
Abstract
Small cell carcinoma of the ovary, hypercalcemic type is a rare, aggressive malignancy which usually occurs in young women and is characterized by mutations in SMARCA4, with few other alterations. We recently encountered uterine tumors with morphologic, immunohistochemical, and genetic similarities to small cell carcinoma of the ovary, hypercalcemic type. Herein we report the clinicopathologic and molecular features (using a targeted massively parallel sequencing [MPS] assay) of these tumors. The cases were diagnosed on cervical and endometrial biopsies (n = 2, 34, and 29 years) or hysterectomy and bilateral salpingo-oophorectomy (n = 3, 25, 33, and 58 years). The tumors were composed of sheets of large atypical epithelioid cells with prominent rhabdoid morphology, indistinguishable from the “large cell” variant of small cell carcinoma of the ovary, hypercalcemic type. In three cases, the ovaries were pathologically examined to exclude a primary ovarian malignancy. Immunohistochemically, four of four cases showed SMARCA4 loss, and were negative or only focally positive for keratins, EMA, and claudin-4. One of three cases was positive for WT-1. Targeted MPS was successfully performed on 4 of 5 tumors, and showed recurrent mutations in SMARCA4, with few other alterations. Of the cases diagnosed on hysterectomy, all had extensive lymphovascular invasion, extra-uterine spread, and marked infiltrative growth. These tumors were uniformly aggressive; all patients died of disease (median survival 7 months, range 1–43 months). We propose this entity be called “SMARCA4-deficient undifferentiated uterine sarcoma (malignant rhabdoid tumor of the uterus)”, a term which describes both the tumor’s underlying molecular abnormality and its morphology. Its unique clinicopathologic and molecular features differentiate it from other related malignancies, including undifferentiated endometrial carcinoma, small cell carcinoma of the ovary (hypercalcemic type), and epithelioid sarcoma. We review and discuss previously reported “rhabdoid tumors of the uterus;” while they are a heterogenous group of tumors, some of them are likely examples of this entity. Correctly identifying cases of SMARCA4-deficient uterine sarcoma from histologic mimics is important as it may have prognostic, predictive, and germline implications.
Similar content being viewed by others


Introduction
Small cell carcinoma of the ovary, hypercalcemic type is a rare, aggressive ovarian tumor that occurs in young females [1]. Recently, germline and/or somatic mutation in SMARCA4, a member of the SWI/SNF chromatin remodeling complex, was found to be its main underlying molecular alteration [2,3,4]. This has facilitated its diagnosis by using widely available immunohistochemistry for SMARCA4 (also known as BRG1), which shows loss of expression in tumor cells. In the ovary, loss of SMARCA4 is a relatively sensitive and specific marker for small cell carcinoma of the ovary, hypercalcemic type [5]. Since the discovery of SMARCA4 mutations as the defining molecular feature of small cell carcinoma of the ovary, hypercalcemic type, some have proposed a shift in nomenclature to “malignant rhabdoid tumor of the ovary” given its similarity to malignant rhabdoid tumors associated with loss of SMARCB1 (also known as INI-1), a related protein [6]. SWI/SNF-deficient sarcomas have been reported in other anatomic sites, including atypical teratoid/rhabdoid tumors of the CNS, and the recently described SMARCA4-deficient thoracic sarcoma [7]. However, a uterine variant has not been previously characterized.
Herein, we provide the first unified description, including immunophenotypic and molecular characterization, of a new member of the SWI/SNF family of tumors: SMARCA4-deficient undifferentiated uterine sarcoma/malignant rhabdoid tumor of the uterus. These aggressive tumors have distinct clinicopathologic and molecular features which differentiate them from histologic mimics including undifferentiated endometrial carcinomas, high-grade endometrial stromal sarcoma, leiomyosarcoma, rhabdomyosarcoma, PEComa, Müllerian adenosarcoma, melanoma, and epithelioid sarcoma. Differentiating cases of SMARCA4-deficient uterine sarcoma from other histologically similar tumors may have prognostic, predictive, and genetic implications for patients.
Materials and methods
An index case of a SMARCA4-deficient uterine sarcoma was identified, which prompted reevaluation of two others cases seen by one of the authors (BH) over a period of 4 years that had similar clinicopathologic features. Two additional cases were identified for inclusion in the study by reviewing pathology reports containing the term “undifferentiated carcinoma,” and then reviewing their morphology and immunohistochemistry, for a total of five cases.
Immunohistochemistry
Immunohistochemistry was performed on 4 μm thick sections from formalin-fixed paraffin embedded tissue. The clone, dilution, and supplier used for each antibody are listed in Table 1. Appropriate positive and negative controls were examined for each immunohistochemical stain.
Targeted massively parallel sequencing (Oncopanel)
The methodology of MPS implementation has been previously reported in detail [8, 9]. In brief, tumor DNA was isolated from formalin-fixed, paraffin embedded tissue and enriched for exonic regions of 447 tumor suppressor genes and oncogenes (Supplemental Material) by solution-based hybrid capture. Sequencing was performed using Illumina HiSeq 2500, and analysis was performed via a custom informatics pipeline.
Results
Five cases for study were identified, three of which had sufficient tissue for complete characterization. Their clinicopathologic features are summarized in Table 2, and presented in greater detail below.
