Case 2 was positive for keratin (e) and EMA (f). Grossly (g), case 2 consisted of a fleshy intramedullary lesion in the metaphysis without a significant soft tissue component. Conventional lateral radiograph (h) and axial CT (h, inset) showed a subtle mixed lytic-sclerotic lesion in the tibial metaphysis; the circumscribed oval lytic lesion on the conventional radiograph is a biopsy cavity
Histologically, intraosseous synovial sarcomas demonstrated a highly cellular population of monomorphic spindle cells growing in short fascicles or broad nests set in a fibrous stroma (Figs 2 and 3). Though neither tumor contained a discrete macroscopic fibrous component, both did contain focal areas of woven bone with osteoblastic activity, whereas tumor from case 2 also contained prominent osteoclasts. In addition, tumor from case 2 infiltrated native trabeculae (Fig. 3d). Mitotic activity was 2 and 5 per 10 high-power fields in cases 1 and 2, respectively, without atypical forms. Nuclei contained finely stippled chromatin and small nucleoli. Overt epithelial (glandular or squamous) differentiation, so-called hemangiopericytomatous (staghorn) vessels or tumor calcifications were not identified in either case. Mast cells were inconspicuous. Both cases were positive for keratin (AE1/AE3 or AE1/AE3 + CAM5.2 cocktail) and EMA, albeit weakly (Figs. 2e, f and 3e, f) and negative for S-100 protein (data not shown). INI1 was completely retained in all cases except the recurrence of case 3, which showed focal loss (data not shown). TLE1 was strongly positive in one of the two synovial sarcomas, and weakly in only the metastasis of the other synovial sarcoma. Weak TLE1 expression was also observed in three of the seven adamantinomas (data not shown).
Clinicopathologic and genetic findings of spindle cell adamantinoma
The remaining seven cases showed an intact (fused) SS18 signal by FISH (e.g., Figs. 1e, f). These are summarized in Table 1 (cases 3–9). The average patient age was 31 (range 11–79), and most (n = 5, 71%) were men. The tumors chiefly involved the diaphysis of the tibia or fibula (n = 6, 86%) and averaged 11 cm (range 3.5–30 cm). One case (case 8) had a reported history of osteofibrous dysplasia decades before presentation but no pathology was available for review. None of the cases had prior history of malignancy elsewhere. Treatment ranged from curettage (case 3, who experienced a local recurrence) to en bloc resection, and amputation (case 9). Three cases received adjuvant chemotherapy. Two developed lung metastasis, one of whom (case 5) died of disease.
Radiographs were available for review in four cases although imaging reports were reviewed on all. In general, conventional radiographs revealed destructive tumors with both cortical and medullary involvement, a mixed sclerotic and lytic pattern on conventional radiograph and cortical disruption with focal soft tissue extension in four cases. Although adamantinoma was raised in the radiographic differential diagnosis of all tibia and fibula cases, the differential also included osteofibrous dysplasia in the more sclerotic examples but other aggressive neoplasms could not be excluded in most cases.
Routine histology revealed highly cellular spindle cell neoplasms most commonly with sheet-like growth, tightly packed short fascicles and scant stroma (Figs. 4, 5, Supplemental Figs. 1, 2). Most tumors showed uniform cytomorphology of plump spindle cells with scant cytoplasm, fine chromatin and occasional small nucleoli. The spindle cell component was predominant in the recurrence of case 3 (Fig. 4c), whereas the initial tumor had the appearance of more conventional adamantinoma (Figs. 4a, b). None of the tumors demonstrated an osteofibrous dysplasia-like component although, as with the above-described synovial sarcomas, reactive woven bone was occasionally present. Mitotic activity ranged from 1 to 10 per 10 high-power fields without atypical forms. One tumor (case 9, Supplemental Fig. 2A) showed focal squamous differentiation. No overt glandular differentiation was noted but occasionally small pools of extracellular matrix formed microcyst-like spaces in two tumors (Supplemental Figs. 1A, 1E).

