
Abstract
Giant cell tumors of soft tissue (GCT-ST) are rare low-grade neoplasms that were at one time thought to represent the soft tissue counterparts of GCT of bone (GCT-B) but are now known to lack the H3F3 mutations characteristic of osseous GCT. We present six distinctive giant cell-rich soft tissue neoplasms that expressed keratins and carried a recurrent HMGA2-NCOR2 gene fusion. Patients were five females and one male aged 14–60 years (median, 29). All presented with superficial (subcutaneous) masses that were removed by conservative marginal (3) or wide (2) local excision. The tumors originated in the upper extremity (2), lower extremity (2), head/neck (1), and trunk (1). Five patients with follow-up (median, 21 months; range, 14–168) remained disease-free. Grossly, all tumors were well-demarcated but not encapsulated with variable lobulation. Histologically, they were composed of bland plump epithelioid or ovoid to spindled mononuclear cells admixed with evenly distributed multinucleated osteoclast-type giant cells. Foci of stromal hemorrhage and hemosiderin were seen in all cases. The mitotic activity ranged from 2 to 14/10 high power fields (median: 10). Foci of necrosis and vascular invasion were seen in one case each. The mononuclear cells were immunoreactive with the AE1/AE3 keratin cocktail and less frequently/less diffusely for K7 and K19 but lacked expression of other lineage-associated markers. RNA-based next-generation sequencing revealed an HMGA2-NCOR2 fusion in all tumors. None of the keratin-negative conventional GCT-ST showed the HMGA2-NCOR2 fusion (0/7). Metaplastic bone (4/9) and SATB2 expression (3/4) were frequent in keratin-negative conventional GCT-ST but were lacking in keratin-positive HMGA2-NCOR2 fusion-positive tumors. The distinctive immunophenotype and genotype of these tumors strongly suggest that they represent a discrete entity, differing from conventional GCT-ST and other osteoclast-rich morphologic mimics. Their natural history appears favorable, although a study of additional cases and longer follow-up are warranted.
Similar content being viewed by others
Introduction
The presence of an evenly distributed osteoclastic giant cell component is a well-known phenomenon in soft tissue and bone neoplasms. This readily recognizable morphological feature occurs in two different settings: (1) neoplasms that are definitionally giant cell-rich and are hence named after this feature such as giant cell tumor of bone (GCT-B) [1] and giant cell tumor of soft tissue (GCT-ST) [2], and (2) neoplasms that only occasionally or rarely display a prominent evenly distributed giant cell component. The latter include subsets of osteosarcoma [3], leiomyosarcoma [4], epithelioid sarcoma [5], and others. Accordingly, the exact subtyping of any giant cell-rich lesion is based on the identification and precise phenotyping of the neoplastic mononuclear component. In routine practice, subtyping is achieved via a set of defined phenotypic (such as demonstration of myoid markers in giant cell-rich leiomyosarcoma) [4], genotypic (e.g., presence of H3F3 mutations by molecular testing or using the mutation-specific H3.3 G34W antibody in GCT-B) [6,7,8,9,10] or both features (e.g. demonstration of epithelial phenotype and SMARCB1 inactivation in epithelioid sarcoma) [5].
Bland giant cell-rich soft tissue lesions are heterogeneous. They encompass extra-osseous aneurysmal cysts of soft tissue [11], a subset of giant cell-rich nodular fasciitis, plexiform fibrohistiocytic tumors [12, 13], and a group of lesions histologically indistinguishable from GCT-B, for which different terminologies have evolved [14,15,16,17,18]. “Giant cell tumor of soft tissue” is the currently accepted WHO terminology for the latter [2].
The molecular pathogenesis of GCT-ST remained obscure as they lack the H3F3 mutations seen in their intraosseous counterparts [19, 20]. We herein describe clinicopathological and molecular features of a distinctive molecular subtype of GCT-ST characterized by keratin immunoreactivity and recurrent gene fusion involving HMGA2 and NCOR2.