Clinicopathologic features
Case 1
The patient was a 25-year-old female who presented with a large mass protruding through the cervix. A biopsy at the outside hospital was reported as most consistent with a high-grade adenosarcoma, associated with large round cell epithelioid areas, hemorrhage, and necrosis.
The patient underwent a hysterectomy, bilateral salpingo-oophorectomy, omentectomy, and pelvic lymph node dissection. Grossly, a 9 cm mass arose from the endomyometrium, invaded through >50% of the myometrial thickness extending to within 0.5 cm of the serosa, involved the cervix and extended to the parametrial and paracervical resection margins. Macroscopic tumor also involved the omentum, and bilateral fallopian tube serosa. Microscopically, the tumor was composed of epithelioid cells displaying a prominent “rhabdoid” morphology reminiscent of epithelioid sarcoma that infiltrated around normal glands that had a vague leaf-like architecture (Fig. 1). The mitotic rate reached 11 per 10 high power fields (HPFs). There was extensive lymphovascular space invasion, involving uterine and adnexal vessels. Although tumor was present in ovarian lymphovascular spaces, there was no tumor in the ovarian parenchyma. Tumor was metastatic to 3 of 4 pelvic lymph nodes, with extranodal extension. Endometriosis was present in parametrial soft tissue. An extensive immunohistochemical work-up at the outside hospital showed that the tumor cells were positive for vimentin, and negative for pan-keratin, EMA, CK7, CD45, HMB-45, S100, and germ cell markers. Subsequently, immunohistochemistry was performed at BWH, and the tumor demonstrated loss of SMARCA4 expression, had retained/intact SMARCB1, MSH6, and PMS2 expression, and was negative for claudin-4, WT-1, SMA, desmin, and caldesmon (Fig. 2). The case was received in consultation and a diagnosis of adenosarcoma with extensive undifferentiated epithelioid component was favored by the submitting pathologist. The patient died of disease 7 months after hysterectomy.

Case 1. a Low power shows a vaguely phyllodiform architecture with tumor infiltrating around benign endometrial glands. b The tumor cells are large, epithelioid, with prominent nucleoli and abundant mitotic activity

Immunohistochemistry shows that SMARCA4-deficient sarcomas show loss of expression of SMARCA4 (a), and at most focal, weak reactivity to claudin-4 (b). Cam5.2 (c) and EMA (d) may also show focal reactivity. Benign glands are included as internal controls for claudin-4, Cam5.2, and EMA stains
Case 2
The patient was a 33-year-old female with a history of anemia, endometriosis, and urinary tract infections. She was brought to the operating room for a pre-operative diagnosis of fibroids. A frozen section performed on a uterine mass was reported as malignant, and a total abdominal hysterectomy, bilateral salpingo-oophorectomy, and omentectomy was performed.
Grossly, the hysterectomy showed a 25.5 cm mass involving her uterus and left ovary. The bulk of tumor was in the uterus, with full thickness involvement of the tumor with nodules in the cervix, and endomyometrium, extending through to the serosa and left ovary. Cut sections of the tumor showed tan-pink to gray-white soft, hemorrhagic tissue.
Microscopically, the tumor appeared to be arising in the endometrium and extending through the myometrium directly into the left ovary. Tumor was metastatic to the cervix, left tubal serosa, bilateral ovarian surfaces, and omentum. Lymphovascular invasion was present in the myometrium and cervix. The tumor showed compressed benign glands with a leaf-like architecture. Most of the tumor cells were large and epithelioid, though monomorphic in appearance (Fig. 3). In some regions, the cells were smaller, lacked rhabdoid morphology and had inconspicuous nucleoli.

Case 2. The tumor is composed of solid sheets of cells (a), with prominent nucleoli, vesicular chromatin, and scant cytoplasm (b). In some areas, the tumor cells are surrounded by hyalinized stroma (c, d)
Immunohistochemistry performed at the outside institution and reviewed showed the tumor cells were positive for vimentin, p16, synaptophysin (focal), and EMA (scattered cells). The cells were negative for chromogranin, p53, PAX8, HMB-45, S100, pan-keratin, CD45, inhibin, CD30, HMWCK, LMWCK, CK7, WT-1, CD10, SMA, and desmin. Immunohistochemistry performed at BWH demonstrated that SMARCA4 was lost, SMARCB1 was intact, and mismatch repair proteins MLH1, MSH2, MSH6, and PMS2 had intact nuclear staining. There was patchy positivity for SALL4 and cyclin D1. The tumor cells were negative for claudin-4, WT-1, inhibin, PAX8, CD34, ERG, BerEP4, NUT and caldesmon. A reticulin stain highlighted a nested growth pattern.
The case was received in consultation and the submitting pathologist favored a diagnosis of a high-grade endometrial stromal sarcoma. The patient died of disease 9 months after hysterectomy.
Case 3
The patient was a 34-year-old female who underwent endocervical curettage and endometrial biopsy. Both samples showed abundant tumor composed of diffuse, solid sheets of atypical epithelioid cells with rhabdoid morphology with extensive necrosis, and no distinct differentiation or architectural pattern (Fig. 4). The tumor involved endocervical tissue in both specimens.