Representative pathologic and radiographic findings of spindle cell adamantinoma (case 4). In case 3, the initial tumor (a, b) had a more conventional adamantinoma appearance but the recurrence (c) was uniformly spindly. The tumor was focally positive for keratin (d) and EMA (e). Conventional AP radiograph (f) showed ill-defined mixed lytic and sclerotic lesion involving the cortex and medulla of the tibia

Representative pathologic and radiographic findings of spindle cell adamantinoma (case 5), which was diffusely spindly (a, b) and positive for keratin (c) but not EMA (d). Conventional AP radiograph (e) showed ill-defined mixed lytic and sclerotic lesion involving the cortex and medulla of the fibula. Additional pathologic findings of adamantinomas are shown in Supplemental Fig. 1 and Supplemental Fig. 2
Immunohistochemistry showed diffusely positive staining for keratins (AE1/AE3) in all cases. By contrast, EMA staining was more variable with only two cases (29%) showing strong diffuse staining, the remainder showing weak or absent staining (Figs. 4, 5, Supplemental Figs. 1, 2). In case 5, only the metastasis was positive for EMA, whereas the primary was negative. S-100 protein was negative in all tumors (data not shown). Additional immunohistochemistry, if evaluated, is summarized in Table 1.
Discussion
In standard textbooks of bone pathology, photomicrographs of spindle cell adamantinoma are virtually indistinguishable from monophasic synovial sarcoma, and the similarity is also acknowledged in the research literature [31, 32]. Based on the presence of SS18 rearrangement in an index case of a synovial sarcoma misclassified as spindle cell adamantinoma, we investigated whether a subset of tumors formerly classified as spindle cell adamantinoma in fact represent intraosseous synovial sarcoma. Although the majority of spindle cell neoplasms of the tibia with epithelial differentiation in our cohort were correctly classified as spindle cell adamantinoma, two of these tumors were in fact synovial sarcoma. The clinical implications are profound. Little is known about the behavior of intraosseous synovial sarcoma, but soft tissue synovial sarcoma is considered an aggressive malignancy that necessitates wide excision followed by radiation and/or chemotherapy. In contrast, the role of radiation or chemotherapy is not established in adamantinoma [15, 28, 29]. Therefore, we recommend genetic or molecular evaluation for SS18 rearrangement prior to making a diagnosis of spindle cell adamantinoma. We acknowledge that nucleic acid-based studies are challenging in acid-decalcified tissue. The result highlights the importance of taking at least one block of undecalcified tissue of all bone tumors, whenever possible.
Our results confirm that distinguishing spindle cell adamantinoma from intraosseous synovial sarcoma based on clinical, radiographic and routine pathology methods may be challenging. Both tumors show monomorphic spindle cells with a fascicular growth pattern, low to intermediate proliferative activity and expression of epithelial markers (keratin, EMA). With the exception of the initial presentation of tumor in one case, none of the spindle cell adamantinomas contained a conspicuous fibro-osseous component (osteofibrous dysplasia-like component), so the absence of such a component does not exclude a diagnosis of spindle cell adamantinoma. Somewhat unexpectedly, the two cases of intraosseous synovial sarcoma demonstrated quite diffuse keratin positivity, not the more common patchy staining seen in soft issue monophasic synovial sarcoma [36]. EMA was less consistently positive in spindle cell adamantinoma than intraosseous synovial sarcoma. Importantly, both synovial sarcomas reported here arose in the tibia diaphysis, which is the typical location of adamantinoma. Furthermore, radiographic findings were not sufficiently specific in any case to differentiate between spindle cell adamantinoma and synovial sarcoma.
We considered whether the two cases reclassified as synovial sarcoma could represent soft tissue tumors involving bone, a feature identified in up to 20% of synovial sarcomas [27]. Preoperatively, this distinction can only be made reliably on imaging or postoperatively if treated with en bloc excision. In the reported cases, both synovial sarcomas were distinctly intraosseous tumors, either entirely confined to bone or centered on bone with only a peripheral soft tissue component similar to previously described cases [18].