Materials and methods
We recently encountered a keratin-positive GCT-ST (Case 1 in Table 1), which we submitted to molecular analyses according to our institution policy of sending soft tissue tumors with unknown genetics for molecular investigation. We detected an HMGA2-NCOR2 fusion in this tumor. Notably, we have not encountered this fusion in >800 other soft tissue neoplasms tested with the same RNA Panel over the last 4 years. To test the hypothesis that this fusion is potentially specific to this rare soft tissue lesion, we retrieved 14 additional tumors diagnosed as GCT-ST or other giant cell-rich soft tissue lesions from our consultation files to test them for keratin expression and presence of the same or other gene fusions using the RNA fusion panel. Immunohistochemistry (IHC) was performed on 3-µm sections cut from paraffin blocks using a fully automated system (“Benchmark XT System”, Ventana Medical Systems Inc., 1910 Innovation Park Drive, Tucson, Arizona, USA) and the following antibodies: keratin cocktail (clones AE1/AE3, 1:40, Zytomed, Berlin, Germany), K5 (clone XM26, 1: 50, Zytomed), K7 (OV-TL, 1:1000, Biogenex), K19 (RCK108, 1:300, Dako), low-molecular weight keratins (clone CAM5.2, ready-to-use, CellMarque), pankeratin (clone OSCAR, ready-to-use, CellMarque), CD68 (clone PGM1, 1:200, Dako), CD163 (clone 10D6, 1:500, Novocastra), p63 (SFI-6, 1:100, DCS), desmin (clone D33, 1:250, Dako), alpha smooth muscle actin (clone 1A4, 1:200, Dako), HMB45 (clone HMB45, 1:50, Enzo), ERG (EPR3864, prediluted, Ventana), S100 protein (polyclonal, 1:2500, Dako), SATB2 (clone EPNCIR130A, 1:200, Abcam) and SMARCB1/INI1 (clone MRQ-27, dilution, 1:50, Zytomed). The H3.3 G34W IHC was performed manually using a mutation-specific antibody (clone RM 263, 1:500, BD Biosciences). Tissue microarray slides containing multiple GCT-B were used as an external control for the H3.3 G34W antibody. Only “clean” nuclear H3.3 G34W antibody staining without background staining was considered positive. Samples were used in accordance with ethical guidelines for the use of retrospective tissue samples provided by the local ethics committee of the Friedrich-Alexander University Erlangen-Nuremberg (ethics committee statements 24.01.2005 and 18.01.2012).
Statistical analysis for the comparison of keratin-positive and keratin-negative giant cell-rich soft tissue tumor subcohorts was performed using JMP SAS V.15.1.0 (SAS). Variables did not show normal distribution, thus the nonparametric Mann–Whitney U test was used for continuous variables. Grouped variables were compared using Fisher’s exact tests.
Next-generation sequencing
RNA was isolated from formalin-fixed paraffin-embedded (FFPE) tissue sections using RNeasy FFPE Kit of Qiagen (Hilden, Germany) and quantified spectrophotometrically using NanoDrop-1000 (Waltham, United States). Molecular analysis was performed using the TruSight RNA Fusion Panel (Illumina, Inc., San Diego, CA, USA) with 500 ng RNA as input according to the manufacturer’s protocol. Libraries were sequenced on a MiSeq (Illumina, Inc., San Diego, CA, USA) with >3 million reads per case, and sequences were analyzed using the RNA-Seq Alignment workflow, version 2.0.1 (Illumina, Inc., San Diego, CA, USA). The RNA-Seq alignment app (Illumina) was employed to call fusions by using the TopHat-Fusion algorithm, and to generate raw counts for each of the targeted 507 genes. Additionally, the Integrative Genomics Viewer (IGV), version 2.2.13 (Broad Institute, REF) was used for data visualization of fusions. For samples 6, 8, and 11, the fusion calling algorithm revealed a failure of quality control parameters, and no fusion was called. However, visual inspection of the automatically generated bam files revealed an HMGA2-NCOR2 gene fusion in sample 6 at low coverage, but no fusion in samples 8 and 11.
Differential expression and gene set enrichment analyses
Samples 6, 8, and 11 were excluded from gene expression analysis as they had total raw read counts <50,000 compared to read counts >1,000,000 in all other cases. Differential expression analysis was performed within the R/Bioconductor environment v.4.0.3 [21, 22] relying on the DESeq2 package v.1.30.0 [23]. Genes with an adjusted p value (Benjamini–Hochberg correction) < 0.01 were considered differentially expressed. Principal component analysis was based on the variance-stabilized transformed counts of the 100 genes with the highest variance across all samples. The heatmap was done using the variance-stabilized transformed count matrix of the differentially expressed genes as input relying on the gplots package v.3.1.0 [24]. Functional enrichment analyses of the differentially expressed genes were performed with DAVID database [25], using the 507 gene list of the panel as background. Although the TruSight RNA fusion panel was not developed for the analysis of differential gene expression, we have recently employed this method successfully in comparing different subgroups of solitary fibrous tumors [26], while a larger version of this panel has been used to classify hematological neoplasms by gene expression [27].
Results
General features of the study cohort
The clinicopathological features of the study cohort are summarized in Table 1. Of 17 initially retrieved tumors, two were excluded upon critical reevaluation. One tumor (1.5 cm) originating in the lower arm of a 10-year-old male represented a giant cell-rich tenosynovial giant cell tumor. Another 12 cm tumor in the left calf and fibula of a 30-year-old male represented a keratin-positive high-grade epithelioid malignancy with a few scattered giant cells and was excluded as well. The remaining 15 cases were then split into a keratin-positive (n = 6) and a keratin-negative (n = 9) subcohorts irrespective of their other histological, immunohistochemical, or molecular features.