Case 3. The tumor forms solid sheets of cells with benign entrapped endocervical glands (a). The tumor cells are atypical, yet monomorphous, with prominent nucleoli, and large nuclei (b). There is abundant mitotic activity
Immunohistochemical stains showed that SMARCA4 was lost in tumor cells, while SMARCB1 was retained. Epithelial markers, including claudin-4, pan-keratin, CAM5.2, and EMA were positive in <1% of tumor cells. SALL4 was weakly positive in scattered cells. WT-1 was diffusely positive. Mismatch repair protein immunohistochemistry for MSH6 and PMS2 was intact. The tumor was negative for SOX10, S100, PTEN, PAX-2, CD30, and CD34. A reticulin stain highlighted both nests of tumor and individual cells.
Immunohistochemical stains performed at the outside institution and reviewed showed the tumor was positive for vimentin, ER (scattered cells), and negative for CK5/6, p16, pCEA, desmin, CD45, synaptophysin, chromogranin, and cytokeratin. p53 showed a patchy, wild-type pattern of expression. Ki-67 expression was increased (~60–70%). The submitting pathologist believed the tumor was a sarcoma but was unable to further classify it. The patient died of disease 1 month after the biopsy.
Case 4
This patient was a 29-year-old female, with a past medical history significant for cervical squamous cell carcinoma, treated 3 years previously with chemoradiation. She presented with vaginal bleeding. A laparoscopy was performed, and biopsies showed tumor involving the cervix, uterine serosa, bilateral ovaries, and omentum. Morphologically, it was composed of sheets of large rhabdoid cells, similar to the other cases (Fig. 5). Immunohistochemistry showed loss of SMARCA4, and negativity for claudin-4, HMB-45, p63 CD45, S100, pan-keratin, and AE1/AE3. CAM5.2 was focally positive. The patient died from disease 4 months after the onset of vaginal bleeding.

Case 4. Similar to the other cases, the tumor forms solid sheets of cells (a) and is composed of discohesive rhabdoid cells with prominent nucleoli (b)
Case 5
This case was a 58-year-old female who presented with post-menopausal bleeding and back pain. An abdominal CT scan showed a 13 cm solid-cystic uterine mass, carcinomatosis, and inguinal lymphadenopathy. Her serum CA-125 was 185. An endometrial biopsy was performed which showed an undifferentiated malignant neoplasm. The patient underwent a hysterectomy, bilateral salpingo-oophorectomy, appendectomy, and omentectomy. Tumor involved nearly the complete thickness of the endomyometrium and cervical stroma. Tumor was metastatic to the uterine serosa, bilateral ovaries, one fallopian tube, omentum, sigmoid colon, and appendix. There was extensive lymphovascular and perineural invasion. Microscopically, the tumor was composed of sheets of large epithelioid cells with prominent nucleoli (Fig. 6). There was brisk mitotic activity (up to 23/10 HPFs). In some areas, the cells were embedded in a fibrous stroma. Immunohistochemistry performed at the outside institution showed that the tumor cells were positive for vimentin, CD10 (weak, focal), and negative for AE1/AE3, desmin, cyclin D1, ER, PR, and c-kit. Immunohistochemistry performed at BWH showed that the tumor was positive for CAM5.2 (rare isolated cells), EMA (focal, weak), S100 (scattered cells), SMA (scattered cells), and CD34 (focal, weak). The tumor was negative for AE1/AE3, pan-keratin, HMB-45, desmin, caldesmon, and LCA. SMARCB1 expression was retained in the tumor cells. No tissue was available for SMARCA4 immunohistochemistry. The patient died from disease 43 months after diagnosis.

Case 5. The tumor is composed of solid sheets of tumor cells (a). In other areas, the tumor cells are embedded in a hyalinized stroma (b), as can be seen in some areas of small cell carcinoma of the ovary, hypercalcemic type. Perineural invasion is also present
Serum calcium levels were not available for any of the cases. Follicle-like spaces and small cell morphology, characteristic of small cell carcinoma of the ovary, hypercalcemic type, were not observed in any of the cases.
Molecular genetics results
MPS quality control metrics
Case 1 failed MPS quality control metrics with poor coverage (mean target coverage was 9×, 0% of the targeted regions had base coverage of at least 30×). Considering cases 2 to 5, the mean number of aligned high quality reads was 9,262,340 (range: 5,766,471–13,048,502). The average mean target coverage was 257× (range: 156×–379×). An average of 98.6% (range: 98%–98.9%) of the targeted regions had base coverage of at least 30×.
SNVs and CNVs identified in SMARCA4-deficient uterine sarcoma
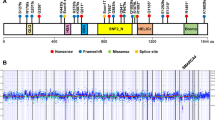
Considering the four cases of SMARCA4-deficient uterine sarcoma with successful sequencing (cases 2–5), case 2 had two truncating SMARCA4 mutations (c.3426delC, p.F1142Lfs*22 and c.4759G>T, p.E1587*), and a TERT promoter mutation. Case 3 harbored a truncating SMARCA4 mutation (c.2554A>T, p.K852*), as well as one missense mutation in ASXL1 (c.1205G>A, p.R402Q). Case 4 had a SMARCA4-PSG8 inversion, and one missense mutation of uncertain significance in ARID1B (c.5687G>A, p.R1896Q). Case 5 harbored two mutations in SMARCA4: a frameshift mutation (c.582_587GCCCCT>G, p.Q194fs) and a missense mutation (c.598C>G, p.L200V) of uncertain significance. MPS did not show evidence of hypermutation or enrichment of mutations at microsatellite sites in any case. Copy number variations included gain of 1q in case 2, single copy loss of CDKN2A in case 3, and scattered low level alterations in case 4. Case 5 did not have any copy number alterations.
