
Abstract
Hereditary leiomyomatosis and renal cell carcinoma syndrome is characterized by an increased risk of agressive renal cell carcinoma, often of type 2 papillary histology, and is caused by FH germline mutations. A prominent eosinophilic macronucleolus with a perinucleolar clear halo is distinctive of hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cell carcinoma according to the 2012 ISUP and 2016 WHO kidney tumor classification. From an immunohistochemistry perspective, tumors are often FH-negative and S-(2-succino)-cysteine (2SC) positive. We performed a pathology review of 24 renal tumors in 23 FH mutation carriers, and compared them to 12 type 2 papillary renal cell carcinomas from FH wild-type patients. Prominent eosinophilic nucleoli with perinucleolar halos were present in almost all FH-deficient renal cell carcinomas (23/24). Unexpectedly, they were also present in 58% of type 2 papillary renal cell carcinomas from wild-type patients. Renal cell carcinoma in mutation carriers displayed a complex architecture with multiple patterns, typically papillary, tubulopapillary, and tubulocystic, but also sarcomatoid and rhabdoid. Such pattern diversity was not seen in non-carriers. FH/2SC immunohistochemistry was informative as all hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinomas were either FH− or 2SC+. For FH and 2SC immunohistochemistries taken separately, sensitivity of negative anti-FH immunohistochemistry was 87.5% and specificity was 100%. For positive anti-2SC immunohistochemistry, sensitivity, and specificity were 91.7% and 91.7%, respectively. All FH wild-type renal cell carcinoma were FH-positive, and all but one were 2SC-negative. In conclusion, multiplicity of architectural patterns, rhabdoid/sarcomatoid components and combined FH/2SC staining, but not prominent eosinophilic nucleoli with perinucleolar halos, differentiate hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinoma from type 2 papillary renal cell carcinoma with efficient FH gene. Our findings are crucial in identifying who should be referred to Cancer Genetics clinics for genetic counseling and testing.
Similar content being viewed by others

Introduction
Hereditary leiomyomatosis and renal cell carcinoma syndrome is an autosomal dominant syndrome induced by germline mutations of the fumarate hydratase (FH) gene located on chromosome 1q42.3 [1]. This gene codes for the enzyme fumarate hydratase, or fumarase, which catabolizes fumarate into malate in the tricarboxylic acid cycle. When FH activity is impaired, the consequent accumulation of fumarate acts as an oncometabolite and as an epigenetic modifier [2]. Several mechanisms of carcinogenesis may contribute to hereditary leiomyomatosis and renal cell carcinoma tumor development, such as inhibition of hypoxia-inducible factor prolyl hydroxylase enzymes that results in hypoxia-inducible factor upregulation [3], changes in glycogen and lipid metabolism [4], and aberrant succination [5].
Loss of FH is associated with an epithelial-to-mesenchymal transition, a phenotypic switch associated with cancer initiation which may explain why hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinoma are high-grade tumors, and why they often and rapidly metastasize [2, 6, 7].
Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cell carcinoma are a distinct entity in both 2012 International Society of Urological Pathology (ISUP) and 2016 World Health Organization (WHO) classifications [8, 9]. According to these classifications, the disctinctive feature of these tumors is a prominent eosinophilic macronucleolus, which represents an actively synthesizing cell [10] with a perinucleolar clear halo (i.e., an inclusion-like nucleolus) [6, 11]. From an immunohistochemistry perspective, tumors are often FH-negative and S-(2-succino)-cysteine (2SC) positive. As a consequence of FH gene mutation and loss of function, staining for FH protein expression is often lost. 2SC production is directly linked to FH inactivation and increased levels of intracellular fumarate [7, 12, 13]. An increase in the level of intracellular fumarate results in its spontaneous reaction with the cysteine residues of many proteins, thereby causing succination and 2SC accumulation. FH antibody is widely available, and is commonly used to confirm post hoc that a renal cell carcinoma in a FH mutation carrier is indeed associated with the hereditary leiomyomatosis and renal cell carcinoma predisposition syndrome. As of 2SC, its limited availability means immunohistochemistry is mainly done in a research context. Until recently, type 2 papillary renal cell carcinoma was the only renal cell carcinoma type associated with hereditary leiomyomatosis and renal cell carcinoma [1, 14, 15]. It is now known that papillary renal cell carcinoma of unspecified type, collecting duct carcinoma, tubulocystic carcinoma, and unclassified renal cell carcinoma are also seen [7, 16, 17]. These observations, mainly published in the past 2–3 years, have undeniably added to the complexity of hereditary leiomyomatosis and renal cell carcinoma, whereas knowledge of the syndrome among urologists, oncologists, and pathologists has not increased accordingly. Considering the benefits of identifying a FH mutation in a patient with renal cell carcinoma, i.e., targeted testing in relatives and yearly renal cell carcinoma screening with magnetic resonance imaging in carriers, complementary easy-to-implement tests in addition to the pathologist’s expertise and attention to characteristic hereditary leiomyomatosis and renal cell carcinoma features, would ensure that all patients with suspected hereditary leiomyomatosis and renal cell carcinoma are appropriately referred to cancer genetics clinics and tested for FH germline mutations.
