
Abstract
Embryonal rhabdomyosarcoma (ERMS) of the uterus has recently been shown to frequently harbor DICER1 mutations. Interestingly, only rare cases of extrauterine DICER1-associated ERMS, mostly located in the genitourinary tract, have been reported to date. Our goal was to study clinicopathologic and molecular profiles of DICER1-mutant (DICER1-mut) and DICER1-wild type (DICER1-wt) ERMS in a cohort of genitourinary tumors. We collected a cohort of 17 ERMS including nine uterine (four uterine corpus and five cervix), one vaginal, and seven urinary tract tumors. DNA sequencing revealed mutations of DICER1 in 9/9 uterine ERMS. All other ERMS of our cohort were DICER1-wt. The median age at diagnosis of patients with DICER1-mut and DICER1-wt ERMS was 36 years and 5 years, respectively. Limited follow-up data (available for 15/17 patients) suggested that DICER1-mut ERMS might show a less aggressive clinical course than DICER1-wt ERMS. Histological features only observed in DICER1-mut ERMS were cartilaginous nodules (6/9 DICER1-mut ERMS), in one case accompanied by foci of ossification. Recurrent mutations identified in both DICER1-mut and DICER1-wt ERMS affected KRAS, NRAS, and TP53. Copy number analysis revealed similar structural variations with frequent gains on chromosomes 2, 3, and 8, independent of DICER1 mutation status. Unsupervised hierarchical clustering of array-based whole-genome DNA methylation data of our study cohort together with an extended methylation data set including different RMS subtypes from genitourinary and extra-genitourinary locations (n = 102), revealed a distinct cluster for DICER1-mut ERMS. Such tumors clearly segregated from the clusters of DICER1-wt ERMS, alveolar RMS, and MYOD1-mutant spindle cell and sclerosing RMS. Only one tumor, previously diagnosed as ERMS arising in the maxilla of a 6-year-old boy clustered with DICER1-mut ERMS of the uterus. Subsequent sequencing analysis identified two DICER1 mutations in the latter case. Our results suggest that DICER1-mut ERMS might qualify as a distinct subtype in future classifications of RMS.
Similar content being viewed by others

Introduction
Rhabdomyosarcoma (RMS) represents the most common soft tissue sarcoma of children and adolescents, accounting for almost 50% of all pediatric soft tissue sarcomas [1]. The current WHO classification of soft tissue tumors defines embryonal rhabdomyosarcoma (ERMS), alveolar rhabdomyosarcoma (ARMS), pleomorphic rhabdomyosarcoma, and spindle cell and sclerosing rhabdomyosarcoma (SRMS) as separate subtypes of RMS [2]. Beyond this classification, recent clinicopathological studies have identified novel molecular subtypes of RMS including two variants of SRMS harboring either MYOD1 mutations or VGLL2/NCOA2 gene fusions, as well as RMS with a predominantly epithelioid phenotype and TFCP2 gene fusions [3].
ERMS represents the most common subtype, being most prevalent within the head and neck region, followed by the genitourinary tract [4]. In the genitourinary tract ERMS most frequently arises in the vagina and the urinary tract of infants [5]. ERMS of the uterine cervix and corpus are uncommon and are usually associated with a later age of onset [6, 7]. While ERMS mostly arises sporadically, it may also develop in the context of various familial tumor predisposition syndromes, such as Li-Fraumeni syndrome (TP53), neurofibromatosis type 1 (NF1), Noonan syndrome (multiple genes), and Costello syndrome (HRAS) [8,9,10,11,12]. Interestingly, uterine ERMS may develop in connection with the pleuropulmonary blastoma (PPB) familial tumor predisposition syndrome, which is characterized by germline mutations in DICER1 (DICER1 syndrome) [6, 13]. Neoplasms associated with the DICER1 syndrome usually arise in young children and adolescents, and may—besides ERMS—include PPB, multinodular goiter, cystic nephroma, Sertoli–Leydig cell tumor of the ovary, and other rare tumor entities [14]. DICER1 is part of the ribonuclease III family and plays an important role in modulating gene expression at the posttranslational level through the processing of miRNA. Mutations in DICER1 may thus contribute to tumorigenesis by activation of oncogenes through dysregulation of miRNA [15].
Recent studies have shown recurrent germline and somatic DICER1 mutations to occur in a majority of uterine ERMS [7, 16, 17]. At the same time, only rare cases of extrauterine DICER1-associated ERMS, mostly located in the genitourinary tract, have been reported [16, 18,19,20]. In addition, molecular studies of larger cohorts of ERMS have failed to identify significant numbers of DICER1 alterations, considering that most analyzed cases were located outside of the genitourinary tract [21,22,23]. Thus, the question arises, if DICER1-mutant (DICER1-mut) ERMS represents a separate clinicopathologic subgroup distinct from DICER1-wild type (DICER1-wt) ERMS.
In an attempt to contribute data to the ongoing discussion on DICER1 alterations in ERMS, we report the clinicopathological characteristics of a cohort of genitourinary ERMS including DICER1-mut and DICER1-wt tumors by applying targeted DNA sequencing and performing comparative, genome-wide DNA methylation and copy number variation (CNV) analyses.