Keratin-positive giant cell-rich soft tissue tumors
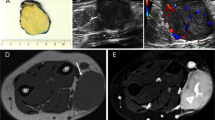
Six cases were diffusely positive with the AE1/AE3 keratin cocktail. These six tumors affected five females and one male aged 14–60 years (median: 29 years). All presented with superficial (subcutaneous) masses that underwent conservative marginal (3) or wide (2) local excision. One case was biopsied only. Two tumors originated in the upper extremity, two in the lower extremity and one each in the head and neck, and the trunk. Their size range was 2–3 cm (median, 2.5). Follow-up was available for five cases and ranged from 6 to 168 months (median, 21); no recurrences or distant metastases have been recorded during this follow-up period. Case 2 exhibited a vascular invasion and the patient presented with an enlarged (1 cm in diameter) PET-positive lymph node in the right inguinal area. She also had a 1 cm nodule at the right arm which was histologically diagnosed (post-chemotherapy) as organizing thrombus. Both findings disappeared after chemotherapy and might have represented tumor manifestations that healed after chemotherapy. None of the patients had evidence of another keratin-positive malignancy. Representative radiological images of one case are depicted in Fig. 1A–C.

Coronal T1-weighted (T1W) pre- (A) and post-contrast (B) and proton-density (C) MR images show an enhancing deep subcutaneous mass that abuts the peripheral fascia at the distal medial aspect of the right thigh. The slight hyperintensity of the tumor relative to muscle on pre-contrast T1W images and the peripheral low signal on all sequences likely represent its hemorrhagic content. Mild reticular edema surrounds the lesion. The epicenter of the tumor is in the subcutaneous tissue with well-circumscribed borders and tan to brown lobulated cut-surface (D).
Pathological findings
The histopathological and immunohistochemical features of the cases are summarized in Table 2. Grossly, all tumors were well-circumscribed, but unencapsulated. The cut-surface was described as tan to brown with vague lobulation (Fig. 1D).
At low power, the tumors were all uninodular, but showed variable lobulation and were occasionally arranged into plexiform lobules of variable size. These lobules were bordered by fibrosclerotic hyaline connective tissue bands (Fig. 2A, B). A variably intense lymphoid reaction at the periphery of the tumor was seen in all cases (Fig. 2C). Foci of ischemic-type necrosis were seen (Fig. 2D). Notably, metaplastic bone or a peripheral shell of mature bone was uniformly absent (Fig. 2E, F). The tumors were predominantly centered within the subcutaneous tissue (Fig. 2A) and were composed of plump epithelioid or ovoid mononuclear cells with vesicular chromatin, variably prominent nucleoli, and a rim of pale eosinophilic cytoplasm with indistinct cell borders (Fig. 3A). Occasional subtle areas with plasmacytoid cytology were observed but only after a careful search. One tumor however showed predominant epithelioid or plasmacytoid morphology (Fig. 3B). The mononuclear neoplastic cells formed diffuse solid sheets interrupted by a prominent component of evenly distributed multinucleated giant cells of osteoclast type. The number of nuclei within the giant cells varied greatly from >10 (Fig. 3A) to few (Fig. 3B). A variable degree of stromal hemorrhage and hemosiderin deposition was seen in all cases. The mitotic activity ranged from 2 to 14 mitoses per 10 high power fields (median: 10). Foci of necrosis and vascular invasion were seen in one case each.

At low-power, the subcutaneous tumor is well circumscribed with vague lobulation and a central zone of ischemic-type necrosis (A). Variable lobulation is seen (B). Pericapsular lymphoid aggregates are consistent findings (C). D Higher magnification of the ischemic-type necrosis. E Cellular areas with pseudoangiomatous or aneurysmal stromal changes. F Numerous multinucleated osteoclast-type giant cells are evenly distributed among the mononuclear bland cell component.

A Higher magnification of the two cell populations. B The neoplastic mononuclear cell component of this tumor (Case 2) had predominantly small epithelioid or plasmacytoid morphology. The AE1/AE3 (C) and OSCAR (D) keratin cocktails highlighted the mononuclear cell component with a variable dendritic pattern. E The case shown in B revealed a prominent paranuclear keratin staining pattern (AE1/AE3) consistent with the plasmacytoid cell morphology. F CD68 was strongly positive in both cell populations highlighting the giant cell component. G On the other hand, CD163 was expressed only in the mononuclear cell component.
Immunohistochemical features
By IHC, all tumors were positive for the AE1/AE3 keratin cocktail in the mononuclear cell population with a diffuse cytoplasmic pattern, occasionally highlighting dendritic-like cytoplasmic extensions (Fig. 3C). Other keratin markers (OSCAR, Cam5.2) were variably positive (Fig. 3D). The one case with plasmacytoid morphology (Case 2) showed distinctive paranuclear reactivity with AE1/E3 (Fig. 3E). K7 was positive in two of three cases. Three of four tumors showed prominent (one case) or single cell (two cases) K19 immunoreactivity. S100 protein was tested in four cases with one case showing limited focal reactivity. The giant cell component was highlighted by CD68 (Fig. 3F) but was negative for CD163 (Fig. 3G). The mononuclear cells revealed variable diffuse reactivity with CD68 (Fig. 3F) and CD163 with more intense staining seen with CD163 (Fig. 3G). SATB2 was negative in all cases (0/5). SMARCB1/INI1 showed retained nuclear reactivity. All six tumors lacked nuclear reactivity with the mutation-specific H3.3 G34W antibody. All other lineage-specific markers including endothelial, melanocytic, myogenic, myoepithelial, and neurogenic (done initially on a single case-based approach) were negative.
