
Abstract
High-grade endometrial stromal sarcomas (HGESSs) are more aggressive and have higher rates of resistance to endocrine therapy than low-grade endometrial stromal sarcomas (LGESSs). The pathogenesis of hormonal resistance in these lesions has yet to be defined. Here we sought to histologically and genetically characterize 3 LGESSs and their recurrences that underwent histologic high-grade transformation following endocrine therapy. For this, DNA from primary tumors and select subsequent recurrences were subject to massively parallel sequencing targeting 468 cancer-related genes. Somatic mutation analyses were performed using validated bioinformatics methods. In addition, RNA from each case was evaluated for the presence of gene fusions using targeted RNA-sequencing. All patients initially presented with LGESS, developed HGESS recurrences, and received at least 2 lines of hormonal suppressive therapy. Gene fusions classically described as associated with LGESS were identified in all 3 cases, including JAZF1-PHF1, EPC1-PHF1 and JAZF1-SUZ12 fusions for Cases 1, 2 and 3, respectively. Targeted sequencing analysis revealed that none of the primary LGESS, however the HGESS recurrences of Cases 1 and 3, and the LGESS and HGESS recurrences of Case 2 post endocrine treatment harbored ESR1 p.Y537S hotspot mutations. These ESR1 ligand-binding domain mutations have been found as a mechanism of acquired endocrine resistance in breast cancer. Also, a reduction in estrogen receptor (ER) expression was observed in recurrences. Our findings suggest that the ESR1 p.Y537S hotspot mutation in LGESS with histologic high-grade transformation may be associated with endocrine resistance in these lesions. Furthermore, our data suggest that genetic analyses may be performed in recurrent LGESS following hormonal therapy, development of high-grade morphology, and/or altered/diminished ER expression.
Similar content being viewed by others

Introduction
Endometrial stromal sarcomas (ESSs) are rare, accounting for ~15% of uterine sarcomas, and are classified as low- (LGESS) or high-grade (HGESS) based on histologic, immunophenotypic, and molecular genetic features1. LGESS are characterized by spindle cells with mild nuclear atypia, low mitotic activity, CD10, estrogen receptor (ER), progesterone receptor (PR) expression, and recurrent chromosomal rearrangements often involving JAZF12,3,4. HGESS consist of round and/or spindle cells demonstrating moderate nuclear atypia, brisk mitotic activity, and loss of CD10 and/or ER and PR expression with cyclin D1 and BCOR overexpression1. Most HGESS harbor YWHAE and BCOR fusions or BCOR internal tandem duplications3,4. Rare tumors harbor LGESS-associated gene fusions and demonstrate increased nuclear atypia and mitotic index warranting classification as HGESS5.
Prognostication of ESS, particularly high-grade variants, is difficult due to the evolving uterine sarcoma classification with increased utility of massively parallel sequencing. LGESS are indolent tumors treated with surgery, hormonal blockade, or a combination thereof6,7. While LGESS has a 5-year disease-free survival rate of >90%, up to 60% of patients develop late recurrences8,9,10. Some LGESS patients develop resistance to hormonal treatment and may benefit from cytotoxic chemotherapy6,11,12. HGESS, including histologically transformed ESS with LGESS-associated fusions, appear to have a more aggressive behavior compared to LGESS and is treated by surgery and chemotherapy with improved response using anthracycline-based regimens in tumors harboring YWHAE fusions13. Mechanisms of hormonal resistance in LGESS have not been previously explored, and the pathogenesis of histologically transformed LGESS is unknown. Here we sought to histologically and genetically characterize 3 LGESS and their recurrences that underwent histologic high-grade transformation following endocrine therapy.