Discussion
We have presented the clinical, morphologic, immunophenotypic, and molecular features of a tumor, which we propose be called “SMARCA4-deficient undifferentiated uterine sarcoma (malignant rhabdoid tumor of the uterus).” Morphologically, it is characterized by diffuse sheets of cells with rhabdoid morphology, with large, epithelioid cells with vesicular chromatin, eosinophilic inclusions, and prominent nucleoli. Immunohistochemically, it shows loss of SMARCA4 expression, retained/intact SMARCB1 and MMR staining, and lack of significant epithelial marker expression including keratin, EMA, and claudin-4. Molecularly, SMARCA4-deficient uterine sarcoma is characterized by truncating mutations (nonsense, frameshift, or inversions) in SMARCA4. The tumor is clinically aggressive, highly infiltrative, and usually with multiple foci of lymphovascular invasion. From this series, it appears to be uniformly fatal. The most salient clinical, pathologic, and genetic features of SMARCA4-deficient uterine sarcoma are presented in Table 3 with a comparison to several closely related tumors.
In case 4, only one H&E slide was available for review, an incomplete immunohistochemical panel had been performed (notably no WT-1), and the tumor involved the ovaries bilaterally. As well, the patient had the confounding factor of prior cervical carcinoma and radiation therapy. Interestingly, there are reports of radiation-induced malignant rhabdoid tumors and atypical teratoid/rhabdoid tumors [10,11,12,13]. The ovarian involvement raises the possibility of small cell carcinoma of the ovary hypercalcemic type. However, small cell carcinoma of the ovary hypercalcemic type is “almost invariably” unilateral [1], and we are not aware of any case presenting with vaginal bleeding, a cervical mass, and bilateral ovarian involvement as in case 4. On balance, we favor this fourth case to be an additional example of SMARCA4-deficient uterine sarcoma.
Case 5 was not available for review in its entirety (only one representative slide), and material was not available for additional immunohistochemical stains, including SMARCA4 and claudin-4. However, given the morphologic features and the molecular profile of the tumor, we believe that this may represent another case of SMARCA4-deficient uterine sarcoma. If so, it suggests that the disease may also occur in older patients, and be associated with a less aggressive course.
Undifferentiated carcinomas of many sites, including the endometrium, may demonstrate discohesive, sheet-like growth with rhabdoid morphology. Like SMARCA4-deficient uterine sarcoma, they may also show loss of SMARCA4 or SMARCB1 expression [14, 15]. However, their clinicopathologic and genetic features are dissimilar to SWI/SNF-deficient sarcomas. For example, SMARCA4-deficient uterine sarcoma, small cell carcinoma of the ovary (hypercalcemic type), and atypical teratoid/rhabdoid tumors occur at a younger age, may be more aggressive, and show fewer genetic abnormalities compared with carcinomas. Specifically, they have recurrent mutations in SMARCA4 or SMARCB1, and few other somatic protein-coding mutations [16].
SMARCA4-deficient uterine sarcoma shares many clinicopathologic features of small cell carcinoma of the ovary (hypercalcemic type), including young age, aggressive clinical behavior, morphologic resemblance to the large cell variant of small cell carcinoma of the ovary, hypercalcemic type, and loss of SMARCA4 expression. Several issues must be considered in both recognizing SMARCA4-deficient uterine sarcoma and distinguishing it from small cell carcinoma of the ovary, hypercalcemic type. (1) SMARCA4-deficient uterine sarcoma arises in the uterus and is frequently negative for WT-1. (2) When there is significant tumor burden in an ovary, the large cell variant of small cell carcinoma of the ovary, hypercalcemic type, involving the uterus, must be considered. However, small cell carcinoma of the ovary, hypercalcemic type is usually a large (mean 15 cm), unilateral mass, whereas SMARCA4-deficient uterine sarcoma may involve both adnexa with smaller deposits [1]. (3) In keeping with the different sites of origin, the clinical presentations also differ: all five cases of SMARCA4-deficient uterine sarcoma presented with a cervical mass or vaginal bleeding. In one series of 124 patients with small cell carcinoma of the ovary, hypercalcemic type, the most common presentations were abdominal pain or swelling, and none presented with a cervical mass or vaginal bleeding [1]. (4) There may be therapeutic implications in recognizing SMARCA4-deficient uterine sarcoma. Recent in vitro studies with small cell carcinoma of the ovary, hypercalcemic type cell lines and in vivo mouse xenograft models have shown antiproliferative and antitumor effects when treated with EZH2 inhibitors [17]. A phase 2 clinical trial is now treating patients with small cell carcinoma of the ovary, hypercalcemic type with the EZH2 inhibitor tazemetostat. Given the aggressive nature of SMARCA4-deficient uterine sarcoma and the shared genetics with small cell carcinoma of the ovary, hypercalcemic type, consideration should be given to including patients with SMARCA4-deficient uterine sarcoma in EZH2 inhibitor trials. There is also a report of four patients with small cell carcinoma of the ovary, hypercalcemic type responding to anti-PD-1 immunotherapy, despite the tumors' low mutational burden [18]. Similarly, there may be a role for PD-1 blockade in SMARCA4-deficient uterine sarcomas.