Molecular findings
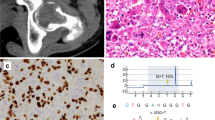
All six keratin-positive tumors revealed an HMGA2-NCOR2 fusion (Table 2), with the 3′- end of HMGA2 exon 3 involved in all six cases. Regarding the NCOR2 fusion partner, four tumors had the breakpoint at the 5′-end of exon 16, and two cases had the breakpoint at the 5′-end of exon 20 (Fig. 4). Regarding the putative chimeric HMGA2-NCOR2 fusion protein, a DNA-binding motif encoded by HMGA2 exons 1–3 was included in the N-terminal part, while two repressor domains, a binding site for histone deacetylase 3 (HDAC3) as well as binding sites for multiple nuclear hormone receptors and transcription factors were included in the C-terminal part encoded by NCOR2 exons 16–47. A SANT/MYB domain encoded by NCOR2 exons 11–17 was partially included in the HMGA2-NCOR2 fusion variant with a breakpoint at NCOR2 exon 16, but not in the two cases with breakpoints at NCOR2 exon 20.

A Case 1 with HMGA2 exon 3—NCOR2 exon 16 gene fusion. Note both split reads and paired reads at the 3′-border of HMGA2 exon 3 (left) corresponding to the 5′-end of NCOR2 exon 16 (right). B Case 2 with HMGA2 exon 3—NCOR2 exon 20 gene fusion. Note both split reads and paired reads at the 3′-border of HMGA2 exon 3 (left) corresponding to the 5′-end of NCOR2 exon 20 (right).
The RNA fusion panel also allows the quantification expression profiles associated with the tumors, as previously shown by us and others [26, 27]. An unsupervised principal component analysis based on the expression of the 100 genes with the highest variability across the whole cohort revealed that tumors with and without the HMGA2-NCOR2 fusion clustered in two distinct groups (Fig. 5A). Differential expression analysis of the 507 genes comprised in the RNA fusion panel showed that, 64 genes had a significantly higher expression in the group of keratin-positive tumors with HMGA2-NCOR2 fusion, while 62 genes were significantly higher expressed in the group of keratin-negative tumors lacking an HMGA2-NCOR2 fusion (Supplementary Table 1, Fig. 5B). Genes that were higher expressed in the tumors with HMGA2-NCOR2 fusion included TMPRSS2, CCND3, and RUNX1, as well as several genes involved in T-cell signaling (e.g. TCL1A, IRF4, and POU2AF1 among the top five higher expressed genes) (Fig. 5C–E). In contrast, FGFR1, FGFR3, and AR encoding for the androgen receptor were significantly higher expressed in the tumors lacking the HMGA2-NCOR2 fusion (Fig. 5D–F). Gene set enrichment analysis revealed a significant enrichment of the Gene Ontology term “skeletal system development” among genes higher expressed in the control group.

A Principal component analysis based on the normalized gene expression levels of 100 genes from the 507 gene panel with the highest variability of expression among the whole cohort. Note that the tumors show distinct clustering correlating to the presence (blue dots) or absence (red dots) of the HMGA2-NCOR2 gene fusion. Each dot represents a single case. B Cluster analysis and heatmap visualization of five tumors with HMGA2-NCOA2 gene fusion (blue) compared to seven tumors without HMGA2-NCOA2 gene fusion (red), based on the expression of 126 significantly differentially expressed genes. Note the homogenous gene expression pattern between both groups. Each row represents a gene and each column represents a tumor, with the normalized gene expression level indicated by color code. C–E Normalized read counts for the genes TMPRSS2, CCND3, and RUNX1 that were significantly higher expressed in the HMGA-NCOR2 fusion-positive tumors. F–H Normalized read counts for the genes FGFR1, FGFR3, and AR that were significantly higher expressed in the HMGA-NCOR2 fusion negative tumors. Each dot represents a single tumor.
Keratin-negative giant cell-rich soft tissue tumors
The keratin-negative tumors (Tables 1 and 2) showed almost an equal sex distribution and affected patients with an age range of 11–78 years (mean, 46.7). Their sizes ranged from 1.5 to 4.5 cm (mean, 2.5). Histologically, they resembled GCT-B, and often showed a shell of mature bone (seen in 44.4% of cases) (Fig. 6A–C). In contrast to the keratin-positive cohort, these keratin-negative tumors lacked the peritumoral lymphoid reaction (p value = 0.01), frequently expressed SATB2 (3/4 cases; Fig. 6D) and lacked HMGA2-NCOR2 fusions (0/7 cases). One case presenting as a minute dermal nodule revealed an ALK-PPFIBP1 fusion and showed ALK immunoexpression (not shown). The H3.3 G34W antibody was negative in all cases. The main clinicopathological and molecular features of the two subgroups are compared in Table 3.

A A subset of cases contained a peripheral shell or islands of mature metaplastic bone. B Higher magnification of A (note close similarity to the case depicted in Fig. 3A). C CD68 highlighted the osteoclastic cells, but was less expressed in the mononuclear cells in this case. D Consistent nuclear immunoreactivity with SATB2 is seen in the majority of these tumors, note lack of expression in the osteoclastic giant cells.