Methods
Cases
The patients provided informed consent for a Memorial Sloan Kettering Cancer Center (MSK) Institutional Review Board-approved tumor genomic profiling study. Between 2014 and 2018, 18 diagnostically confirmed LGESS and HGESS were subjected to clinical sequencing at our institution. Three cases with ESR1 mutations and available tissues were identified. Formalin-fixed paraffin-embedded (FFPE) tissues of primary tumors and matched recurrences were obtained. All available hematoxylin and eosin and ER, PR, BCOR, and cyclin D1 immunohistochemical stained slides were reviewed by expert pathologists (S.C., R.A.S.). Cases were histologically subtyped using the World Health Organization classification system14. ER and PR expression were interpreted as positive if ≥1% of tumor cells showed nuclear staining, and intensity of staining was recorded2. ER and PR Allred scores were calculated as the sum of proportion and intensity scores. The proportion score was assigned as 0 (0%), 1 (<1%), 2 (1–10%), 3 (11–33%), 4 (34–66%), and 5 (67–100%). The intensity score was assigned as 0 (negative), 1 (weak), 2 (moderate), and 3 (strong)15. BCOR and cyclin D1 were interpreted as positive if ≥50% of tumor cells showed nuclear staining3,4. Clinical data, including demographics, treatment, and follow-up, were extracted from the electronic medical record.
Targeted massively parallel sequencing and validation
Representative 8 μm-thick sections from representative LGESS and HGESS FFPE tumor blocks were subjected to microdissection using a sterile needle under a stereomicroscope when appropriate to ensure >80% tumor cell content, as previously described16,17. Genomic DNA was extracted from the tumor samples and matched normal blood or normal tissue devoid of neoplastic cells using the DNeasy Blood and Tissue Kit (Qiagen) according to manufacturers’ instructions18. Tumor and matched normal DNA samples were subjected to massively parallel sequencing targeting 468 genes using MSK-Integrated Mutational Profiling of Actionable Cancer Targets (MSK-IMPACT)16,19 at a median depth of 595x (range, 584-625x), and sequencing data were analyzed using validated bioinformatics tools as previously described16,17. In brief, somatic single nucleotide variants (SNVs) were identified using MuTect (v1.17)20, and small insertions and deletions (indels) using Strelka (v1.0.15), VarScan 2 (v2.3.7), Lancet (v1.0.0) and Scalpel (v0.5.3)21,22,23,24. Mutations identified in the primary LGESS or recurrence from a given patient were interrogated in the respective LGESS and/or recurrence(s) by manual inspection of BAM files using mpileup files (SAMtools version 1.2 htslib 1.2.1)25. Copy number alterations (CNAs) and loss of heterozygosity (LOH) were defined using FACETS26, as previously described16,17. Cancer cell fractions (CCFs) of somatic mutations identified were defined using ABSOLUTE (v1.0.6)27, and a mutation was classified as clonal if its probability of being clonal was >50% or if the lower bound of the 95% confidence interval of its CCF was >90%, as previously described16,17. Mutational hotspot annotation was performed according to Chang et al28.
ESR1 p.Y537S mutations identified by MSK-IMPACT sequencing were subjected to orthogonal validation using Sanger sequencing, as previously described29. In brief, PCR amplification was performed using the AmpliTaq Gold 360 Master Mix kit (Life Technologies, ThermoFisher Scientific; primers: forward 5’-CCTTTCTGTGTCTTCCCACCT, reverse 3’-AGTGGCTTTGGTCCGTCTC), and PCR fragments were cleaned using ExoSAP-IT (ThermoFisher Scientific).
Targeted fusion gene detection
Tumor samples were subjected to the MSK-Fusion Solid Panel assay (Archer FusionPlex) utilizing Archer Anchored Multiplex PCR technology to detect 85 validated gene fusions in solid tumors, as previously described5. For this, tumor RNA was extracted from 5-μm-thick tumor sections, followed by cDNA synthesis and library preparation. Final targeted amplicons were sequenced, and data analyzed using the Archer analysis software V5.0.