We studied two groups of renal cell carcinomas: 24 renal cell carcinomas from proven mutations carriers and 12 type 2 papillary renal cell carcinomas from patients without FH mutations. We compared their pathological features and assessed more specifically whether characteristics deemed distinctive of hereditary leiomyomatosis and renal cell carcinoma are indeed exclusively seen in tumors from mutation carriers. We also calculated the sensitivity and specificity of FH/2SC immunohistochemistries and association of both in predicting a patient’s genetic status. This study complements a previous publication reassessing the clinical spectrum associated with hereditary leiomyomatosis and renal cell carcinoma [16].
Materials and methods
Patients
Renal cell carcinoma from FH germline mutations carriers, and papillary renal cell carcinoma type 2 from patients who had been tested negatively for mutation of FH were identified through the National Cancer Institute (INCa) “Inherited Predisposition to Kidney Cancer” database located at the Kremlin-Bicêtre PREDIR Centre. In most cases, detailed clinical and pathological reports were obtained from the PREDIR database. For the remaining cases, data were collected either by the physicians who prescribed the FH genetic testing or by review of the institutional records.
We contacted each pathologist involved in order to get a representative tissue block of the renal neoplasm. In keeping with legal requirements, written consent for genetic testing had been obtained from all of the patients.
Genetic testing and evaluation of FH mutations
FH germline genetic testing was performed in the Gustave Roussy Cancer Genetics Laboratory (Villejuif). Mutations and large rearrangements were identified using Sanger sequencing complemented by multiplex ligation probe-dependent amplification for the most recent cases. With the exception of three first degree relatives from proven hereditary leiomyomatosis and renal cell carcinoma families who died from metastatic renal cell carcinoma before genetic testing could take place, all of our patients underwent FH mutation analysis. These three cases were considered obligatory carriers.
All frameshift mutations, nonsense mutations, and large deletions were considered deleterious. For other types of genetic alterations (e.g., missense and splice-site mutations), assessment of FH enzyme activity was performed using spectrophotometry [18]. An enzymatic activity of ≤78% reflects loss of FH function [19]. If FH activity could not be measured, information taken from the LOVD database was used as a reference (http://databases.lovd.nl/shared/variants/FH/unique).
Tumor analysis
An uropathologist (SF) reviewed all tumors, and compiled the following pathological features: architectural pattern, Internal Society of Urological Pathology (ISUP) grade, pTN stage, and the presence or not of prominent macronucleoli and a perinucleolar clear halo. A perinucleolar halo was defined as an entirely clear area (i.e., devoid of any chromatin) between the nucleus and the cytoplasm as seen by microscopy at high magnification. The ISUP grade and the TNM classification were evaluated according to the most recent ISUP/World Health Organization (WHO)/American Joint Committee on Cancer (AJCC) classifications [8].
Immunohistochemistry
Immunostaining for FH and 2SC was performed on formalin-fixed paraffin embedded sections in two different French laboratories (i.e., the Department of Pathology of Bicêtre Hospital and the Anatomical Pathology Laboratory of the Gustave Roussy Institute, Campus Cancer Grand Paris, Villejuif), and they were scored independently by two pathologists in a single-blind manner (SF and CG). We used a commercially available primary anti-FH mouse monoclonal antibody (1:1000 dilution, clone J-13, reference sc-100743, Santa Cruz Biotechnology) with an automated staining platform (Leica Bond III Autostainer, Leica Biosystems) at the Bicêtre Hospital. Anti-2SC rabbit polyclonal antibody was kindly provided by Dr Norma Frizzell (University of South Carolina, USA), and it was used at a 1:5000 dilution. Automated staining with the 2SC antibody was performed with a BenchMark ULTRA system (Roche/Ventana Biosystems). FH-negative staining in tumor cells was considered negative in the presence of an internal positive control in inflammatory or stromal cells, or when FH-positive staining in the non-tumoral kidney parenchyma was obvious. 2SC-positive staining was considered positive when there was 2SC-negative staining in the adjacent non-neoplastic cells (i.e., an internal negative control). For each case, cytoplasmic staining was assessed as being either absent (negative) or present (positive).