Discussion
The nosologic classification of osteoclast-rich soft tissue neoplasms has been a matter of ongoing controversy and terminological evolution over the decades. This is due to the wide histogenetic spectrum of “osteoclastoma”-like lesions presenting within soft tissue, which encompasses metastatic giant cell-rich anaplastic carcinoma, giant cell-rich bone tumors extending into soft tissue (GCT-B, extra-osseous aneurysmal cysts, brown tumors, giant cell-rich osteosarcoma, and other rare variants), and osteoclast-rich variants of different specific soft tissue sarcoma types [4, 5].
Historically, the term “malignant giant cell tumor of soft parts” was used loosely to refer to a variety of soft tissue neoplasms, irrespective of the presence and the degree of cytological atypia and other features of malignancy [14]. However, due to the significant biological heterogeneity of the giant cell tumor category, these tumors underwent continuous refinement of diagnostic criteria [16]. When all the above entities and mimics are excluded, a subset of soft tissue GCTs cannot be otherwise classified into any specific sarcoma category. However, these definitionally bland-looking GCT-STs remain poorly characterized and the question whether they form a specific entity or merely represent a morphological pattern remains unsolved. The current WHO classification adopted for them the noncommitted term “giant cell tumor of soft tissue/GCT-ST” [2]. Older terminologies such as “malignant giant cell tumor of soft parts”, “giant cell tumor of low malignant potential” and “giant cell-type sarcomas” have been abandoned.
GCT-ST originates predominantly in the superficial soft tissues of the extremities followed by the trunk, head and neck, and other rare sites [16,17,18]. Adults in their fifth decade are predominantly affected with a wide age range (5–89 years) without sex predilection. Defining histological criteria of GCT-ST are multinodular superficial growth of uniform histiocytoid mononuclear cells lacking nuclear pleomorphism, admixed bland osteoclastic giant cells, variable hemosiderin deposits, and frequent metaplastic bone formation [2, 16,17,18]. A peripheral rim (shell) of woven bone is seen in almost half of the cases [2, 16,17,18]. Vascular invasion has been reported in up to 30% and aneurysmal features in a subset of cases [16,17,18]. Foamy macrophages may be observed in some cases. If strictly diagnosed, the biological behavior of GCT-ST is indolent with infrequent local recurrences (12%) and very rare or no metastases [2, 16]. The neoplastic mononuclear cells of GCT-ST display a similar immunophenotype as their osseous counterparts. They express monocyte-macrophage-associated antigen CD68, tartrate-resistant acid phosphatase, and smooth muscle actin, but not CD45, desmin, S100 protein, and lysozyme [16,17,18,19,20]. The osteoclastic giant cells express CD68, but not CD163 [28].
Although GCT-ST has traditionally been considered the soft tissue counterpart of GCT-B, based on the morphological similarities between the two entities, they lack the H3F3 mutations, that characterize their osseous counterparts [6, 7, 19, 20] indicating different molecular pathogenesis. Further supporting this concept, studies have shown GCT-ST to share RANK and RUNX2 expression with GCT-B, but to show RANKL and SATB2 expression in only 25% of cases [20, 29]. The H3.3 G34W mutation-specific antibody, which has been developed as a valuable surrogate marker with high sensitivity and specificity for GCT-B, was negative in our cases, which practically rules out the H3F3 H3.3 G34W mutation (observed in 85–90% of GCT-B cases) [8, 10, 19, 20]. However, other H3.3 mutations (seen in the remaining 10% of cases) are not excluded by negative H3.3 G34W immunostaining.
Keratin expression has been previously studied to only a limited degree in GCT-ST. Oliveira et al. reported variable keratin expression in 3 of 19 cases [17]. In the current study, keratin expression separated GCT-ST into two subgroups: one group (40% of all cases) displayed keratin expression and was uniformly positive for the HMGA2-NCOR2 fusion, while the other subgroup lacked both features. Comparing the two subgroups by keratin expression, the keratin-positive tumors tend to present at a younger mean age (34.7 vs. 46.7 years), affect predominantly females (83% vs. 44.4%), lack mature bone formation (0% vs. 44.4%), display peritumoral lymphoid reaction (66.7% vs. 0%) and harbor the HMGA2-NCOR2 fusion (100% vs. 0%), respectively. However, only peritumoral lymphoid reaction and the presence of HMGA2-NCOR2 fusion appeared to be statistically significant due to the low number of cases. These distinctive demographic, clinicopathological and genetic features suggest two independent and separate tumor entities.
The high mobility group AT-hook 2 (HMGA2) encodes for a member of the non-histone transcriptional regulatory proteins involved in early developmental stages during embryogenesis [30]. Rearrangements involving HMGA2 at chromosomal region 12q14.3 have been detected in a variety of benign mesenchymal and mixed tumors including lipomas and osteochondrolipomas, chondromas of soft tissue, uterine leiomyomas, salivary gland pleomorphic adenomas, and many other entities [31, 32].