Results
Case 1
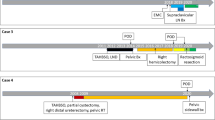
Case 1 was a 47-year-old woman who, after resection, was found to have FIGO stage IIIB LGESS with residual miliary disease (Fig. 1A). The primary uterine (Case1-Prim) and extrauterine tumor demonstrated predominately conventional LGESS morphology characterized by small monomorphic spindle cells with round to oval nuclei, inconspicuous nucleoli, scant cytoplasm, and a mitotic index of <1 per 10 high power fields (HPF; Fig. 1B); focal variant decidual change was present. Strong ER and PR expression was observed in >90% of tumor cells (ER and PR Allred scores 8 and 8, respectively; Fig. 1B). She received post-resection megestrol acetate and remained radiographically disease-free for 6.5 years. A large recurrent jejunal mass was partially resected in the setting of a small bowel obstruction (Case1-Recur1), and she received post-resection anastrozole for 3.5 years, at which time attempted resection of persistent disease was not successful (Case1-Recur2). Treatment was changed to letrozole with disease control for 2 years, after which the patient had successful complete resection of the intra-colonic mass (Case1-Recur3) and peritoneal disease.

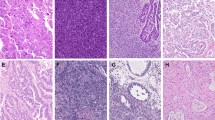
A Clinical, surgical and treatment history including histology and estrogen receptor (ER)/progesterone receptor (PR) status of the primary low-grade endometrial stromal sarcoma (LGESS) (Case1-Prim) and recurrences (Case1-Recur1/Recur2/Recur3). B Micrographs of representative hematoxylin and eosin (H&E)-stained sections and ER/PR expression by immunohistochemistry of the primary LGESS (Case1-Prim) and each recurrence (Case1-Recur1/Recur2/Recur3). Magnification ×20. C Fusion gene and non-synonymous somatic mutations identified in the primary LGESS (Case1-Prim) and the second (Case1-Recur2) and third (Case1-Recur3) recurrences. Mutation types (middle), including the variant allele fraction for the ESR1 mutation, and cancer cell fraction of mutations identified (bottom) are color-coded according to the legend. Note the clonal ESR1 hotspot mutation was only detected in the last recurrence (Case1-Recur3). The Allred scores for ER and PR are provided in the phenobar. LGESS, low-grade endometrial stromal sarcoma; HGESS, high-grade endometrial stromal sarcoma.
All recurrences (Case1-Recur1/Recur2/Recur3) demonstrated LGESS morphology with foci of high-grade histologic transformation characterized by nuclear enlargement, prominent nucleoli, and increased mitotic activity of 9–11 mitoses per 10 HPF (Fig. 1B). The first and second recurrences (Case1-Recur1/Recur2) demonstrated ER and PR expression with variable intensity in >90% of tumor cells, including histologically high-grade foci (ER and PR Allred scores 7 and 7 in Case1-Recur1, respectively; ER and PR Allred scores 8 and 7 in Case1-Recur2, respectively). The third recurrence (Case1-Recur3) demonstrated strong PR but weak ER expression in 90% of tumor cells (ER and PR Allred scores 6 and 8, respectively). Cyclin D1 and BCOR were negative in all recurrences (Case1-Recur1/Recur2/Recur3).
A JAZF1-PHF1 fusion was detected. Targeted massively parallel sequencing revealed the presence of a somatic KDM5C frameshift mutation in the primary and second and third recurrent tumors (Case1-Prim/Recur2/Recur3; Fig. 1C); the first recurrence (Case1-Recur1) had insufficient tissue for sequencing. Notably, the third recurrence (Case1-Recur3) acquired a STK40 missense p.R128W mutation and an ESR1 p.Y537S hotspot mutation (variant allele fraction (VAF), 40%) not present in the primary tumor or second recurrence (Case1-Prim/Recur2; Fig. 1C). We next defined the cancer cell fraction (CCF; i.e., the bioinformatically inferred percentage of tumor cells harboring the mutation in the sample) of the ESR1 mutation, taking into account the tumor purity, ploidy and local copy number27, which revealed that the ESR1 p.Y537S hotspot mutation was clonal (i.e., present in 100% of the cancer cells; Fig. 1C). With the finding of an ESR1 mutation in the third recurrence (Case1-Recur3), the patient was treated with fulvestrant and continues to have no measurable disease for 48 months, 15 years from initial diagnosis (Fig. 1A).