Material and methods
Study cohort and pathology review
A total of 17 ERMS of the genitourinary tract were collected, including nine uterine (four uterine cervix and five uterine corpus), one vaginal, and seven urinary-tract tumors (four bladders, one prostate, and two bladder/prostate not other specified). Uterine tumors were collected from the referral center archives of two of the authors (DSc, FK, n = 9). The remaining ERMS were allocated from the study archives of two of the authors (JM, ME, n = 3), as well as from the INFORM study cohort (n = 5) [24]. For all but one tumor, Hematoxylin & Eosin (H&E) slides from either fresh frozen (n = 2) or formalin-fixed and paraffin-embedded (FFPE) (n = 14) material was available. Additional desmin (Dako, mouse monoclonal, clone D33) and myogenin (Cell Marque, Rocklin, CA, USA, mouse monoclonal, clone F5D) immunohistochemistry (IHC) was only performed in cases were additional FFPE material was available (uterine tumors only, n = 9). All tumors with available H&E slides were subject to expert pathology review. A diagnosis of ERMS was made applying the current WHO criteria [2]. In cases of uterine tumors, special attention was given to the distinction from and the exclusion of uterine adenosarcoma, a well-known differential diagnosis of uterine ERMS [6]. Because this distinction is exceptionally difficult, for this study, uterine tumors where no consensus diagnosis of ERMS could be reached were excluded. Molecular features of two of the cases presented herein (ERMS 3 and 4) have previously been described elsewhere [25]. This study was approved by the institutional ethics committee and performed in accordance with the Declaration of Helsinki.
DNA extraction
DNA was extracted from either fresh-frozen or FFPE tumor tissue. The Maxwell® 16 FFPE Plus LEV DNA Kit or the Maxwell® 16 Tissue DNA Purification Kit (for frozen tissue) was applied on the automated Maxwell device (Promega, Madison, WI, USA) according to the manufacturer’s instructions. A minimum of 100 ng DNA was extracted in every case and provided for subsequent DNA sequencing and array-based DNA methylation analysis.
DNA sequencing
DNA was sequenced either as whole-exome using Agilent SureSelectXT Human V5 or V7 kit (n = 5, including from germline) or using a customized SureSelect XT technology (Agilent) panel (n = 14, no germline available) covering the coding regions of 130 genes. Library preparation, quality control, sequencing on a NextSeq or HiSeq sequencer (Illumina), and data processing were performed as previously described [24, 26]. Reads were aligned to the reference genome hg19 and variants were annotated using ANNOVAR software [27]. For samples without a matching germline control, synonymous and stop-loss variants, variants with a frequency exceeding 1% in the healthy population as well as variants described as known polymorphisms in the single nucleotide polymorphism database were excluded.
Genome‑wide DNA methylation data generation, plotting and pre‑processing
DNA was also analyzed using the Illumina Infinium HumanMethylation450 (450 k) BeadChip or the EPIC/850k BeadChip (Illumina, San Diego, USA) at the Genomics and Proteomics Core Facility of the German Cancer Research Center (DKFZ) in Heidelberg. DNA methylation data were normalized by applying background correction and dye bias correction (shifting of negative control probe mean intensity to zero and scaling of normalization control probe mean intensity to 20,000, respectively). Probes targeting sex chromosomes, probes containing multiple single nucleotide polymorphisms, and probes that could not be uniquely mapped were removed.
Copy number analysis, unsupervised clustering and t-SNE analysis
Copy number assessment was performed on methylation array data using the R-package conumee and copy number variants were identified by manual inspection [28]. For subsequent DNA methylation analyses, we included a previously compiled methylation data set of a large cohort of RMS of genitourinary and extra-genitourinary locations, including ARMS (n = 43), MYOD1-mutant SRMS (n = 12), as well as additional cases of ERMS (n = 39) and non-neoplastic striated muscle (control, n = 8), which has previously been published in part [29]. For unsupervised hierarchical clustering of DNA methylation data, 10,000 probes with the highest median absolute deviation across beta values were selected. Samples were hierarchically clustered using Euclidean distance and Ward’s linkage method. Methylation probes were reordered by hierarchical clustering using Euclidean distance and complete linkage. The unscaled methylation levels were shown in a heat map from the unmethylated state (blue color) to the methylated state (red color). For the unsupervised 2D representation of pairwise sample correlations, dimensionality reduction by t-distributed stochastic neighbor embedding (t-SNE) was performed using the 10,000 most variable probes, a perplexity of 10 and 3000 iterations. The stability of methylation groups was tested by varying the number of the most variable probes.