HMGA2 chromosomal breakages mainly involve the third long intron between exon 3 and exon 4 of the gene. The translocation frequently results in the formation of a truncated form of fusion transcript [31,32,33]. Loss of the C-terminal domain as a consequence of the chromosomal breakage is likely related to tumorigenesis. Although a plethora of fusion partners (LPP, RAD51L1, NFIB, EBF1, PPAP2B, LHFP, NCOA2, and others) have been reported [31,32,33,34], we are aware of only one recent case report of an HMGA2 fusion involving NCOR2 as a fusion partner (see below) [35].
The NCOR2 gene, mapped to chromosomal region 12q24.31, encodes the nuclear receptor co-repressor 2 (NCOR2), a member of the thyroid hormone- and retinoic acid receptor-associated co-repressors family [36, 37]. As a member of a multisubunit complex, NCOR2 (AKA: silencing mediator for retinoid or thyroid-hormone receptors = SMRT and T3 receptor-associating cofactor 1 = TRAC-1) is involved in transcriptional silencing of certain target genes via chromatin remodeling [36, 37]. The putative chimeric HMGA2-NCOR2 fusion protein combines a DNA-binding motif encoded by HMGA2 exons 1–3 in the N-terminal part with two repressor domains, a binding site for histone deacetylase 3 (HDAC3) as well as binding sites for multiple nuclear hormone receptors and transcription factors encoded by NCOR2 exons 16–47 in the C-terminal part [36, 37]. Although the exact mechanisms through which the novel HMGA2-NCOR2 fusion initiates tumorigenesis are unknown, a de-regulating effect on gene expression through interaction with other transcription factors is likely expected.
To the best of our knowledge, the novel HMGA2-NCOR2 gene fusion has been reported only once before [35]. Brahmi et al. reported on a rapidly growing tenosynovial giant cell tumor of the hand of a 61-year-old female that responded to pexidartinib. Molecular profiling revealed HMGA2-NCOR2 and NCOR2-SUPT3H fusions, but not the expected COL6A3-CSF1 fusion [35]. The single histological image suggests a tenosynovial giant cell tumor and keratin expression is not mentioned in the case description [35]. Notably, we have never encountered this fusion among >800 soft tissue neoplasms we have analyzed routinely in our laboratory using the same TruSight RNA fusion panel since 2017, including >10 tenosynovial giant cell tumors (data not shown). This strongly suggests that this novel fusion represents a specific genetic marker defining this subset of keratin-positive GCT-ST. Although a comprehensive survey of other giant cell containing soft tissue and bone neoplasms is beyond the scope of the current study, it is remarkable that keratin expression appears to be a feature only of tumors harboring HMGA2-NCOR2 fusions, among osteoclast-rich soft tissue tumors.
It is unclear what keratin expression in HMGA2-NCOR2-positive GCT-ST signifies. Certainly, they show no morphologic features to suggest epithelial origin, lack the cytologic atypia seen in osteoclast-rich carcinomas, and do not appear to arise in association with the epidermis or cutaneous adnexa. Furthermore, with the exception of keratins, these tumors were entirely negative for all other tested markers, corresponding to markers of endothelial, melanocytic, myogenic, myoepithelial, and neurogenic lines of differentiation. Similarly, the morphologic features of these lesions are wholly dissimilar from those of epithelioid sarcoma, and they show retained SMARCB1 expression. Finally, the distinctive genetic features of these keratin-positive tumors and absent expression of SATB2 would suggest that this does not represent simply aberrant keratin immunoreactivity in “garden variety” GCT-ST. Our cases share some clinical (predilection for young women) and immunohistochemical (keratin expression) features with the entity reported recently by Fritchie et al. as “xanthogranulomatous epithelial tumor”, but the two tumor types are morphologically very different [38].
Although limited by the panel-based approach, the expression profiling of our two cohorts showed significant differences, with a clear separation of the tumors on the basis of presence or absence of the HMGA2-NCOR2 gene fusion. Genes significantly overexpressed in the tumors with HMGA2-NCOR2 fusion included TMPRSS2, CCND3 and RUNX1. Additionally, genes physiologically expressed in lymphocytes (e.g. TCL1A, IRF4, and POU2AF1) were significantly overexpressed, likely related to the lymphoid infiltrate seen in the HMGA2-NCOR2 group. In contrast, the keratin-negative “control” group overexpressed FGFR1 and FGFR3 (among others) and were significantly enriched for the Gene Ontology term “skeletal formation”, findings consonant with the bone formation and SATB2 expression observed more frequently in this group. The one tumor showing ALK rearrangement clustered with the keratin-negative “control” group. Although no definite conclusions on the biological relevance of deregulated pathways can be drawn from this method, the pattern clearly shows differences on a larger scale level of gene expression comparing these two subgroups of tumors.
In summary, we have described a distinctive osteoclast-rich, keratin-positive tumor of the subcutaneous tissues, differing morphologically and immunohistochemically from conventional GCT-ST, and consistently harboring HMGA2-NCOR2 fusions. We propose the descriptive term “keratin-positive giant cell-rich soft tissue tumors with HMGA2-NCOR2 fusion” for these unusual lesions. HMGA2-NCOR2-positive osteoclast-rich tumors should be carefully distinguished from other osteoclast-rich and keratin-positive superficial soft tissue tumors, in particular carcinoma, germ cell tumor metastasis and epithelioid sarcoma. The natural history of these rare tumors appears quite favorable, although additional study and longer clinical follow-up are needed.