Case 2
A 56-year-old woman was found to have FIGO stage IB LGESS (Fig. 2A), with a mass resected from the vaginal cuff (Case2-Prim). The patient was treated with post-resection letrozole and remained disease-free for 23 months, at which time imaging showed recurrent pelvic disease. Megestrol acetate was added to the letrozole, but there was rapid progression of disease. She was subsequently treated with gemcitabine-docetaxel, but the disease continued to progress. She underwent resection of multi-site peritoneal and retroperitoneal disease (Case2-Recur1) followed by observation for 11 months. For subsequent progression, she underwent another resection (Case2-Recur2), and then progressed 4 months later. She was treated with doxorubicin but had progression of disease.

A Clinical, surgical and treatment history including histology and estrogen receptor (ER)/ progesterone receptor (PR) status of the primary low-grade endometrial stromal sarcoma (LGESS) (Case2-Prim) and recurrences (Case2-Recur1/Recur2). B Micrographs of representative hematoxylin and eosin (H&E)-stained sections and ER/PR expression by immunohistochemistry of the primary LGESS (Case2-Prim) and each recurrence (Case2-Recur1/Recur2). For Case2-Recur2, the low-grade component of the HGESS was subjected to ER and PR immunohistochemical analysis separately. Magnification ×20. C Fusion gene and non-synonymous somatic mutations identified in the primary LGESS (Case2-Prim) and first (Case2-Recur1) and second (Case2-Recur2) recurrences. Mutation types (middle), including the variant allele fraction for the ESR1 mutation, and cancer cell fraction of mutations identified (bottom) are color-coded according to the legend. Note the clonal ESR1 hotspot mutation was identified in both recurrences (Case2-Recur1/Recur2). The Allred scores for ER and PR are provided in the phenobar. LGESS low-grade endometrial stromal sarcoma, HGESS high-grade endometrial stromal sarcoma, POD progression of disease, PR partial response.
The primary tumor and first recurrence (Case2-Prim/Recur1) showed conventional and variant sex cord-like LGESS morphology and a mitotic index of 2/10 HPF (Fig. 2B). PR expression was strong in >90% of tumor cells in the primary tumor and first recurrence (Case2-Prim/Recur1; PR Allred scores of 8 and 8, respectively); however, ER expression was strong in >90% of tumor cells in the primary tumor (ER Allred score 8; Case2-Prim) and weak in 10% of tumor cells of the first recurrence (ER Allred score 3; Case2-Recur1; Fig. 2B). The second recurrence (Case2-Recur2) involved multiple peritoneal and retroperitoneal sites mostly demonstrating conventional and variant sex cord-like LGESS morphology with moderate ER expression in 60% of tumor cells and strong PR expression in >90% of tumor cells (ER and PR Allred scores of 6 and 8, respectively). The para-aortic recurrence (Case2-Recur2), however, demonstrated round cells with enlarged nuclei, abnormal chromatin, and 12 mitoses per 10 HPF consistent with histologic high-grade transformation (Fig. 2B). These foci demonstrated strong ER/PR expression in >90% of tumor cells (ER and PR Allred scores of 8 and 8, respectively; Fig. 2B) and were positive for BCOR, while negative for cyclin D1.
An EPC1-PHF1 fusion was detected. Massively parallel sequencing revealed the presence of an ESR1 p.Y537S hotspot mutation in the recurrences (Case2-Recur1, VAF 43%; Case2-Recur2, VAF 29%) but not in the primary tumor (Case2-Prim; Fig. 2C). Analysis to define the CCFs showed that the ESR1 hotspot mutation was clonal and present in 100% of the cancer cells of both recurrences. Noting the ESR1 mutation on the most recent pathology (Case2-Recur2), the patient was treated with fulvestrant with radiographic response sustained for 1 year. At further progression, she was changed to anastrozole, then deceased 6 months later, 6 years from initial diagnosis.
Case 3
A 56-year-old was found to have FIGO stage IIIB LGESS after resection (Case3-Prim; Fig. 3A). She was treated with post-resection megestrol acetate for 11 years. Over 8 years since her initial surgery, she underwent resection of an intrabdominal mass, followed by splenectomy and a small bowel resection. While no recurrent disease was found in the splenectomy specimen, the pathology from the other 2 surgeries were not reviewed. She presented to our institution 11 years after initial diagnosis with 2 large recurrent abdominal masses, which were debulked (Case3-Recur1), and she remained disease-free while on post-resection letrozole for 6 years. She underwent surgery, including removal of an infected mesh and small bowel partial resection, with no evidence of recurrent disease intraoperatively. Letrozole was resumed, but she was found to have peritoneal nodules on imaging a year later, and underwent resection for recurrent disease (Case3-Recur2).