Given the above information, the distinction between undifferentiated endometrial carcinoma and SMARCA4-deficient uterine sarcoma is important. Lack of SWI/SNF complex expression by immunohistochemical staining is a necessary but not sufficient requirement for the diagnosis of SMARCA4-deficient uterine sarcoma, because dedifferentiated and undifferentiated carcinomas may commonly also show lack of expression of SMARCA4 or SMARCB1 [14, 19]. SMARCA4-deficient uterine sarcoma is uniformly fatal (albeit with small numbers in this study) and may be even more aggressive than undifferentiated endometrial carcinoma. However, given the rarity of these tumors, a direct comparison of survival is difficult. The average age of patients with SMARCA4-deficient uterine sarcoma in this limited series is 36 years old (median 33 years), which is far closer to the average age of small cell carcinoma of the ovary, hypercalcemic type (24 years) [1] than undifferentiated endometrial carcinoma (59 years, range 40–69) [20]. Further work is needed to determine the full age range of affected patients. Morphologically, undifferentiated carcinomas may also show rhabdoid morphology, albeit usually only focally when present [20], and distinguishing them SWI/SNF-deficient sarcomas can be challenging. Undifferentiated carcinomas may show increased nuclear pleomorphism, characterized by occasional large, highly atypical cells. In contrast, SMARCA4-deficient uterine sarcoma is composed of a population of uniformly atypical cells. The reticulin staining in SMARCA4-deficient uterine sarcoma highlights a nested pattern, which also may overlap with reticulin patterns in undifferentiated carcinomas. Immunohistochemistry may be helpful in this regard. SMARCA4-deficient uterine sarcomas are microsatellite stable and lack significant expression of epithelial markers. Undifferentiated carcinomas usually express claudin-4 [21]. Claudin-4 has recently been shown to be a marker of epithelial differentiation, which may remain expressed in undifferentiated and poorly differentiated carcinomas which have rhabdoid morphology, but is negative in small cell carcinoma (hypercalcemic type), SWI/SNF-deficient sarcomas, and in our series of SMARCA4-deficient uterine sarcomas [7, 21]. Four of the cases presented here were negative for claudin-4, suggesting that it may be an effective marker to distinguish them from rhabdoid carcinomas and indicating that the term “sarcoma” is more appropriate than “carcinoma” to describe these tumors. However, claudin-4 has not been extensively studied in undifferentiated endometrial carcinomas, so its specificity in this setting is unknown. Cyclin D1 immunohistochemistry may also be helpful; it is diffuse and strong in half of undifferentiated endometrial carcinomas [22], but is negative or patchy in SMARCA4-deficient uterine sarcoma. We suggest that when faced with a poorly differentiated uterine neoplasm with rhabdoid cells, if genomic studies are unavailable, claudin-4, cyclin D1, mismatch repair, and SMARCA4 immunohistochemistry may be used, in combination with histology and the appropriate clinical setting, to differentiate SMARCA4-deficient uterine sarcoma from mimics, including undifferentiated carcinomas with rhabdoid morphology.
Undifferentiated uterine carcinomas have many mutations, some of which occur in genes frequently mutated in endometrioid carcinoma (e.g., KRAS, PTEN, CTNNB1, PIK3CA). They may also have mutations in SWI/SNF complex members. In contrast, SMARCA4 is the sole driver mutation in SMARCA4-deficient uterine sarcoma.
Although proximal-type epithelioid sarcoma and SMARCA4-deficient uterine sarcoma share SWI/SNF complex abnormalities and a common age range, they have several differences. (1) SMARCA4-deficient uterine sarcoma is a more aggressive disease with dismal prognosis, while epithelioid sarcoma follows a protracted course with frequent local recurrences. (2) The anatomical sites affected also differ: while SMARCA4-deficient uterine sarcoma occurs in the uterus, proximal-type epithelioid sarcoma is usually in the inguinal region or vulva. CD34 is positive in approximately half of cases of epithelioid sarcoma, but is negative in SMARCA4-deficient uterine sarcoma. (3) Finally, epithelioid sarcoma is typically characterized by loss of SMARCB1, but SMARCA4 is altered in SMARCA4-deficient uterine sarcoma.
The morphologic differential diagnosis of malignant epithelioid tumors in the uterus also includes epithelioid leiomyosarcoma and malignant PEComa. Both entities show immunoreactivity to smooth muscle markers, but PEComas are also immunoreactive to HMB-45 and/or melanA. In contrast, SMARCA4-deficient uterine sarcoma is negative for markers of both smooth muscle and melanocytic differentiation. Loss of SMARCA4 by immunohistochemistry effectively excludes leiomyosarcoma, as it was intact in all cases in one series of 21 leiomyosarcomas [5]. Histologically, leiomyosarcomas and malignant PEComas often demonstrate striking nuclear pleomorphism; in contrast, SMARCA4-deficient uterine sarcoma has a more uniform cytologic atypia without significant pleomorphism. Molecularly, both leiomyosarcoma and PEComa show complex chromosomal abnormalities, whereas SMARCA4-deficient uterine sarcoma usually has a diploid karyotype with a small number of CNVs [23, 24]. Additionally, PEComas often show loss of heterozygosity of the TSC2 locus; [24] this aberration was not observed in any cases of SMARCA4-deficient sarcoma in this series.