References
Flanagan AM, Larousserie F, O´Donnell PG, Yoshida A. Giant cell tumour of bone. In: WHO Classification of Tumours, Soft Tissue and Bone Tumours, 5th Edition, International Agency for Research on Cancer, Lyon, France, Vol. 3. 2020. p. 440–46.
Oliveira AM, Lee JC. Giant cell tumour of soft tissue. In: In: WHO Classification of Tumours, Soft Tissue and Bone Tumours, 5th Edition, Vol. 3. 2020. p. 141–42.
Chow LT. Giant cell rich osteosarcoma revisited-diagnostic criteria and histopathologic patterns, Ki67, CDK4, and MDM2 expression, changes in response to bisphosphonate and denosumab treatment. Virchows Arch. 2016;468:741–55.
Mentzel T, Calonje E, Fletcher CD. Leiomyosarcoma with prominent osteoclast-like giant cells. Analysis of eight cases closely mimicking the so-called giant cell variant of malignant fibrous histiocytoma. Am J Surg Pathol. 1994;18:258–65.
Rekhi B, Verma A, Jambhekar NA, Menon S, Laskar S, Merchant N, et al. Osteoclast-rich, proximal-type epithelioid sarcoma: clinicopathologic features of 3 unusual cases expanding the histomorphological spectrum. Ann Diagn Pathol. 2016;21:39–43.
Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet. 2013;45:1479–82.
Presneau N, Baumhoer D, Behjati S, Pillay N, Tarpey P, Campbell PJ, et al. Diagnostic value of H3F3A mutations in giant cell tumour of bone compared to osteoclast-rich mimics. J Pathol Clin Res. 2015;1:113–23.
Lüke J, von Baer A, Schreiber J, Lübbehüsen C, Breining T, Mellert K, et al. H3F3A mutation in giant cell tumour of the bone is detected by immunohistochemistry using a monoclonal antibody against the G34W mutated site of the histone H3.3 variant. Histopathology. 2017;71:125–33.
Righi A, Mancini I, Gambarotti M, Picci P, Gamberi G, Marraccini C, et al. Histone 3.3 mutations in giant cell tumor and giant cell-rich sarcomas of bone. Hum Pathol. 2017;68:128–35.
Yamamoto H, Iwasaki T, Yamada Y, Matsumoto Y, Otsuka H, Yoshimoto M, et al. Diagnostic utility of histone H3.3 G34W, G34R, and G34V mutant-specific antibodies for giant cell tumors of bone. Hum Pathol. 2018;73:41–50.
Nielsen GP, Fletcher CD, Smith MA, Rybak L, Rosenberg AE. Soft tissue aneurysmal bone cyst: a clinicopathologic study of five cases. Am J Surg Pathol. 2002;26:64–9.
Enzinger FM, Zhang RY. Plexiform fibrohistiocytic tumor presenting in children and young adults. An analysis of 65 cases. Am J Surg Pathol. 1988;12:818–26.
Moosavi C, Jha P, Fanburg-Smith JC. An update on plexiform fibrohistiocytic tumor and addition of 66 new cases from the Armed Forces Institute of Pathology, in honor of Franz M. Enzinger, MD. Ann Diagn Pathol. 2007;11:313–9.
Guccion JG, Enzinger FM. Malignant giant cell tumor of soft parts. An analysis of 32 cases. Cancer. 1972;29:1518–29.
Angervall L, Hagmar B, Kindblom LG, Merck C. Malignant giant cell tumor of soft tissues: a clinicopathologic, cytologic, ultrastructural, angiographic, and microangiographic study. Cancer. 1981;47:736–47.
Folpe AL, Morris RJ, Weiss SW. Soft tissue giant cell tumor of low malignant potential: a proposal for the reclassification of malignant giant cell tumor of soft parts. Mod Pathol. 1999;12:894–902.
Oliveira AM, Dei Tos AP, Fletcher CD, Nascimento AG. Primary giant cell tumor of soft tissues: a study of 22 cases. Am J Surg Pathol. 2000;24:248–56.
O’Connell JX, Wehrli BM, Nielsen GP, Rosenberg AE. Giant cell tumors of soft tissue: a clinicopathologic study of 18 benign and malignant tumors. Am J Surg Pathol. 2000;24:386–95.
Lee JC, Liang CW, Fletcher CD. Giant cell tumor of soft tissue is genetically distinct from its bone counterpart. Mod Pathol. 2017;30:728–33.
Mancini I, Righi A, Gambarotti M, Picci P, Dei Tos AP, Billings SD, et al. Phenotypic and molecular differences between giant-cell tumour of soft tissue and its bone counterpart. Histopathology. 2017;71:453–60.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80.
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: 2020. https://www.R-project.org/.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, et al. gplots: Various R Programming Tools for Plotting Data. R package version 3.1.0. 2020. https://CRAN.R-project.org/package=gplots.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat Protoc. 2009;4:44–57.