A Clinical, surgical and treatment history including histology and estrogen receptor (ER)/ progesterone receptor (PR) status of the primary low-grade endometrial stromal sarcoma (LGESS) (Case3-Prim) and recurrences (Case3-Recur1/Recur2). B Micrographs of representative hematoxylin and eosin (H&E)-stained sections and ER/PR expression by immunohistochemistry of the primary LGESS (Case3-Prim) and each recurrence (Case3-Recur1/Recur2). Magnification ×20. C Fusion gene and non-synonymous somatic mutations identified in the primary LGESS (Case3-Prim) and first (Case3-Recur1) and second (Case3-Recur2) recurrences. Mutation types (middle), including the variant allele fraction for the ESR1 mutation, and cancer cell fraction of mutations identified (bottom) are color-coded according to the legend. Note the clonal ESR1 hotspot mutation was identified only the second recurrence (Case3-Recur2). The Allred scores for ER and PR are provided in the phenobar. LGESS, low-grade endometrial stromal sarcoma; HGESS, high-grade endometrial stromal sarcoma.
The primary tumor demonstrated LGESS morphology and a mitotic index of <1/10 HPF; strong ER and PR staining was seen in >90% of tumor cells (ER and PR Allred scores of 8 and 8, respectively; Fig. 3B). The first recurrence (Case3-Recur1) demonstrated epithelioid and spindled cells with enlarged nuclei with moderate to severe atypia, abundant eosinophilic cytoplasm, prominent myxoid change, and <1 mitotic figure per 10 HPF, associated with a prominent lymphocytic infiltrate; PR expression was strong in >90% of tumor cells, while ER was weak in <5% of cells in the first recurrence (PR and ER Allred scores of 8 and 3, respectively; Case3-Recur1; Fig. 3B). The second recurrence (Case3-Recur2) demonstrated spindle cells with uniform moderate cytologic atypia, including nuclear enlargement and prominent nucleoli, and a mitotic index of 10/10 HPF, consistent with high-grade transformation; no epithelioid or myxoid change was present (Fig. 3B). ER and PR expression was strong in 80% and >90% of cells, respectively (ER and PR Allred scores of 8 and 8, respectively).
A JAZF1-SUZ12 fusion was detected. Massively parallel sequencing revealed the presence of a frameshift mutation in SMARCB1 in the first recurrence (Case3-Recur1) and a clonal ESR1 p.Y537S hotspot mutation in the second recurrence (Case3-Recur2, VAF 43%; Fig. 3C). Akin to Case 1 and Case 2, the ESR1 hotspot mutation was clonal in the second recurrence (Case3-Recur2), with all cancer cells harboring this mutation (CCF 100%; Fig. 3C). Noting the ESR1 mutation on the most recent pathology (Case3-Recur2), the patient completed 3 cycles of fulvestrant and is alive with disease, 19 years from initial diagnosis.
Discussion
Here we report on likely de novo ESR1 p.Y537S hotspot mutations in the recurrences of 3 patients who were initially diagnosed with LGESS and subsequently developed recurrent disease demonstrating histologic high-grade transformation after hormonal treatment.
The ESR1 gene codes for the estrogen receptor 1 (ERalpha), a ligand-activated transcription factor implicated in the tumorigenesis of many ER-positive cancers including ESS30,31. ESR1 p.Y537S hotspot mutations are activating and enable ER coactivator binding in the absence of estrogen32,33. Mutations in the ESR1 ligand-binding domain have been identified as an important mechanism of acquired endocrine resistance and occur under the selective pressure from hormonal therapies34,35. Particularly in breast cancer, ESR1 mutations have been found in ~1% of primary ER-positive breast cancers and 10–50% of metastatic breast cancers previously treated with hormonal blockade34,36,37. Also, a de novo ESR1 hotspot mutation in a patient with endometrial carcinoma treated with an aromatase inhibitor has been reported38.