Case 1 showed focal phyllodiform growth (Fig. 1a), raising the possibility of an adenosarcoma. However, no adenosarcoma to date has shown loss of SMARCA4 by immunohistochemistry (which would indicate biallelic inactivation). Single SMARCA4 mutations are uncommonly seen in adenosarcomas, but they are always in the setting of other pathogenic alterations [25]. Also, many tumors (such as carcinosarcoma and benign endometrial polyps) may mimic adenosarcoma by showing focal phyllodiform growth, and this pattern is not diagnostic of adenosarcoma [26]. In our case, it was only focal, and no spindled growth was present. Epithelioid areas of adenosarcoma are rarely seen [27]. However, these epithelioid areas in adenosarcoma have only been reported in tumors with smooth muscle differentiation, and only as a focal finding. The tumor cells in case 1 were negative for SMA, desmin, and caldesmon, and the epithelioid morphology was seen throughout the tumor, ruling out this possibility.
While small cell carcinoma of the ovary, hypercalcemic type and malignant rhabdoid tumor are associated with germline mutations in 8–50% and 15–30% of cases, respectively, SMARCA4-deficient thoracic sarcoma—which occurs in a much older age group—is not. Because the age distribution of SMARCA4-deficient uterine sarcoma more closely mirrors that of small cell carcinoma of the ovary, hypercalcemic type than SMARCA4-deficient thoracic sarcoma, it is conceivable that a subset of our cases is associated with a germline mutation in SMARCA4. Association with germline mutations in SMARCA4 should be investigated, as it may have implications for family members who may be carriers (e.g., consideration of hysterectomy and bilateral oophorectomy). Of note, patients with rhabdoid tumor predisposition syndrome 2 (OMIM: 613325, caused by heterozygous germline mutations in SMARCA4), have not been reported to have uterine tumors.
Others have previously postulated that uterine tumors with rhabdoid cells may be genetically related to malignant rhabdoid tumors [28,29,30]. While we feel the unqualified term “malignant rhabdoid tumor” is not completely accurate (SMARCA4-deficient uterine sarcoma does not occur in infants, is not currently associated with SMARCB1 mutations, and has been used historically to describe a variety of tumors of many anatomic sites), their idea of linking rhabdoid tumors in the uterus with other SWI/SNF-deficient tumors ultimately proved correct, and was enabled by the availability of immunohistochemical and genomic assays to study SMARCA4 in tumor biology.
In other SWI/SNF-deficient tumors, there is usually not total exclusivity between tumors caused by SMARCA4 and SMARCB1 deficiencies. For example, malignant rhabdoid tumors are usually associated with mutations in SMARCB1, but may rarely be caused by SMARCA4 loss. Although the cases presented here have SMARCA4 loss-of-function, other cases of SMARCA4-deficient uterine sarcoma may be shown to have SMARCB1 loss.
Unlike SMARCA4-deficient thoracic sarcoma, which is usually associated with heavy smoking [7], we have identified no known risk factors for SMARCA4-deficient uterine sarcoma, similar to small cell carcinoma of the ovary, hypercalcemic type in its lack of known non-genetic risk factors. Although two of our cases had endometriosis, the extra-uterine location of endometriosis would make it unlikely to have a role in the pathogenesis of this disease. Notably, the exposure to radiation therapy in case 4 may have had a role in tumorigenesis.
SMARCA4-deficient uterine sarcomas have not been previously reported as a distinct clinicopathologic entity. However, there have been reports of uterine tumors, diagnosed as undifferentiated carcinomas, sarcomas, or malignant rhabdoid tumors, which may be examples of SMARCA4-deficient uterine sarcoma (Table 4; cases 1–8). Although immunohistochemistry for SMARCA4 and claudin-4 and molecular studies would be needed to establish a diagnosis of SMARCA4-deficient uterine sarcoma, the constellation of clinical, morphologic and immunophenotypic features of the above cases are suggestive of this entity. There are also reports of other uterine rhabdoid tumors (Table 4; cases 9–14) which likely represent other tumors or have insufficient morphologic or immunohistochemical work-up to assess as candidate cases. Some of these may be better classified as another tumor type (e.g., carcinosarcoma or dedifferentiated carcinoma) which happen to display rhabdoid morphologic features.
In summary, recognizing and differentiating SMARCA4-deficient uterine sarcoma from its histologic mimics is important, as it has a dismal prognosis. This tumor type may be a rational candidate for targeted therapy (such as EZH2 inhibitors and anti-PD-1 therapy), and the possible association with germline mutations in SMARCA4 should be elucidated in future studies.
References
Young RH, Oliva E, Scully RE. Small cell carcinoma of the ovary, hypercalcemic type. A clinicopathological analysis of 150 cases. Am J Surg Pathol. 1994;18:1102–16.
Witkowski L, Carrot-Zhang J, Albrecht S, et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet. 2014;46:438–43.