For tumors with a peculiar architectural pattern, staining with additional antibodies was performed in order to establish the histological type according to the 2016 WHO classification. The following antibodies were used: anti-vimentin (Clone V9, Dako, at 1:100 dilution), anti-CD10 (Clone 56C6, Leica, at 1:200 dilution), anti-P504S (Polyclonal, Biocare, ready to use), anti-cytokeratin 7 (CK7) (Clone OV-TL 12/30, Dako, at 1:100 dilution), anti-CA9 (Polyclonal, Novusbio, 1:800), and anti-TFE3 (Polyclonal, tebu-bio, 1:800).
Statistical analysis
Statistical analyses were performed in regard to the sensitivity and specificity of the following strategies: presence of a clear perinucleolar halo, FH− or 2SC+ immunostaining, and combined FH−/2SC+ immunohistochemistries. The sensitivity of each strategy corresponds to the number of positive cases according to this strategy out of the 24 renal cell carcinomas from proven mutations carriers. The specificity of each strategy corresponds to the number of negative cases according to this strategy out of the 12 papillary renal cell carcinomas from patients without FH mutations. Comparison of sensitivities between combined FH−/2SC+ immunohistochemistries and anti-FH immunostaining was performed with a Mac Nemar test by comparing the proportion of positive cases for each strategy among the 24 renal tumors from proven mutations carriers. The threshold for statistical significance was set to p < 0.05. Statistical analysis was performed using SAS software (SAS Institute Inc., Cary, NC).
Results
Renal cell carcinoma in FH mutation carriers
We identified 42 hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinomas in 39 FH mutation carriers, and obtained tissue blocks for 24 renal cell carcinomas from 23 patients. Out of these 24 tumors, 23 were included in our latest publication, which had a mainly clinical focus, did not include an in-depth pathology exploration of tumors, and above all did not compare renal cell carcinoma in carriers with type 2 papillary renal cell carcinoma in proven non-carriers [16]. The 23 renal cell carcinomas had been classified according to their primary pathological feature, but secondary components were not detailed. A majority of renal cell carcinomas were papillary type 2 (n = 13). There were also papillary renal cell carcinomas of unspecified type (non-type 1, non-type 2, n = 4), unclassified carcinoma (n = 3), tubulocystic carcinoma (n = 2), and one collecting carcinoma. A 29-year-old woman with hereditary leiomyomatosis and renal cell carcinoma has since been diagnosed with type 2 papillary renal cell carcinoma, and was therefore included in this study.
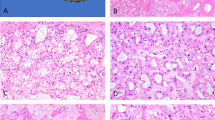
When reviewing tissue blocks, we observed prominent eosinophilic nucleoli with a perinucleolar halo in 23/24 renal cell carcinomas (96%, Table 1). These nuclear characteristics were observed either in many of the tumor cells or sometimes only in a small fraction of the cells, even at small magnification (Fig. 1). Most renal cell carcinomas displayed a complex architecture with multiple pathological patterns. Eighteen renal cell carcinomas had at least two components, and ten had three components or more. In addition to the patterns reported previously by our group (e.g., papillary, tubulopapillary, tubulocystic), cribriform, sarcomatoid, and rhabdoid components were also observed in 13 cases (Table 2; Fig. 2a–d). Multiplicity of components and sarcomatoid/rhabdoid features were seen across the spectrum of hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinoma, regardless of the primary type (type 2 papillary renal cell carcinoma vs. other).

Hereditary leiomyomatosis and renal cell carcinoma tumor cells had a very prominent eosinophilic nucleus with a large nucleolus and perinucleolar halo at high magnification (hematoxylin and eosin)

Various architectural patterns of hereditary leiomyomatosis and renal cell carcinoma kidney tumors: papillary (a), tubulopapillary (b), sarcomatoid (c), and tubulocystic (d) patterns
FH immunohistochemistry was negative in 21 renal cell carcinoma, and positive in three (Table 1). 2SC immunohistochemistry was positive in 22 renal cell carcinomas and negative in two (Fig. 3a, b). Overall, 19 renal cell carcinomas were FH−/2SC+, three were 2SC+/FH+ and two were 2SC−/FH− (Tables 3 and 4).