Other mechanisms of endocrine resistance include ESR1 amplification/ fusion and alterations in receptor tyrosine kinases, PI3K and MAPK pathway components, regulators of gene expression, and DNA repair genes. We explored these alterations in the remaining 15 ESS of the 18 diagnostically confirmed ESS subjected to clinical sequencing at our institution (see Methods). Somatic genetic alterations associated with endocrine resistance were detected in only 2 cases: a recurrent HGESS harboring a YWHAE rearrangement showed TSC2 and MTOR deep deletions as well as AKT2 amplification. AKT1 amplification and PMS2 deletion were detected in a primary LGESS with a JAZF1-SUZ12 fusion. Among the 15 ESS with sequencing data, 3 were histologically confirmed LGESS lacking ESR1 mutations, and none of these 3 patients had evidence of hormonal resistance.
The association between ER expression and ESR1 mutation in our cohort is unclear. In Cases 1 and 2, ER expression was significantly attenuated in the recurrences that harbored ESR1 mutations. In Case 3, however, ER expression was diminished in the first recurrence with wild-type ESR1. Interestingly, in Cases 2 and 3, the recurrences that initially showed reduced ER expression were followed by recurrences with strong levels of ER expression.
In all cases studied here, histologically high-grade transformation of LGESS was observed during disease progression as evidenced by increased mitotic rates and nuclear atypia in the recurrences, which paralleled the detection of ESR1 p.Y537S hotspot mutations. While Case 2 displayed endocrine resistance at first recurrence, in Cases 1 and 3 prolonged disease control was observed on a selective ER degrader (SERD; fulvestrant) following resistance to a selective ER modulator (SERM)/aromatase inhibitor (AI). In fact, work in breast cancer has shown that the mechanism of resistance to SERMs/AIs is distinct from that of SERDs39,40. All 3 patients had some degree of clinical benefit from fulvestrant, which was administered after detection of ESR1 hotspot mutations in the recurrences. Further studies are warranted to assess whether patients with ESR1-mutant ESS may benefit from SERDs over other therapies.
High-grade transformation of LGESS is rare, and data on the molecular underpinnings of these lesions are limited. The majority of LGESS are driven by PHF1 and JAZF1 translocations5,41, as the 3 cases described in this study, whereas YWHAE and BCOR genetic abnormalities are associated with HGESS42. In Case 1, this occurred prior to the identification of the ESR1 mutation and in Cases 2 and 3, the high-grade transformation was identified after the emergence of an ESR1 mutation. Notably in Case 3, however, there was increased nuclear atypia despite low mitotic activity in the first recurrence (Case3-Recur1), suggesting early transformation at that time. The interplay between ER-alpha signaling and histologic high-grade transformation of LGESS remains unknown and warrants further investigation.
Ligand independent activation of ER receptors results in increased translational activity outside of the downstream affects typically attributed to estrogen binding43,44. For example, ESR1 mutations were found to result in increased expression of non-ER regulation genes associated with breast cancer cell migration such as WNT1145 and RET46, suggesting that these mutations may contribute to increased metastatic potential of these tumors33. This theory is supported by identification of these mutations preferentially in distant metastatic sites in comparison to locoregional recurrences37,47. Thus, the presence of an ESR1 mutation may have prognostic implications separate from the limitation on effective therapies.
This study has several limitations. Given the rarity of LGESS in general, and of those undergoing high-grade transformation in particular, the number of cases studied is small, however the identification of potential mechanisms of endocrine resistance contributes to the body of knowledge in this disease. In addition, ER and PR immunohistochemical analyses were performed at the referring institution (Case 1) or at our clinical and research laboratories (Cases 1–3), which may contribute to differences in staining intensities.
In summary, de novo ESR1 hotspot mutations may occur in LGESS following histologic high-grade transformation and resistance to endocrine treatment. Larger series are required to further investigate the frequency of ESR1 mutations and their role in endocrine treatment resistance. Our findings suggest that genetic analyses may be performed in recurrent LGESS following hormonal therapy, development of high-grade morphology, and/or altered/ diminished ER expression.