Ramos P, Karnezis AN, Craig DW, et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat Genet. 2014;46:427–9.
Jelinic P, Mueller JJ, Olvera N, et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat Genet. 2014;46:424–6.
Karnezis AN, Wang Y, Ramos P, et al. Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcaemic type. J Pathol. 2016;238:389–400.
Foulkes WD, Clarke BA, Hasselblatt M, et al. No small surprise—small cell carcinoma of the ovary, hypercalcaemic type, is a malignant rhabdoid tumour. J Pathol. 2014;233:209–14.
Yoshida A, Kobayashi E, Kubo T, et al. Clinicopathological and molecular characterization of SMARCA4-deficient thoracic sarcomas with comparison to potentially related entities. Mod Pathol. 2017;30:797–809.
Sholl LM, Do K, Shivdasani P, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight. 2016;1:e87062.
Garcia EP, Minkovsky A, Jia Y, et al. Validation of OncoPanel: a targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch Pathol Lab Med. 2017;141:751–8.
Litman DA, Bhuta S, Barsky SH. Synchronous occurrence of malignant rhabdoid tumor two decades after Wilms’ tumor irradiation. Am J Surg Pathol. 1993;17:729–37.
Gorayski P, Boros S, Ong B, et al. Radiation-induced primary cerebral atypical teratoid/rhabdoid tumour in an adult. J Clin Neurosci. 2013;20:1466–8.
Oliveira E, Lavrador JP, Roque L, et al. Atypical teratoid/rhabdoid tumour following craniopharyngioma radiotherapy during childhood. Child’s Nerv Syst. 2016;32:1573–5.
Kuge A, Sato S, Sakurada K, et al. Atypical teratoid rhabdoid tumor located in the pineal region following prophylactic irradiation for acute lymphoblastic leukemia. Brain Tumor Pathol. 2012;29:177–81.
Köbel M, Hoang LN, Tessier-Cloutier B, et al. Undifferentiated endometrial carcinomas show frequent loss of core switch/sucrose nonfermentable complex proteins. Am J Surg Pathol. 2018;42:76–83.
Strehl JD, Wachter DL, Fiedler J, et al. Pattern of SMARCB1 (INI1) and SMARCA4 (BRG1) in poorly differentiated endometrioid adenocarcinoma of the uterus: analysis of a series with emphasis on a novel SMARCA4-deficient dedifferentiated rhabdoid variant. Ann Diagn Pathol. 2015;19:198–202.
Fahiminiya S, Witkowski L, Nadaf J, et al. Molecular analyses reveal close similarities between small cell carcinoma of the ovary, hypercalcemic type and atypical teratoid/rhabdoid tumor. Oncotarget. 2016;7:1732–40.
Chan-Penebre E, Armstrong K, Drew A, et al. Selective killing of SMARCA2- and SMARCA4-deficient small cell carcinoma of the ovary, hypercalcemic type cells by inhibition of EZH2: in vitro and in vivo preclinical models. Mol Cancer Ther. 2017;16:850–60.
Jelinic P, Ricca J, Van Oudenhov E, et al. Immune-Active Microenvironment in Small Cell Carcinoma of the Ovary, Hypercalcemic Type: Rationale for Immune Checkpoint Blockade. NCI J Natl Cancer Inst. 2018 Jan 22. https://doi.org/10.1093/jnci/djx277. [Epub ahead of print].
Karnezis AN, Hoang LN, Coatham M, et al. Loss of switch/sucrose non-fermenting complex protein expression is associated with dedifferentiation in endometrial carcinomas. Mod Pathol. 2016;29:302–14.
Altrabulsi B, Malpica A, Deavers MT, et al. Undifferentiated carcinoma of the endometrium. Am J Surg Pathol. 2005;29:1316–21.
Schaefer I-M, Agaimy A, Fletcher CD, et al. Claudin-4 expression distinguishes SWI/SNF complex-deficient undifferentiated carcinomas from sarcomas. Mod Pathol. 2017;30:539–48.
Shah VI, McCluggage WG. Cyclin D1 does not distinguish YWHAE-NUTM2 high-grade endometrial stromal sarcoma from undifferentiated endometrial carcinoma. Am J Surg Pathol. 2015;39:722–4.
Quade BJ. Pathology, cytogenetics and molecular biology of uterine leiomyomas and other smooth muscle lesions. Curr Opin Obstet Gynecol. 1995;7:35–42.
Conlon N, Soslow RA, Murali R. Perivascular epithelioid tumours (PEComas) of the gynaecological tract. J Clin Pathol NIH Public Access. 2015;68:418–26.
Howitt BE, Sholl LM, Dal Cin P, et al. Targeted genomic analysis of Müllerian adenosarcoma. J Pathol. 2015;235:37–49.
McCluggage WG. A practical approach to the diagnosis of mixed epithelial and mesenchymal tumours of the uterus. Mod Pathol. 2016;29:S78–91.
Clement PB, Scully RE. Mullerian adenosarcoma of the uterus: a clinicopathologic analysis of 100 cases with a review of the literature. Hum Pathol. 1990;21:363–81.
Al-Hussaini M, Hirschowitz L, McCluggage WG. Uterine neoplasms composed of rhabdoid cells do not exhibit loss of INI1 immunoreactivity and are not related to childhood malignant rhabdoid tumor. Int J Gynecol Pathol. 2008;27:236–42.