Results
DICER1 mutations in ERMS of the genitourinary tract
DNA sequencing of our cohort revealed a total of 14 DICER1-mutations in 9/9 (100%) uterine ERMS (Table 1, Fig. 1a, c). In detail, we identified ribonuclease III (RNase IIIb) domain hotspot mutations of DICER1 in nine uterine ERMS. Four cases harbored additional nonsense or frameshift mutations of DICER1. In one case we identified an additional non-hotspot DICER1 missense alteration (p.T1474A), which has previously been reported in hepatocellular carcinoma [30]. In four uterine ERMS, only the single RNase IIIb domain hotspot mutation of DICER1 was identified. Analysis of the allelic fraction of the alteration indicated homozygous mutations in three of the latter tumors (ERMS 1, 3, and 4). Due to low tumor cell content in the fourth tumor (ERMS 9) the allelic fraction of the DICER1 alteration was not informative of zygosity (Fig. 1b). Unfortunately, no germline data was available for DICER1-mut ERMS. No DICER1 alteration was identified in any of the extrauterine genitourinary ERMS of our cohort (Fig. 1a).

a Age at diagnosis, gender distribution, sites of origin and DNA sequencing data, (b) variant allele frequency (VAF) of DICER1 alterations suggesting either retained (ROH) or loss (LOH) of heterozygosity and (c) mutational spectrum of DICER1 mutations in 9 uterine ERMS. Cumulative copy-number profiles of (d) 9 DICER1-mut ERMS and (e) 8 DICER1-wt ERMS. Molecular data of ERMS 3 and 4 have previously been reported elsewhere [25].
Clinicopathological features of DICER1-mut and DICER1-wt ERMS
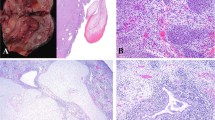
Median patient age at primary diagnosis was 36 years for DICER1-mut ERMS (mean: 38.3 years, range: 28–67 years) and 5 years in cases of DICER1-wt ERMS (mean: 7 years, range: 0.5–19 years). While all DICER1-mut ERMS arose in women, the male-to-female ratio for DICER1-wt tumors was 1.6 (63% male and 37% female). Classical histomorphological features of ERMS were present in both DICER1-mut (Fig. 2) and DICER1-wt (Fig. 3) tumors. These included polypoid growth of hyper- and hypocellular areas of small blue cells with scant cytoplasm and varying foci of rhabdomyoblastic differentiation exhibiting tumor cells with abundant eosinophilic cytoplasm (so-called “strap cells”). Perivascular condensation of tumor cells was seen in hypocellular areas. A subset of ERMS exhibited polypoid exophytic growth with a cambium layer consisting of a linear subepithelial tumor cell condensation, resembling the botryoid variant of ERMS (sarcoma botryoides). Marked anaplasia in the form of poorly differentiated spindle cells with high-grade nuclear atypia was focally detected in DICER1-mut (1/9) and DICER1-wt (3/8) tumors. The majority of DICER1-mut ERMS (6/9) showed small foci of cartilaginous differentiation, while such differentiation did not occur in any of the DICER1-wt ERMS. Interestingly, one DICER1-mut ERMS (ERMS 2) arising in a 67-year-old patient showed focal ossification (formation of osteoid deposits with associated multinucleated osteoclast-like giant cells), abutting atypical nodular cartilaginous differentiation (Fig. 2a, b). IHC of DICER1-mut tumors showed immunoreactivity for desmin and at least focal positivity for myogenin in all DICER1-mut ERMS (n = 9). Limited clinical follow-up was obtained in 15/17 of selected cases. 6/7 DICER1-mut ERMS showed an uneventful clinical course (median follow-up time: 8 years, range: 3–9 years). The remaining patient died of disease after 3 years. Among DICER1-wt ERMS 7/9 patients of our series suffered tumor recurrences or died of disease (median follow-up time: 5.5 years, range: 1–15 years). Clinicopathological data are summarized in Table 1.

a Nodular growth of hypocellular areas resembling the botryoid variant of ERMS, (b) entrapment of epithelium with cuffing of adjacent small blue cells with scant cytoplasm, as well as (c) desmin and (d) myogenin positivity. e Focal rhabdomyoblasts, and (f) anaplasia may be present. g Nodules of chondroid matrix as well as (h) areas of abutting ossification with osteoid matrix and multinucleated osteoclast-like giant cells may be suggestive of DICER1-association.

a Botryoid variant of ERMS with (b) a distinct cambium layer and (c) proliferation of small blue cells with scant cytoplasm. d Anaplasia and rhabdomyoblasts may be present, however, in contrast to DICER1-mut ERMS no nodules of cartilage or osteoid are present.
Recurrent alterations affecting TP53 and the RAS-pathway in DICER1-mut and DICER1-wt ERMS
Mutations identified in both DICER1-mut and DICER1-wt ERMS (Fig. 1a) affected KRAS and NRAS (3/9 DICER1-mut and 1/8 DICER1-wt ERMS) and TP53 (1/9 DICER1-mut and 2/8 DICER1-wt ERMS). Alterations only identified in either DICER1-mut or DICER1-wt ERMS affected KTM2D (1/9 DICER1-mut ERMS), BRAF (1/8 DICER1-wt ERMS), and NF1 (1/8 DICER1-wt ERMS). CNV identified in both DICER1-mut and DICER1-wt ERMS included frequent gains on chromosomes 2, 3, and 8 (Fig. 1d, e). While no amplifications were noted in any ERMS of our cohort, one DICER1-wt tumor harbored a somatic deletion including the NF1 locus. Detailed DNA sequencing results are provided in Supplementary Table 1.