On immunohistochemistry, hereditary leiomyomatosis and renal cell carcinoma tumor cells demonstrated FH-negative staining, whereas non-neoplastic cells showed granular cytoplasmic staining (a). 2SC showed diffuse and strong staining in the neoplastic cells (b)
Interestingly, three benign unilocular cysts were observed in one patient. Lining cells exhibited strong 2SC staining and FH-expression loss (Fig. 4a–c).

Unilocular cyst lined by single cells in the non-tumoral kidney (a). 2SC immunohistochemistry showed a cytoplasmic staining pattern (b), whereas FH staining was negative (c)
Renal cell carcinoma in FH wild-type patients
Twelve type 2 papillary renal cell carcinomas from individuals with no germline FH mutation were reviewed. Prominent clear perinucleolar halos were observed in 7/12 cases (58%) (Fig. 5a, b). Pathological patterns were few, and consisted only of papillary, tubular, and solid components (Table 5). Only one renal cell carcinoma displayed all three patterns. Aggressive sarcomatoid or rhabdoid features were absent. All renal cell carcinomas were FH-positive on immunohistochemistry, and all but one were 2SC-negative (Tables 1 and 4).

Papillary renal cell carcinoma type 2 with efficient FH gene. Papillary pattern (a) with tumor cells showing nuclear features (b) similar to hereditary leiomyomatosis and renal cell carcinoma tumors
Sensitivity and specificity of FH/2SC immunohistochemistries
Table 1 Sensitivity of negative anti-FH immunohistochemistry was 87.5% (21/24) and specificity was 100% (12/12). For positive anti-2SC immunohistochemistry, sensitivity and specificity were 91.7% (22/24) and 91.7% (11/12), respectively. Combined FH−/2SC+ immunohistochemistry had 100% sensitivity (24/24), i.e., all tumors were FH−, 2SC+ or both, and 91.7% specificity (11/12). This combination seems to improve the sensitivity of the test compared to anti-FH only immunostaining (without reaching the statistical significance, p = 0.083).
Discussion
On the basis of our comparison of renal cell carcinoma in FH mutation carriers and in wild type patients, we conclude that multiplicity of architectural patterns, rhabdoid/sarcomatoid components and FH/2SC staining, but not prominent eosinophilic nucleoli with perinucleolar halos, differentiate hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinoma from type 2 papillary renal cell carcinoma without FH alteration. To our knowledge, this is the first time that a formal comparison between proven hereditary leiomyomatosis and renal cell carcinoma patients and individuals tested negative for FH germline mutations is done.
Our most unexpected finding was the presence of prominent eosinophilic nucleoli with perinucleolar halos in 58% of type 2 papillary renal cell carcinomas from patients not carrying FH germline mutations. Indeed, this feature is distinctive of hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinoma in both ISUP and WHO kidney tumors classifications [8]. More precisely, pathologists who observe it are expected to conclude in their report that the patient is likely to have hereditary leiomyomatosis and renal cell carcinoma. We would therefore urge caution against premature conclusions, given the anxiety such a genetic diagnosis could trigger in patients and their relatives. We would also suggest future versions of kidney tumor classifications put less emphasis on these prominent nucleoli and halos in diagnosing hereditary leiomyomatosis and renal cell carcinoma, as this is apparently a sensitive, but not very specific feature of the syndrome. We did observe it in most (23/24) hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinomas. Admittedly, the twelve wild type cases had been referred for germline testing because clinical and pathological features, prominent nucleoli with halos included, suggested hereditary leiomyomatosis and renal cell carcinoma, and this might have led to selection bias. The ideal control group would have been large consecutive institutional, non-referral case series of type 2 papillary renal cell carcinoma and renal cell carcinoma with rare or complex histologies, with subsequent negative FH mutational germline testing.