Cho KR, Rosenshein NB, Epstein JI. Malignant rhabdoid tumor of the uterus. Int J Gynecol Pathol. 1989;8:381–7.
Hsueh S, Chang T-C. Malignant rhabdoid tumor of the uterine corpus. Gynecol Oncol. 1996;61:142–6.
Brennan B, Stiller C, Bourdeaut F. Extracranial rhabdoid tumours: what we have learned so far and future directions. Lancet Oncol. 2013;14:e329–36.
Heck JE, Lombardi CA, Cockburn M, et al. Epidemiology of rhabdoid tumors of early childhood. Pediatr Blood Cancer. 2013;60:77–81.
Florell SR, Bruggers CS, Matlak M, et al. Ovarian small cell carcinoma of the hypercalcemic type in a 14 month old: the youngest reported case. Med Pediatr Oncol. 1999;32:304–7.
Estel R, Hackethal A, Kalder M, et al. Small cell carcinoma of the ovary of the hypercalcaemic type: an analysis of clinical and prognostic aspects of a rare disease on the basis of cases published in the literature. Arch Gynecol Obstet. 2011;284:1277–82.
Witkowski L, Goudie C, Ramos P, et al. The influence of clinical and genetic factors on patient outcome in small cell carcinoma of the ovary, hypercalcemic type. Gynecol Oncol. 2016;141:454–60.
Sultan I, Qaddoumi I, Rodríguez-Galindo C, et al. Age, stage, and radiotherapy, but not primary tumor site, affects the outcome of patients with malignant rhabdoid tumors. Pediatr Blood Cancer. 2010;54:35–40.
Hasselblatt M, Gesk S, Oyen F, et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol. 2011;35:933–5.
Eaton KW, Tooke LS, Wainwright LM, et al. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer. 2011;56:7–15.
Hasegawa T, Matsuno Y, Shimoda T, et al. Proximal-type epithelioid sarcoma: a clinicopathologic study of 20 cases. Mod Pathol. 2001;14:655–63.
Le Loarer F, Watson S, Pierron G, et al. SMARCA4 inactivation defines a group of undifferentiated thoracic malignancies transcriptionally related to BAF-deficient sarcomas. Nat Genet. 2015;47:1200–5.
Cattani MG, Viale G, Santini D, et al. Malignant rhabdoid tumour of the uterus: an immunohistochemical and ultrastructural study. Virchows Arch A Pathol Anat Histopathol. 1992;420:459–62.
Tsuda H, Maed K, Hashiguchi Y, et al. Malignant rhabdoid tumour of the uterine cervix. BJOG. 2001;108:120–3.
Knapik J, Yachnis AT, Ripley D, et al. Aggressive uterine sarcoma with rhabdoid features: diagnosis by peritoneal fluid cytology and absence of INI1 gene mutation. Hum Pathol. 2001;32:884–6.
Niemann T. Malignant rhabdoid tumor of the uterine corpus. Gynecol Oncol. 1997;64:181–2.
Mount SL, Lee KR, Taatjes DJ. Carcinosarcoma (malignant mixed müllerian tumor) of the uterus with a rhabdoid tumor component. An immunohistochemical, ultrastructural, and immunoelectron microscopic case study. Am J Clin Pathol. 1995;103:235–9.
Donner LR, Wainwright LM, Zhang F, et al. Mutation of the INI1 gene in composite rhabdoid tumor of the endometrium. Hum Pathol. 2007;38:935–9.
Baschinsky DY, Niemann TH, Eaton LA, et al. Malignant mixed Müllerian tumor with rhabdoid features: a report of two cases and a review of the literature. Gynecol Oncol. 1999;73:145–50.
Gaertner EM, Farley JH, Taylor RR, et al. Collision of uterine rhabdoid tumor and endometrioid adenocarcinoma: a case report and review of the literature. Int J Gynecol Pathol. 1999;18:396–401.
Acknowledgements
We thank Jeff Sanborn and the staff at the Center for Advanced Molecular Diagnostics, Brigham and Women’s Hospital as well as Dr. Jason Hornick, Mei Zheng, and the staff of the BWH immunohistochemistry lab.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Kolin, D.L., Dong, F., Baltay, M. et al. SMARCA4-deficient undifferentiated uterine sarcoma (malignant rhabdoid tumor of the uterus): a clinicopathologic entity distinct from undifferentiated carcinoma. Mod Pathol 31, 1442–1456 (2018). https://doi.org/10.1038/s41379-018-0049-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0049-z
This article is cited by
-
Pathological characteristics and immune microenvironment of SMARCA4-deficient undifferentiated uterine sarcoma
Diagnostic Pathology (2023)
-
Thoracic SMARCA4-deficient undifferentiated tumor
Discover Oncology (2023)
-
Alanine supplementation exploits glutamine dependency induced by SMARCA4/2-loss
Nature Communications (2023)
-
Unusual lung tumors—from morphology to genetics
Modern Pathology (2022)
-
Mullerian adenosarcoma: clinicopathologic and molecular characterization highlighting recurrent BAP1 loss and distinctive features of high-grade tumors
Modern Pathology (2022)