The small number of cases in the present series limits any statistical comparison between intraosseous synovial sarcoma and adamantinoma, in particular with respect to clinical outcome. However, the results do provide insight into the rarity of both tumors. Specifically, we extensively searched the archives of four academic centers with busy orthopedic oncology practices and bone pathology consultation services, but identified only two genetically confirmed intraosseous synovial sarcomas and seven spindle cell adamantinomas. To date, descriptions of synovial sarcoma of bone have been restricted to case reports [18,19,20, 22,23,24,25, 37]. Only four of these have documented t(X;18) or SS18 rearrangement [18,19,20, 25]. Although three have been previously described involving the tibia, all were metaphyseal, distinct from the diaphyseal location of the two cases presented here [18, 23, 25]. Studies of spindle cell adamantinoma are limited to larger studies on adamantinoma as a whole [3, 4, 15, 31].
Some examples of high-grade spindle cell sarcoma arising in adamantinoma may represent so-called “dedifferentiation.” [38] We cannot exclude this possibility in two cases in our series. Specifically, case 3 showed a relatively conventional adamantinoma histomorphology at initial presentation (Figs. 4a, b) and case 8 was reported at initial presentation as osteofibrous dysplasia (though histology was not available); both had spindled morphology noted only at recurrence. However, the remaining cases displayed no clinical or histologic evidence of a precursor lesion. Unlike previous descriptions of dedifferentiation with increased pleomorphism, the cases presented here showed a relatively uniform pattern without significant pleomorphism [38]. We propose, therefore, that most spindle cell adamantinomas are a unique subtype rather than a progression phenomenon. Additional study, in particular investigation of the genetic or epigenetic features of spindle cell adamantinoma compared with other patterns, is required to better characterize the pathogenesis.
In summary, spindle cell adamantinoma shows significant clinical, radiographic and pathologic overlap with intraosseous synovial sarcoma, including tibia diaphyseal location, aggressive imaging findings in some cases, monotonous spindle cells and expression of epithelial antigens. Therefore, we recommend genetic or molecular evaluation for SS18 rearrangement prior to making a diagnosis of spindle cell adamantinoma.
References
Hogendoorn PC, Kanamori M. Adamantinoma. In: Fletcher CD, Bridge JA, Hogendoorn PC, Mertens F, editors. WHO classification of tumours of soft tissue and bone. Lyon: IARC Press; 2013. p. 343–5.
Moon NF, Mori H. Adamantinoma of the appendicular skeleton--updated. Clin Orthop Relat Res. 1986:215-37.
Keeney GL, Unni KK, Beabout JW, Pritchard DJ. Adamantinoma of long bones. A clinicopathologic study of 85 cases. Cancer. 1989;64:730–7.
Weiss SW, Dorfman HD. Adamantinoma of long bone. An analysis of nine new cases with emphasis on metastasizing lesions and fibrous dysplasia-like changes. Hum Pathol. 1977;8:141–53.
Hazelbag HM, Taminiau AH, Fleuren GJ, Hogendoorn PC. Adamantinoma of the long bones. A clinicopathological study of thirty-two patients with emphasis on histological subtype, precursor lesion, and biological behavior. J Bone Jt Surg Am. 1994;76:1482–99.
Gleason BC, Liegl-Atzwanger B, Kozakewich HP, Connolly S, Gebhardt MC, Fletcher JA, et al. Osteofibrous dysplasia and adamantinoma in children and adolescents: a clinicopathologic reappraisal. Am J Surg Pathol. 2008;32:363–76.
Taylor RM, Kashima TG, Ferguson DJ, Szuhai K, Hogendoorn PC, Athanasou NA. Analysis of stromal cells in osteofibrous dysplasia and adamantinoma of long bones. Mod Pathol. 2012;25:56–64.
Khanna M, Delaney D, Tirabosco R, Saifuddin A. Osteofibrous dysplasia, osteofibrous dysplasia-like adamantinoma and adamantinoma: correlation of radiological imaging features with surgical histology and assessment of the use of radiology in contributing to needle biopsy diagnosis. Skelet Radiol. 2008;37:1077–84.