Bieg M, Moskalev EA, Will R, Hebele S, Schwarzbach M, Schmeck S, et al. Bieg M, Moskalev EA, Will R, Hebele S, Schwarzbach M, Schmeck S. et al. Gene expression in Solitary Fibrous Tumors (SFTs) correlates with anatomical localization and NAB2-STAT6 gene fusion variants. 2021. https://doi.org/10.1016/j.ajpath.2020.12.015.
de Lange K, de Boer EN, Bosga A, Alimohamed MZ, Johansson LF, Mulder AB, et al. Targeted RNA-sequencing enables detection of relevant translocations and single nucleotide variants and provides a method for classification of hematological malignancies-RANKING. Clin Chem. 2021. https://doi.org/10.1093/clinchem/hvaa221.
Maggiani F, Forsyth R, Hogendoorn PC, Krenacs T, Athanasou NA. The immunophenotype of osteoclasts and macrophage polykaryons. J Clin Pathol. 2011;64:701–5.
de la Roza G. p63 expression in giant cell-containing lesions of bone and soft tissue. Arch Pathol Lab Med. 2011;135:776–9.
Reeves R, Beckerbauer L. HMGI/Y proteins: flexible regulators of transcription and chromatin structure”. Biochim Biophys Acta. 2001;519:13–29.
Dahlén A, Mertens F, Rydholm A, Brosjö O, Wejde J, Mandahl N, et al. Fusion, disruption, and expression of HMGA2 in bone and soft tissue chondromas. Mod Pathol. 2003;16:1132–40.
Stenman G. Fusion oncogenes in salivary gland tumors: molecular and clinical consequences. Head Neck Pathol. 2013;7:S12–9.
Bianchini L, Birtwisle L, Saâda E, Bazin A, Long E, Roussel JF, et al. Identification of PPAP2B as a novel recurrent translocation partner gene of HMGA2 in lipomas. Genes Chromosomes Cancer. 2013;52:580–90.
Panagopoulos I, Gorunova L, Bjerkehagen B, Lobmaier I, Heim S. The recurrent chromosomal translocation t(12;18)(q14~15;q12~21) causes the fusion gene HMGA2-SETBP1 and HMGA2 expression in lipoma and osteochondrolipoma. Int J Oncol. 2015;47:884–90.
Brahmi M, Alberti L, Tirode F, Karanian M, Eberst L, Pissaloux D, et al. Complete response to CSF1R inhibitor in a translocation variant of teno-synovial giant cell tumor without genomic alteration of the CSF1 gene. Ann Oncol. 2018;29:1488–9.
Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–7.
Sande S, Privalsky ML. Identification of TRACs (T3 receptor-associating cofactors), a family of cofactors that associate with, and modulate the activity of, nuclear hormone receptors. Mol Endocrinol. 1996;10:813–25.
Fritchie KJ, Torres-Mora J, Inwards C, Thway K, Vaiyapuri S, Jackson R, et al. Xanthogranulomatous epithelial tumor: report of 6 cases of a novel, potentially deceptive lesion with a predilection for young women. Mod Pathol. 2020;33:1889–95.
Acknowledgements
The authors thank Prof. Dr. Ustun Aydingoz (Department of Radiology, Hacettepe University Faculty of Medicine, Turkey) for his radiological interpretation and comments on case #2. We also thank Dr. Veronica Weyerer (Institute of Pathology, Erlangen University Hospital) for help with the statistical analysis.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
AA, KK, ALF: conception and design of the work, acquisition, analysis and interpretation of data, drafting the MS and revising it critically for important intellectual content and scientific integrity. AA, Michael M, RS, FF, PF, MM, AF, FH, ALF, KK: acquisition, analysis and interpretation of data, reading and revising the MS critically for important intellectual content and scientific integrity. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics statement
The authors confirm that the study has been performed in accordance with accepted principles of ethical and professional conduct for biomedical scientific research. This study is covered by the ethical vote for retrospective translational research studies of the Ethical Committee of the Medical Faculty of the Friedrich-Alexander-Universität Erlangen-Nürnberg (ethics committee statements 24.01.2005 and 18.01.2012).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Agaimy, A., Michal, M., Stoehr, R. et al. Recurrent novel HMGA2-NCOR2 fusions characterize a subset of keratin-positive giant cell-rich soft tissue tumors. Mod Pathol 34, 1507–1520 (2021). https://doi.org/10.1038/s41379-021-00789-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00789-8
This article is cited by
-
Bone and soft tissue tumors: clinicoradiologic-pathologic molecular-genetic correlation of novel fusion spindled, targetable-ovoid, giant-cell-rich, and round cell sarcomas
Skeletal Radiology (2023)
-
Diagnostic utility of CSF1 immunohistochemistry in tenosynovial giant cell tumor for differentiating from giant cell-rich tumors and tumor-like lesions of bone and soft tissue
Diagnostic Pathology (2022)
-
Xanthogranulomatous epithelial tumors and keratin-positive giant cell-rich soft tissue tumors: two aspects of a single entity with frequent HMGA2-NCOR2 fusions
Modern Pathology (2022)
-
GLI1-altered mesenchymal tumor: a clinicopathological and molecular analysis of ten additional cases of an emerging entity
Virchows Archiv (2022)