References
WHO Classification of Tumours Editorial Board. Female genital tumours: WHO classification of tumours, 5th ed, Vol. 4 (International Agency for Research on Cancer: Lyon, 2020).
Toki, T., Shimizu, M., Takagi, Y., Ashida, T. & Konishi, I. CD10 is a marker for normal and neoplastic endometrial stromal cells. Int. J. Gynecol. Pathol. 21, 41–47 (2002).
Chiang, S. & Oliva, E. Cytogenetic and molecular aberrations in endometrial stromal tumors. Hum. Pathol. 42, 609–617 (2011).
Momeni-Boroujeni, A. & Chiang, S. Uterine mesenchymal tumours: recent advances. Histopathology 76, 64–75 (2020).
Zou, Y. et al. High-grade transformation of low-grade endometrial stromal sarcomas lacking YWHAE and BCOR genetic abnormalities. Mod. Pathol. 33, 1861–1870 (2020).
Chu, M. C. et al. Low-grade endometrial stromal sarcoma: hormonal aspects. Gynecol. Oncol. 90, 170–176 (2003).
Yamaguchi, M. et al. Long-term outcome of aromatase inhibitor therapy with letrozole in patients with advanced low-grade endometrial stromal sarcoma. Int. J. Gynecol. Cancer 25, 1645–1651 (2015).
Amant, F. et al. Transition of endometrial stromal sarcoma into high-grade sarcoma. Gynecol. Oncol. 103, 1137–1140 (2006).
Kurihara, S. et al. Endometrial stromal sarcomas and related high-grade sarcomas: immunohistochemical and molecular genetic study of 31 cases. Am. J. Surg. Pathol. 32, 1228–1238 (2008).
Leath, C. A. 3rd et al. A multi-institutional review of outcomes of endometrial stromal sarcoma. Gynecol. Oncol. 105, 630–634 (2007).
Lee, C. H. et al. The clinicopathologic features of YWHAE-FAM22 endometrial stromal sarcomas: a histologically high-grade and clinically aggressive tumor. Am. J. Surg. Pathol. 36, 641–653 (2012).
Pink, D. et al. Harm or benefit of hormonal treatment in metastatic low-grade endometrial stromal sarcoma: single center experience with 10 cases and review of the literature. Gynecol. Oncol. 101, 464–469 (2006).
Hemming, M. L. et al. YWHAE-rearranged high-grade endometrial stromal sarcoma: two-center case series and response to chemotherapy. Gynecol. Oncol. 145, 531–535 (2017).
Cree, I. A., White, V. A., Indave, B. I. & Lokuhetty, D. Revising the WHO classification: female genital tract tumours. Histopathology 76, 151–156 (2020).
Allred, D. C., Harvey, J. M., Berardo, M. & Clark, G. M. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod. Pathol. 11, 155–168 (1998).
Da Cruz Paula, A. et al. Genomic profiling of primary and recurrent adult granulosa cell tumors of the ovary. Mod. Pathol. 33, 1606–1617 (2020).
da Silva, E. M. et al. Mesonephric and mesonephric-like carcinomas of the female genital tract: molecular characterization including cases with mixed histology and matched metastases. Mod. Pathol. 34, 1570–1587 (2021).
Martelotto, L. G. et al. Genomic landscape of adenoid cystic carcinoma of the breast. J. Pathol. 237, 179–189 (2015).
Cheng, D. T. et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 17, 251–264 (2015).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Saunders, C. T. et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817 (2012).
Koboldt, D. C. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012).
Narzisi, G. et al. Genome-wide somatic variant calling using localized colored de Bruijn graphs. Commun. Biol. 1, 20 (2018).
Narzisi, G. et al. Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat. Methods 11, 1033–1036 (2014).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Shen, R. & Seshan, V. E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 44, e131 (2016).
Carter, S. L. et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–421 (2012).
Chang, M. T. et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Disco 8, 174–183 (2018).
Weinreb, I. et al. Hotspot activating PRKD1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nat. Genet 46, 1166–1169 (2014).