Hazelbag HM, Fleuren GJ, Cornelisse CJ, van den Broek LJ, Taminiau AH, Hogendoorn PC. DNA aberrations in the epithelial cell component of adamantinoma of long bones. Am J Pathol. 1995;147:1770–9.
Kanamori M, Antonescu CR, Scott M, Bridge RS, Jr., Neff JR, Spanier SS, et al. Extra copies of chromosomes 7, 8, 12, 19, and 21 are recurrent in adamantinoma. J Mol Diagn. 2001;3:16–21.
Camp MD, Tompkins RK, Spanier SS, Bridge JA, Bush CH. Best cases from the AFIP: adamantinoma of the tibia and fibula with cytogenetic analysis. Radiographics. 2008;28:1215–20.
Giannoulis DK, Gantsos A, Giotis D, Paschos NK, Vagionas A, Arnaoutoglou CM, et al. Multiple recurrences and late metastasis of adamantinoma in the tibia: a case report. J Orthop Surg (Hong Kong). 2014;22:420–2.
Ray R, Beesley M, Patton JT, Salter D, Porter D. Unusual late metastasis of adamantinoma presenting thirty years after resection of original tumor. JBJS Case Connect. 2014;4:e31–e4.
Schowinsky JT, Ormond DR, Kleinschmidt-DeMasters BK. Tibial adamantinoma: late metastasis to the brain. J Neuropathol Exp Neurol. 2015;74:95–7.
Jain D, Jain VK, Vasishta RK, Ranjan P, Kumar Y. Adamantinoma: a clinicopathological review and update. Diagn Pathol. 2008;3:8.
Suurmeijer AJ, De Bruijn DR, Geurts van Kessel A, Miettinen M. Synovial sarcoma. In: Fletcher CD, Bridge JA, Hogendoorn PC, Mertens F, editors. WHO classification of tumours of soft tissue and bone. 4th ed. Lyon, France: IARC Press; 2013. p. 213–5.
Zulkarnaen M, Pan K, Shanmugam P, Ibrahim Z, Chan W. Intraosseous synovial sarcoma of the proximal femur: case report. Malays Orthop J. 2012;6:49–52.
Beck SE, Nielsen GP, Raskin KA, Schwab JH. Intraosseous synovial sarcoma of the proximal tibia. Int J Surg Oncol. 2011;2011:184891.
Cohen IJ, Issakov J, Avigad S, Stark B, Meller I, Zaizov R, et al. Synovial sarcoma of bone delineated by spectral karyotyping. Lancet. 1997;350:1679–80.
Hiraga H, Nojima T, Isu K, Yamashiro K, Yamawaki S, Nagashima K. Histological and molecular evidence of synovial sarcoma of bone. A case report. J Bone Jt Surg Am. 1999;81:558–63.
Nakajo M, Ohkubo K, Nandate T, Shirahama H, Yanagi M, Anraku M, et al. Primary synovial sarcoma of the sternum: computed tomography and magnetic resonance imaging findings. Radiat Med. 2005;23:208–12.
O’Donnell P, Diss TC, Whelan J, Flanagan AM. Synovial sarcoma with radiological appearances of primitive neuroectodermal tumour/Ewing sarcoma: differentiation by molecular genetic studies. Skelet Radiol. 2006;35:233–9.
Scully SP, Temple HT, Harrelson JM. Synovial sarcoma of the foot and ankle. Clin Orthop Relat Res 1999:220-6.
Tilakaratne WM. Synovial sarcoma of the mandible. J Oral Pathol Med. 2006;35:61–3.
Jung SC, Choi JA, Chung JH, Oh JH, Lee JW, Kang HS. Synovial sarcoma of primary bone origin: a rare case in a rare site with atypical features. Skelet Radiol. 2007;36:67–71.
Milchgrub S, Ghandur-Mnaymneh L, Dorfman HD, Albores-Saavedra J. Synovial sarcoma with extensive osteoid and bone formation. Am J Surg Pathol. 1993;17:357–63.