Considering the benefits of identifying a FH mutation in a patient with renal cell carcinoma, complementary easy-to-implement tests in addition to the pathologist’s expertise, would ensure that all patients with suspected hereditary leiomyomatosis and renal cell carcinoma are appropriately referred to cancer genetics clinics and tested for FH germline mutations. We have demonstrated the limits of prominent nucleoli and adjacent halos in predicting a patient’s genetic status. According to our results, the multiplicity and variety of patterns, and FH/2SC immunohistochemistry are much more informative in identifying hereditary leiomyomatosis and renal cell carcinoma patients. Not only did we see that the vast majority of renal cell carcinoma in carriers contained multiple components, we also observed sarcomatoid or rhabdoid patterns in eight tumors, reflecting the aggressive behavior of hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinoma [20]. These features were absent in non-carriers, although cannot exclude the possibility that a review of a larger number of tissue blocks would have shown more pattern diversity. All hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinomas were either FH− or 2SC+, and the sensitivity of combined immunohistochemistry was therefore 100%, higher than FH immunohistochemistry only. The use of both antibodies is thus necessary to identify all mutation carriers. We acknowledge the limited availability of anti-2SC antibody, and the fact that it has mainly been used in in a research context so far. There is, however, obvious commercial interest in marketing such an antibody considering its use not only in renal cell carcinoma but also in both uterine and cutaneous leiomyomas [13, 21]
Trpkov et al. [7] performed FH/2SC immunohistochemistry on a large series of renal cell carcinomas. They did show optimal sensitivity and specificity for combined immunohistochemistry, but their conclusions were limited by the low number of proven FH germline mutations carriers (n = 9, vs. 24 in this study). Like us, they observed that FH-deficient renal cell carcinomas were complex and made of several distinct patterns. But again, the number of proven mutation carriers was low. Furthermore, they did not report renal cell carcinoma detailed morphological patterns in proven non-carriers, while we reviewed 12 type 2 papillary renal cell carcinomas from wild-type patients.
Finally, one of our cases presented three FH−/2SC+ unilocular cysts in the renal parenchyma adjacent to a renal cell carcinoma (Fig. 4a–c), confirming observations already made in a mouse model and in two hereditary leiomyomatosis and renal cell carcinoma cases [5, 6, 22]. The hypothesis is that cysts act as precursor lesions, although the pathogenesis of the malignant transformation remains unclear.
In conclusion, our study shows that prominent eosinophilic nucleoli with perinucleolar halos are not a distinctive feature of hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinoma, as they are often seen in type 2 papillary renal cell carcinoma from patients with no FH germline mutation. Both ISUP and WHO renal cell carcinoma classification should be revised accordingly. Multiple growth patterns in renal cell carcinoma, in particular rhabdoid and sarcomatoid, and combined FH−/2SC+ immunohistochemistries are crucial in identifying which patients should be referred to Cancer Genetics clinics for genetic counseling and germline analysis. One must always keep in mind the benefits of identifying a FH mutation, i.e., targeted testing in relatives and subsequent yearly renal cell carcinoma screening in carriers, with the aim of diagnosing potentially life-threatening tumors before it is too late.
References
Tomlinson IPM, Alam NA, Rowan AJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406–10.
Sciacovelli M, Gonçalves E, Johnson TI, et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature. 2016;537:544–7.
Isaacs JS, Jung YJ, Mole DR, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–53.
Frizzell N, Lima M, Baynes JW. Succination of proteins in diabetes. Free Radic Res. 2011;45:101–9.
Bardella C, El-Bahrawy M, Frizzell N, et al. Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J Pathol. 2011;225:4–11.
Merino MJ, Torres-Cabala C, Pinto P, et al. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol. 2007;31:1578–85.
Trpkov K, Hes O, Agaimy A, et al. Fumarate hydratase-deficient renal cell carcinoma is strongly correlated with fumarate hydratase mutation and hereditary leiomyomatosis and renal cell carcinoma syndrome. Am J Surg Pathol. 2016;40:865–75.
Moch H, Cubilla AL, Humphrey PA, et al. The 2016 WHO classification of tumours of the urinary system and male genital organs—part a: renal, penile, and testicular tumours. Eur Urol. 2016;70:93–105.
Srigley JR, Delahunt B, Eble JN, et al. The International Society of Urological Pathology (ISUP) Vancouver Classification of renal neoplasia. Am J Surg Pathol. 2013;37:1469–89.
Bavle R. Eosinophilic nucleoli. J Oral Maxillofac Pathol. 2014;18:152.
Chen Y-B, Brannon AR, Toubaji A, et al. Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cancer: recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am J Surg Pathol. 2014;38:627–37.