Distinct patterns of DNA methylation in DICER1-mut ERMS
Unsupervised hierarchical clustering analysis of 17 ERMS of our cohort revealed two distinct methylation clusters corresponding to DICER1-mut and DICER1-wt ERMS (Supplementary Fig. 1a). Unsupervised hierarchical clustering (Fig. 4a) and t-SNE analysis (Fig. 4b) of 17 ERMS of our cohort, together with a large methylation data set of RMS of genitourinary and extra-genitourinary locations, confirmed a distinct DICER1-mut ERMS cluster. Such tumors clearly segregated from clusters of DICER1-wt ERMS, alveolar RMS, MYOD1-mutant spindle cell, and sclerosing RMS and non-neoplastic striated muscle samples. Methylation clusters remained stable when varying the analyzed number of CpG sites (data not shown). Interestingly, one ERMS from the reference set, arising in the maxillary region of a 6-year-old boy clustered with DICER1-mut ERMS. Histomorphological evaluation of this case revealed classical features of ERMS with focal anaplasia, however, no foci of cartilaginous differentiation were noted. Subsequent targeted DNA sequencing identified a missense (p.E844X) and an RNase IIIb domain hotspot mutation of DICER1 (p.D1709N). The patient remains without evidence of disease 8 years after the initial diagnosis. Detailed information on the latter case is given in Supplementary Fig. 2.

Unsupervised hierarchical clustering (a) and t-SNE analysis (b) of 9 DICER1-mut and 8 DICER1-wt ERMS, together with a large methylation data set of RMS of genitourinary and extra-genitourinary locations shows distinct cluster formation for ARMS (n = 43), MYOD1-mut SRMS (n = 12), ERMS (n = 46), non-neoplastic striated muscle tissue (control; n = 8) and DICER1-mut ERMS (n = 10).
Discussion
Herein we present the clinicopathologic characteristics of a cohort of DICER1-mut and DICER1-wt ERMS of the genitourinary tract. We found DICER1 alterations to be present in all ERMS of the uterine corpus and cervix, while no such mutations were identified in the remaining cases of our series. To date, only rare cases of DICER1-mut ERMS of the vagina and the urinary tract have been published [16, 31,32,33]. In keeping with the latter findings, most published series of molecularly analyzed ERMS arising in the vagina or urinary tract have lacked any tumors with DICER1 mutations [16, 21, 22]. In contrast, most cases of molecularly analyzed ERMS of the uterine corpus and cervix harbor DICER1 mutations [7]. In addition, few cases of ERMS arising in the broad ligament, the ovary, and the fallopian tube are also DICER1 mutation-positive [18, 20]. In combination with published evidence, our results suggest that ERMS of the inner female genital tract—maybe with the exception of most tumors arising in the vagina—are associated with DICER1 mutations, in contrast to other ERMS arising in the urinary tract which is usually DICER1-wt. In their recent paper, Appellaniz-Ruiz et al. hypothesized that DICER1 mutations could predispose to tumor development in Müllerian-derived tissues [34]. The Müllerian ducts ontogenetically give rise to the upper portion of the vagina, the uterine cervix, and corpus, as well as the fallopian tubes, potentially providing a developmental explanation for the predilection of DICER1-mut ERMS at these sites.
Our results further indicate clinical differences between DICER1-mut and DICER1-wt ERMS. Here, the presence of DICER1 alterations in ERMS was associated with older patient age at diagnosis (median age 36 years vs. 5 years for DICER1-wt ERMS). These results are in line with a recent report by de Kock et al. who reported a series of 19 DICER1-associated uterine ERMS with a median age at diagnosis of 30 years [7]. In contrast, a younger median age of 16 years at diagnosis of patients with DICER1-associated ERMS arising anywhere in the female genital tract was published in a recent meta-analysis [34]. Our results suggest that on the other hand most extrauterine ERMS, more specifically DICER1-wt ERMS, usually do not occur in older patients [5]. Although potentially limited by a selection bias, in our study, follow-up data suggest that in the genitourinary tract, DICER1-mut ERMS might show a more favorable clinical course as compared to DICER1-wt ERMS. Interestingly, genitourinary RMS arising in so-called non-bladder/non-prostate sites, such as the vagina or the uterus was reported to behave less aggressively than those with primary location in the urinary tract, irrespective of DICER1 mutation status in two studies [35, 36]. While clinical information on DICER1-associated ERMS has rarely been published to date, potentially aggressive behavior of these tumors is exemplified by one of our patients who died of disease 3 years after initial diagnosis. Further, McCluggage et al. recently reported a DICER1-mut ERMS of the ovary, which relapsed only 3 months after surgery [20]. Given the above, further investigations of the clinical behavior of DICER1-mut ERMS are clearly needed.