Blanchard, Z., Vahrenkamp, J. M., Berrett, K. C., Arnesen, S. & Gertz, J. Estrogen-independent molecular actions of mutant estrogen receptor 1 in endometrial cancer. Genome Res. 29, 1429–1441 (2019).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Disco 2, 401–404 (2012).
Li, S. et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 4, 1116–1130 (2013).
Fanning S. W., et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife 5, e12792 (2016)
Toy, W. et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet 45, 1439–1445 (2013).
Chandarlapaty, S. et al. Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol. 2, 1310–1315 (2016).
Spoerke, J. M. et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat. Commun. 7, 11579 (2016).
Jeselsohn, R., Buchwalter, G., De Angelis, C., Brown, M. & Schiff, R. ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol. 12, 573–583 (2015).
Morel, A. et al. De novo ESR1 hotspot mutation in a patient with endometrial cancer treated with an aromatase inhibitor. JCO Precis Oncol. 3, 1–3 (2019).
Ingle, J. N. et al. Fulvestrant in women with advanced breast cancer after progression on prior aromatase inhibitor therapy: North Central Cancer Treatment Group Trial N0032. J. Clin. Oncol. 24, 1052–1056 (2006).
Toy, W. et al. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Disco 7, 277–287 (2017).
Ali, R. H. & Rouzbahman, M. Endometrial stromal tumours revisited: an update based on the 2014 WHO classification. J Clin Pathol 68, 325–332 (2015).
Sciallis, A. P. et al. High-grade endometrial stromal sarcomas: a clinicopathologic study of a group of tumors with heterogenous morphologic and genetic features. Am. J. Surg. Pathol. 38, 1161–1172 (2014).
Rothenberger N. J., Somasundaram A., Stabile L. P. The role of the estrogen pathway in the tumor microenvironment. Int. J. Mol. Sci. 19, 611 (2018)
Bahreini, A. et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res. 19, 60 (2017).
Dwyer, M. A. et al. WNT11 expression is induced by estrogen-related receptor alpha and beta-catenin and acts in an autocrine manner to increase cancer cell migration. Cancer Res. 70, 9298–9308 (2010).
Gattelli, A. et al. Ret inhibition decreases growth and metastatic potential of estrogen receptor positive breast cancer cells. EMBO Mol. Med. 5, 1335–1350 (2013).
Niu, J. et al. Incidence and clinical significance of ESR1 mutations in heavily pretreated metastatic breast cancer patients. Onco. Targets Ther. 8, 3323–3328 (2015).
Acknowledgements
This work was funded in part the NIH/ NCI Cancer Center Support Grant P30-CA008748. B.W. was funded in part by Cycle for Survival and Breast Cancer Research Foundation grants.
Author information
Authors and Affiliations
Contributions
Conception and design: M.L.H., B.W., S.C. Financial support: N.R.A.-R. Provision of study material or patients: S.C. Collection and assembly of data: K.D., K.M.M., E.K., A.D.C.P., Y.Z., E.M.d.S., S.D., R.B., C.W.A., R.A.S., M.L.H., B.W., S.C. Data analysis and interpretation: K.D., K.M.M., E.K., A.D.C.P., P.S., B.W., S.C. Manuscript writing: K.D., K.M.M., E.K., M.L.H., B.W., S.C. Final approval of manuscript: All authors.
Corresponding authors
Ethics declarations
Competing interests
N.R.A.-R. reports institutional grants from Stryker/Novadaq and GRAIL, outside the submitted work. M.L.H. reports spouse employment by Sanofi, advisory board consulting for Tesaro, Glaxo Smith Kline. B.W. reports ad hoc membership of the scientific advisory board of Repare Therapeutics, outside the current study. S.C. reports consulting for AstraZeneca. The remaining authors have no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dessources, K., Miller, K.M., Kertowidjojo, E. et al. ESR1 hotspot mutations in endometrial stromal sarcoma with high-grade transformation and endocrine treatment. Mod Pathol 35, 972–978 (2022). https://doi.org/10.1038/s41379-021-01003-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-01003-5