Llamas-Velasco M, Requena L, Kutzner H, et al. Fumarate hydratase immunohistochemical staining may help to identify patients with multiple cutaneous and uterine leiomyomatosis (MCUL) and hereditary leiomyomatosis and renal cell cancer (HLRCC) syndrome. J Cutan Pathol. 2014;41:859–65.
Joseph NM, Solomon DA, Frizzell N, et al. Morphology and immunohistochemistry for 2SC and FH aid in detection of fumarate hydratase gene aberrations in uterine leiomyomas from young patients. Am J Surg Pathol. 2015;39:1529–39.
Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637–44.
Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci. 2001;98:3387–92.
Muller M, Ferlicot S, Guillaud-Bataille M, et al. Reassessing the clinical spectrum associated with hereditary leiomyomatosis and renal cell carcinoma syndrome in French FH mutation carriers. Clin Genet. 2017;92:606–15.
Smith SC, Trpkov K, Chen Y-B, et al. Tubulocystic carcinoma of the kidney with poorly differentiated foci: a frequent morphologic pattern of fumarate hydratase-deficient renal cell carcinoma. Am J Surg Pathol. 2016;40:1457–72.
Gardie B, Remenieras A, Kattygnarath D, et al. Novel FH mutations in families with hereditary leiomyomatosis and renal cell cancer (HLRCC) and patients with isolated type 2 papillary renal cell carcinoma. J Med Genet. 2011;48:226–34.
Alam NA, Rowan AJ, Wortham NC, et al. Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum Mol Genet. 2003;12:1241–52.
Grubb RL, Franks ME, Toro J, et al. Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol. 2007;177:2074–80.
Buelow B, Cohen J, Nagymanyoki Z, et al. Immunohistochemistry for 2-succinocysteine (2SC) and fumarate hydratase (FH) in cutaneous leiomyomas may aid in identification of patients with HLRCC (hereditary leiomyomatosis and renal cell carcinoma syndrome). Am J Surg Pathol. 2016;40:982–8.
Ristau BT, Kamat SN, Tarin TV. Abnormal cystic tumor in a patient with hereditary leiomyomatosis and renal cell cancer syndrome: evidence of a precursor lesion? Case Rep Urol. 2015;2015:1–5.
Acknowledgements
We thank the following colleagues and collaborators for their support or contributions: Sophie Couvé and Sophie Gad (Laboratoire de Génétique Oncologique EPHE, INSERM U1186, Gustave Roussy Cancer Campus, Villejuif), Olivier Morassi (CHU Bicêtre), Norma Frizzell (University of South Carolina), Joël Cucherousset (CH Montfermeil), Viorel Vasiliu (Hôpital Necker, Paris), Yves Allory (Hôpital Henri Mondor, Créteil), Karine Renaudin-Autin (CHU Nantes), Camelia Radulescu (Hôpital Foch, Suresnes), Jean-Michel Goujon (CHU Poitiers), Isabelle Serre (CHU Montpellier), Marilyn Augros (CH Valence), Eric Bonte (Compiègne), Gilles Pugens (CYPATH Villeurbanne), and Sophie Domingues. We also thank the Plateforme de Ressources Biologiques at the Hôpital Henri Mondor for providing us pathology samples for one of the cases (BB-0033-00021).
Funding
The PREDIR Centre is supported by the French National Cancer Institute (INCa) and the French Department of Health. The Laboratoire de Génétique Oncologique EPHE is supported by grants from the French league against Cancer (Comités du Cher et de l’Indre).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Muller, M., Guillaud-Bataille, M., Salleron, J. et al. Pattern multiplicity and fumarate hydratase (FH)/S-(2-succino)-cysteine (2SC) staining but not eosinophilic nucleoli with perinucleolar halos differentiate hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinomas from kidney tumors without FH gene alteration. Mod Pathol 31, 974–983 (2018). https://doi.org/10.1038/s41379-018-0017-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0017-7
This article is cited by
-
Onkozytäre Tumoren der Niere – neue Differenzialdiagnosen
Der Pathologe (2021)
-
Histologische Subtypen des Nierenzellkarzinoms
Der Pathologe (2021)
-
Das FH-defiziente Nierenzellkarzinom erweitert das Spektrum der papillären Tumoren in der Niere
Der Pathologe (2021)
-
New developments in existing WHO entities and evolving molecular concepts: The Genitourinary Pathology Society (GUPS) update on renal neoplasia
Modern Pathology (2021)
-
Renal cell carcinoma in young FH mutation carriers: case series and review of the literature
Familial Cancer (2020)