Histologically, ERMS of the genitourinary tract involving epithelial-lined viscera such as the vagina, the bladder, and the uterus, often resemble botryoid ERMS [37,38,39]. Despite a similar or in some instances even identical morphology, the presence of cartilaginous nodules has previously been described in DICER1-associated ERMS, a finding that is confirmed by the current study [7, 20]. Similarly, we report osteoid formation in conjunction with DICER1 mutations in a uterine ERMS, which has previously been reported in two cases of DICER1-associated ERMS of the female genital tract [34, 40]. Thus, identification of cartilaginous nodules and/or osteoid formation in ERMS of the genitourinary tract, including the uterus, may suggest a DICER1-association. Therefore, patients with this diagnosis should be referred to genetic counseling.
DICER1-wt ERMS are known to frequently harbor alterations impacting the RAS–RAF–MAPK (mitogen-activated protein kinase 1) pathway, including mutations in RAS genes (NRAS, KRAS, and HRAS) and TP53 [21, 41]. Furthermore, genetic hallmarks of DICER1-wt ERMS may include aneuploidy with chromosome gains most frequently involving chromosomes 2, 3, and 8, as well as copy number neutral loss of heterozygosity (cnLOH) of chr11p alleles (11p15.5) [21, 42]. Interestingly, we identified similar patterns of aneuploidy as well as mutations affecting TP53 and activating the RAS signaling cascade in both DICER1-mut and DICER1-wt ERMS. Preliminary analysis of single nucleotide polymorphism data from gene panel sequencing suggests that cnLOH of chr11p, typically a hallmark of ERMS, may not be a common feature of DICER1-mut ERMS. The panel data, however, do not cover sufficient loci in this region to be conclusive, and the 11p status in this subgroup will need confirming with more detailed analysis in the future.
A key finding of our study is that DICER1-mut and DICER1-wt ERMS are defined by distinct DNA methylation profiles, irrespective of their primary location. Recent studies have shown whole-genome DNA methylation profiling to be helpful in the identification of novel tumor entities and to be able to reliably assign CNS and mesenchymal tumors to diagnostic groups, including RMS and various uterine neoplasms [22, 29, 43,44,45,46]. The rationale behind this approach is the assumption that cancer cell-specific DNA methylation patterns to some extent recapitulate the DNA methylation patterns of their originating precursor cell, which are retained through cell division and tumorigenesis [47, 48]. Developmentally, RMS is believed to originate from myogenic progenitors, a concept supported by the expression of myogenic genes such as desmin and myogenin in RMS tumor cells [49,50,51]. Yet, studies have suggested that RMS may also develop from transdifferentiating non-myogenic cells, such as endothelial progenitors [52]. While the cell of origin of RMS remains elusive and may vary contextually, our results may suggest a differing cell lineage of DICER1-mut and DICER1-wt ERMS.
In DICER1 syndrome-associated neoplasms, predisposing loss-of-function mutations in DICER1 typically occur together with a characteristic somatic missense hotspot mutation on the second allele [13]. However, also sporadic DICER1-associated neoplasms with biallelic DICER1 alterations have been identified in the absence of germline alterations [53]. Although no germline DNA was available for study in the current paper, four DICER1-mut ERMS showed a combination of a DICER1 frameshift or nonsense, and a DICER1 hotspot mutation, consistent with alterations previously reported in DICER1 syndrome associated ERMS [7, 16]. The remaining DICER1-associated ERMS of our cohort showed multiple missense mutations or homozygous hotspot mutations in DICER1. Interestingly, the DICER1 locus (14q) in the latter neoplasms of our cohort did not show any copy number alterations, particularly no deletion/microdeletion. However, we cannot rule out that a cnLOH may inactivate the second DICER1 wild-type allele in these cases [54].
In the uterus, DICER1 alterations are not limited to ERMS but have also been identified in a significant number of uterine adenosarcomas and few cases of carcinosarcoma [55, 56]. Adenosarcoma is a mesenchymal neoplasm typically exhibiting a phyllodes-like growth which may show significant morphological overlap with ERMS, especially when exhibiting sarcomatous overgrowth and/or rhabdomyoblastic differentiation [57]. A recent paper by de Kock et al. has elegantly shown that although DICER1 alterations may be significantly more frequent in uterine ERMS, a distinction between uterine ERMS and adenosarcoma, based on DICER1 mutations status alone is not always possible [7]. In keeping with this finding, few cases of uterine tumors resembling ERMS were excluded from our study after pathology review, as no consensus diagnosis could be reached due to equivocal histologic features, thus limiting the number of uterine cases in our series. Future studies investigating the potential of DNA methylation-based profiling in clarifying the relationship of the latter neoplasms are warranted.
Apart from uterine sarcomas, DICER1 alterations have also been reported in other rare sarcoma entities, such as anaplastic sarcoma of the kidney and a recently described primary intracranial sarcoma with DICER1 mutation [25, 58, 59]. Furthermore, DICER1-associated sarcomas of various locations show a significant morphological overlap with other DICER1-associated neoplasms such as PPB and Sertoli–Leydig-cell tumors [60, 61]. Shared histological features may include undifferentiated small blue cells, poorly differentiated spindle cells with areas of rhabdomyoblastic differentiation, a subepithelial cambium layer, chondroid differentiation as well as bone/osteoid formation [58]. Consequently, a novel unifying nomenclature for such neoplasms has been proposed [62]. However, more detailed comparative studies investigating the clinical and molecular characteristics of such rare DICER1-associated neoplasms arising in different anatomical locations are needed in order to clarify whether such tumors are in fact part of the same biological tumor spectrum.