O’Donnell P. Soft tissue tumours. In: Grainger AJ, O’Connor P, editors. Grainger and Allison’s diagnostic radiology: the musculoskeletal system. 6th ed. New York, NY: Elsevier; 2016. p. 113.
Lewis JJ, Antonescu CR, Leung DH, Blumberg D, Healey JH, Woodruff JM, et al. Synovial sarcoma: a multivariate analysis of prognostic factors in 112 patients with primary localized tumors of the extremity. J Clin Oncol. 2000;18:2087–94.
Eilber FC, Brennan MF, Eilber FR, Eckardt JJ, Grobmyer SR, Riedel E, et al. Chemotherapy is associated with improved survival in adult patients with primary extremity synovial sarcoma. Ann Surg. 2007;246:105–13.
Sultan I, Rodriguez-Galindo C, Saab R, Yasir S, Casanova M, Ferrari A. Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005: an analysis of 1268 patients. Cancer. 2009;115:3537–47.
Unni KK, Inwards C, Bridge JA, Kindblom JM, Wold LE. Adamantinoma. In: Silverberg SJ, editor. AFIP atlas of tumor pathology: tumors of the bones and joints. Vol. 2. Washington, DC: American Registry of Pathology; 2005. p. 299–307.
Shirai T, Tsuchida S, Terauchi R, Mizoshiri N, Konishi E, Tomita Y et al. Primary pulmonary synovial sarcoma requiring differentiation from pulmonary metastasis of tibial adamantinoma: a case report. BMC Res Notes. 2014;7:736.
Verbeke SL, Fletcher CD, Alberghini M, Daugaard S, Flanagan AM, Parratt T, et al. A reappraisal of hemangiopericytoma of bone; analysis of cases reclassified as synovial sarcoma and solitary fibrous tumor of bone. Am J Surg Pathol. 2010;34:777–83.
Schrijver WA, van der Groep P, Hoefnagel LD, Ter Hoeve ND, Peeters T, Moelans CB, et al. Influence of decalcification procedures on immunohistochemistry and molecular pathology in breast cancer. Mod Pathol. 2016;29:1460–70.
Vergara-Lluri ME, Stohr BA, Puligandla B, Brenholz P, Horvai AE. A novel sarcoma with dual differentiation: clinicopathologic and molecular characterization of a combined synovial sarcoma and extraskeletal myxoid chondrosarcoma. Am J Surg Pathol. 2012;36:1093–8.
Pelmus M, Guillou L, Hostein I, Sierankowski G, Lussan C, Coindre JM. Monophasic fibrous and poorly differentiated synovial sarcoma: immunohistochemical reassessment of 60 t(X;18)(SYT-SSX)-positive cases. Am J Surg Pathol. 2002;26:1434–40.
Sar C, Eralp L. Surgical treatment of primary tumors of the sacrum. Arch Orthop Trauma Surg. 2002;122:148–55.
Nouri H, Jaafoura H, Bouaziz M, Ouertatani M, Abid L, Meherzi MH, et al. Dedifferentiated adamantinoma associated with fibrous dysplasia. Orthop Traumatol Surg Res. 2011;97:770–5.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Horvai, A., Dashti, N.K., Rubin, B.P. et al. Genetic and molecular reappraisal of spindle cell adamantinoma of bone reveals a small subset of misclassified intraosseous synovial sarcoma. Mod Pathol 32, 231–241 (2019). https://doi.org/10.1038/s41379-018-0115-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0115-6
This article is cited by
-
The rare primary bone sarcomas: imaging-pathological correlation
Skeletal Radiology (2021)
-
Molekulare Pathologie in der Diagnostik der Knochentumoren: Aktueller Stand
Der Pathologe (2020)
-
Primary intraosseous hybrid epithelioid schwannoma/perineurioma in the proximal tibia: a case report of benign hybrid neoplasm with local hypercellularity
Diagnostic Pathology (2019)