Herein we describe the clinicopathologic features of a cohort of genitourinary ERMS in relation to DICER1 mutation status. While all genitourinary ERMS showed overlapping morphological features, diverging clinicopathological characteristics and distinct DNA methylation profiles imply that DICER1-mut ERMS might qualify as a distinct subtype in future classifications of RMS.
Data availability
The data for this study data are available upon reasonable request.
References
Siegel DA, King J, Tai E, Buchanan N, Ajani UA, Li J. Cancer incidence rates and trends among children and adolescents in the United States, 2001–2009. Pediatrics. 2014;134:e945–955.
Board WCoTE. WHO classification of tumours of soft tissue and bone. 5th ed. Lyon: IARC Press; 2020.
Leiner J, Le Loarer F. The current landscape of rhabdomyosarcomas: an update. Virchows Arch. 2020;476:97–108.
Hawkins DS, Chi YY, Anderson JR, Tian J, Arndt CAS, Bomgaars L, et al. Addition of vincristine and irinotecan to vincristine, dactinomycin, and cyclophosphamide does not improve outcome for intermediate-risk rhabdomyosarcoma: a report from the children’s oncology group. J Clin Oncol. 2018;36:2770–7.
Nasioudis D, Alevizakos M, Chapman-Davis E, Witkin SS, Holcomb K. Rhabdomyosarcoma of the lower female genital tract: an analysis of 144 cases. Arch Gynecol Obstet. 2017;296:327–34.
Dehner LP, Jarzembowski JA, Hill DA. Embryonal rhabdomyosarcoma of the uterine cervix: a report of 14 cases and a discussion of its unusual clinicopathological associations. Mod Pathol. 2012;25:602–14.
de Kock L, Yoon JY, Apellaniz-Ruiz M, Pelletier D, McCluggage WG, Stewart CJR, et al. Significantly greater prevalence of DICER1 alterations in uterine embryonal rhabdomyosarcoma compared to adenosarcoma. Mod Pathol. 2020;33:1207–19.
Li FP, Fraumeni JF Jr. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med. 1969;71:747–52.
Lupo PJ, Danysh HE, Plon SE, Curtin K, Malkin D, Hettmer S, et al. Family history of cancer and childhood rhabdomyosarcoma: a report from the children’s oncology group and the Utah population database. Cancer Med. 2015;4:781–90.
Sung L, Anderson JR, Arndt C, Raney RB, Meyer WH, Pappo AS. Neurofibromatosis in children with Rhabdomyosarcoma: a report from the Intergroup Rhabdomyosarcoma study IV. J Pediatr. 2004;144:666–8.
Moschovi M, Touliatou V, Papadopoulou A, Mayakou MA, Nikolaidou-Karpathiou P, Kitsiou-Tzeli S. Rhabdomyosarcoma in a patient with Noonan syndrome phenotype and review of the literature. J Pediatr Hematol Oncol. 2007;29:341–4.
Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, Tanaka Y, et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005;37:1038–40.
Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009;325:965.
Schultz KAP, Williams GM, Kamihara J, Stewart DR, Harris AK, Bauer AJ. et al. DICER1 and associated conditions: identification of at-risk individuals and recommended surveillance strategies. Clin Cancer Res. 2018;24:2251–61.
Foulkes WD, Priest JR, Duchaine TF. DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer. 2014;14:662–72.
Doros L, Yang J, Dehner L, Rossi CT, Skiver K, Jarzembowski JA, et al. DICER1 mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr Blood Cancer. 2012;59:558–60.
Cowan M, Suntum T, Olivas AD, Perpich M, Applebaum MA, Lastra RR, et al. Second primary rhabdomyosarcoma of the uterine cervix presenting with synchronous ovarian Sertoli–Leydig cell tumor: an illustrative case of DICER1 syndrome. Gynecol Oncol Rep. 2018;25:94–97.
B-Teche’b LDK, Druker H, Weber E, Hamel N, Traubici J, Malkin D, et al. Ovarian embryonal rhabdomyosarcoma is a rare manifestation of the DICER1 syndrome. Hum Pathol. 2015;46:917–22.
de Kock L, Rivera B, Revil T, Thorner P, Goudie C, Bouron-Dal Soglio D, et al. Sequencing of DICER1 in sarcomas identifies biallelic somatic DICER1 mutations in an adult-onset embryonal rhabdomyosarcoma. Br J Cancer. 2017;116:1621–6.
McCluggage WG, Apellaniz-Ruiz M, Chong AL, Hanley KZ, Vega JEV, McVeigh TP, et al. Embryonal rhabdomyosarcoma of the ovary and fallopian tube rare neoplasms associated with germline and somatic DICER1 mutations. Am J Surg Pathol. 2020;44:738–47.
Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4:216–31.
Seki M, Nishimura R, Yoshida K, Shimamura T, Shiraishi Y, Sato Y, et al. Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nat Commun. 2015;6:7558.
Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555:321.
Worst BC, van Tilburg CM, Balasubramanian GP, Fiesel P, Witt R, Freitag A, et al. Next-generation personalised medicine for high-risk paediatric cancer patients—the INFORM pilot study. Eur J Cancer. 2016;65:91–101.
Koelsche C, Mynarek M, Schrimpf D, Bertero L, Serrano J, Sahm F et al. Primary intracranial spindle cell sarcoma with rhabdomyosarcoma-like features share a highly distinct methylation profile and DICER1 mutations. 136 (2018).
Sahm F, Schrimpf D, Jones DT, Meyer J, Kratz A, Reuss D. et al. Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol. 2016;131:903–10.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Hovestadt V, Zapatka M. Conumee: enhanced copy-number variation analysis using Illumina DNA methylation arrays. R package version 1.9.0, http://bioconductor.org/packages/conumee/.
Koelsche C, Schrimpf D, Stichel D, Sill M, Sahm F, Reuss DE, et al. Sarcoma classification by DNA methylation profiling. Nat Commun. 2021;12:498.
Caruso S, Calderaro J, Letouze E, Nault JC, Couchy G, Boulai A, et al. Germline and somatic DICER1 mutations in familial and sporadic liver tumors. J Hepatol. 2017;66:734–42.
Fremerey J, Balzer S, Brozou T, Schaper J, Borkhardt A, Kuhlen M. Embryonal rhabdomyosarcoma in a patient with a heterozygous frameshift variant in the DICER1 gene and additional manifestations of the DICER1 syndrome. Fam Cancer. 2017;16:401–5.
Chen KS, Stuart SH, Stroup EK, Shukla AS, Wang J, Rajaram V, et al. Distinct DICER1 hotspot mutations identify bilateral tumors as separate events. JCO Precis Oncol. 2018;2:1–9.
Nakano Y, Hasegawa D, Stewart DR, Schultz KAP, Harris AK, Hirato J, et al. Presacral malignant teratoid neoplasm in association with pathogenic DICER1 variation. Mod Pathol. 2019;32:1744–50.
Apellaniz-Ruiz M, McCluggage WG, Foulkes WD. DICER1-associated embryonal rhabdomyosarcoma and adenosarcoma of the gynecologic tract: pathology, molecular genetics, and indications for molecular testing. Genes Chromosom Cancer. 2020. https://doi.org/10.1002/gcc.22913.
Crist W, Gehan EA, Ragab AH, Dickman PS, Donaldson SS, Fryer C, et al. The third intergroup rhabdomyosarcoma study. J Clin Oncol. 1995;13:610–30.
Koscielniak E, Harms D, Henze G, Jurgens H, Gadner H, Herbst M, et al. Results of treatment for soft tissue sarcoma in childhood and adolescence: a final report of the German cooperative soft tissue sarcoma study CWS-86. J Clin Oncol. 1999;17:3706–19.
Newton WA Jr., Gehan EA, Webber BL, Marsden HB, van Unnik AJ, Hamoudi AB, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification-an intergroup rhabdomyosarcoma study. Cancer. 1995;76:1073–85.
Daya DA, Scully RE. Sarcoma botryoides of the uterine cervix in young women: a clinicopathological study of 13 cases. Gynecol Oncol. 1988;29:290–304.
Leuschner I, Harms D, Mattke A, Koscielniak E, Treuner J. Rhabdomyosarcoma of the urinary bladder and vagina: a clinicopathologic study with emphasis on recurrent disease: a report from the Kiel pediatric tumor registry and the German CWS study. Am J Surg Pathol. 2001;25:856–64.
McCluggage WG, Apellaniz-Ruiz M, Chong AL, Hanley KZ, Velazquez Vega JE, McVeigh TP, et al. Embryonal rhabdomyosarcoma of the ovary and fallopian tube: rare neoplasms associated with Germline and somatic DICER1 mutations. Am J Surg Pathol. 2020;44:738–47.
Taylor AC, Shu L, Danks MK, Poquette CA, Shetty S, Thayer MJ, et al. P53 mutation and MDM2 amplification frequency in pediatric rhabdomyosarcoma tumors and cell lines. Med Pediatr Oncol. 2000;35:96–103.
Scrable H, Cavenee W, Ghavimi F, Lovell M, Morgan K, Sapienza C. A model for embryonal rhabdomyosarcoma tumorigenesis that involves genome imprinting. Proc Natl Acad Sci USA. 1989;86:7480–4.
Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D, et al. New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell. 2016;164:1060–72.
Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555:469–74.
Koelsche C, Hartmann W, Schrimpf D, Stichel D, Jabar S, Ranft A. et al. Array-based DNA-methylation profiling in sarcomas with small blue round cell histology provides valuable diagnostic information. Mod Pathol. 2018;31:1246–56.
Kommoss FKF, Stichel D, Schrimpf D, Kriegsmann M, Tessier-Cloutier B, Talhouk A, et al. DNA methylation-based profiling of uterine neoplasms: a novel tool to improve gynecologic cancer diagnostics. J Cancer Res Clin Oncol. 2020;146:97–104.
Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–98.
Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153:38–55.
Morotti RA, Nicol KK, Parham DM, Teot LA, Moore J, Hayes J, et al. An immunohistochemical algorithm to facilitate diagnosis and subtyping of rhabdomyosarcoma: the children’s oncology group experience. Am J Surg Pathol. 2006;30:962–8.
Blum JM, Ano L, Li Z, Van Mater D, Bennett BD, Sachdeva M, et al. Distinct and overlapping sarcoma subtypes initiated from muscle stem and progenitor cells. Cell Rep. 2013;5:933–40.
Boscolo Sesillo F, Fox D, Sacco A. Muscle stem cells give rise to rhabdomyosarcomas in a severe mouse model of Duchenne muscular dystrophy. Cell Rep. 2019;26:689–701.e686.
Drummond CJ, Hanna JA, Garcia MR, Devine DJ, Heyrana AJ, Finkelstein D, et al. Hedgehog pathway drives fusion-negative rhabdomyosarcoma initiated from non-myogenic endothelial progenitors. Cancer Cell. 2018;33:108–24.e105.
Seki M, Yoshida K, Shiraishi Y, Shimamura T, Sato Y, Nishimura R, et al. Biallelic DICER1 mutations in sporadic pleuropulmonary blastoma. Cancer Res. 2014;74:2742–9.
de Kock L, Sabbaghian N, Plourde F, Srivastava A, Weber E, Bouron-Dal Soglio D, et al. Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol. 2014;128:111–22.
Howitt BE, Sholl LM, Dal Cin P, Jia Y, Yuan L, MacConaill L, et al. Targeted genomic analysis of Mullerian adenosarcoma. J Pathol. 2015;235:37–49.
Chen J, Wang Y, McMonechy MK, Anglesio MS, Yang W, Senz J, et al. Recurrent DICER1 hotspot mutations in endometrial tumours and their impact on microRNA biogenesis. J Pathol. 2015;237:215–25.
McCluggage WG. Mullerian adenosarcoma of the female genital tract. Adv Anat Pathol. 2010;17:122–9.
Warren M, Hiemenz MC, Schmidt R, Shows J, Cotter J, Toll S, et al. Expanding the spectrum of dicer1-associated sarcomas. Mod Pathol. 2020;33:164–74.
Wu MK, Vujanic GM, Fahiminiya S, Watanabe N, Thorner PS, O’Sullivan MJ, et al. Anaplastic sarcomas of the kidney are characterized by DICER1 mutations. Mod Pathol. 2018;31:169–78.
Karnezis AN, Wang Y, Keul J, Tessier-Cloutier B, Magrill J, Kommoss S. et al. DICER1 and FOXL2 mutation status correlates with clinicopathologic features in ovarian Sertoli–Leydig cell tumors. Am J Surg Pathol. 2019;43:628–38.
Hill DA, Jarzembowski JA, Priest JR, Williams G, Schoettler P, Dehner LP. Type I pleuropulmonary blastoma: pathology and biology study of 51 cases from the international pleuropulmonary blastoma registry. Am J Surg Pathol. 2008;32:282–95.
McCluggage WG, Foulkes WD. DICER1-associated sarcomas: towards a unified nomenclature. Mod Pathol. 2020. https://doi.org/10.1038/s41379-020-0602-4.
Funding
We want to thank the Microarray Unit of the Genomics and Proteomics Core Facility, German Cancer Research Center (DKFZ) for providing excellent technical support. This work was supported by the German Cancer Aid (Grant: 70112499), the Fondation ISREC, Lausanne, Switzerland, and Stiftung zur Krebsbekämpfung, Zurich, Switzerland. FKFK is funded by the Physician Scientist-Program of Heidelberg University. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
FKFK and CK conceptualized the project. FKFK, CK, and AvD coordinated the project and ran data generation. Pathology review was performed by FKFK, TM, and FK. FS and AvD supervised array-based analysis. D Stichel and DTWJ analyzed DNA-seq and methylation data. FKFK, JM, ME, DTWJ, SMP, EB, WM, PKB, HPS, D Schmidt, and FK provided tumor samples and corresponding metadata. FKFK wrote the original draft, which was reviewed and approved by all authors.
Corresponding authors
Ethics declarations
Ethics approval/consent to participate
This study was approved by the institutional ethics committee and performed in accordance with the Declaration of Helsinki.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kommoss, F.K.F., Stichel, D., Mora, J. et al. Clinicopathologic and molecular analysis of embryonal rhabdomyosarcoma of the genitourinary tract: evidence for a distinct DICER1-associated subgroup. Mod Pathol 34, 1558–1569 (2021). https://doi.org/10.1038/s41379-021-00804-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00804-y
This article is cited by
-
Early-onset gynecological tumors in DNA repair-deficient xeroderma pigmentosum group C patients: a case series
Communications Medicine (2023)
-
Genomic characterization of DICER1-associated neoplasms uncovers molecular classes
Nature Communications (2023)
-
DICER1-sarcoma: an emerging entity
Modern Pathology (2021)
