
Abstract
Most succinate dehydrogenase (SDH)-deficient renal cell carcinomas (RCCs) demonstrate stereotypical morphology characterized by bland eosinophilic cells with frequent intracytoplasmic inclusions. However, variant morphologic features have been increasingly recognized. We therefore sought to investigate the incidence and characteristics of SDH-deficient RCC with variant morphologies. We studied a multi-institutional cohort of 62 new SDH-deficient RCCs from 59 patients. The median age at presentation was 39 years (range 19–80), with a slight male predominance (M:F = 1.6:1). A relevant family history was reported in 9 patients (15%). Multifocal or bilateral tumors were identified radiologically in 5 patients (8%). Typical morphology was present at least focally in 59 tumors (95%). Variant morphologies were seen in 13 (21%) and included high-grade nuclear features and various combinations of papillary, solid, and tubular architecture. Necrosis was present in 13 tumors, 7 of which showed variant morphology. All 62 tumors demonstrated loss of SDHB expression by immunohistochemistry. None showed loss of SDHA expression. Germline SDH mutations were reported in all 18 patients for whom the results of testing were known. Among patients for whom follow-up data was available, metastatic disease was reported in 9 cases, 8 of whom had necrosis and/or variant morphology in their primary tumor. Three patients died of disease. In conclusion, variant morphologies and high-grade nuclear features occur in a subset of SDH-deficient RCCs and are associated with more aggressive behavior. We therefore recommend grading all SDH-deficient RCCs and emphasize the need for a low threshold for performing SDHB immunohistochemistry in any difficult to classify renal tumor, particularly if occurring at a younger age.
Similar content being viewed by others
Introduction
Autosomal dominant germline mutations of the succinate dehydrogenase (SDH) genes (SDHA, SDHB, SDHC, SDHD, or SDHAF2) cause a hereditary tumor syndrome characterized by multiple different neoplasms, all of which are driven by dysfunction of the mitochondrial complex 2 (also known as the SDH complex)1,2. Immunohistochemistry (IHC) for SDHB is negative (i.e., lost) whenever there is biallelic inactivation of any component of the mitochondrial complex 2 (SDHA, SDHB, SDHC, SDHD, or SDHAF2)1,2. In contrast to most tumor-suppressor genes, biallelic inactivation of the SDH genes is rarely a somatic-only event, and almost always occurs in the setting of germline mutation with the subsequent acquisition of a somatic second hit in the tumor1,2,3,4,5,6. Tumors that show loss of SDHB expression by IHC are termed SDH-deficient1,2, and include SDH-deficient pheochromocytoma/paraganglioma, SDH-deficient gastrointestinal stromal tumor (GIST), SDH-deficient pituitary adenoma, and SDH-deficient renal cell carcinoma (RCC)1,2,3,4,5,6,7,8,9,10,11.
Most RCCs arising in this syndrome demonstrate distinctive morphology characterized by sheet-like or compact nested growth of bland cuboidal cells with eosinophilic (but not oncocytic) cytoplasm, with frequent microcysts, and entrapment of non-neoplastic tubules4,8,12,13,14,15,16. Cytoplasmic inclusions containing eosinophilic or pale flocculent material are a clue to the diagnosis but may be inconspicuous or absent. Most cases occur in the setting of an SDHB mutation, while SDHC and SDHA mutations are uncommon but well-reported, and SDHD mutations are very rare6,7,13,14,15. Tumors with biallelic inactivation of SDHA also show loss of SDHA expression by IHC in addition to loss of SDHB17.
Most cases are indolent, but high-grade transformation (found in up to one third of cases), tumor necrosis, and sarcomatoid change are associated with a high risk of metastasis—up to 70%7. High-grade cases may be unrecognizable by morphology, justifying a low threshold for screening IHC in any unusual or difficult to classify renal tumor, particularly those with eosinophilic cytoplasm6,13,16. It has been reported that SDHA-deficient RCCs more frequently demonstrate variant morphologies, including higher nuclear grade and a combination of variable growth patterns, including papillary, solid, cribriform, and desmoplastic18,19,20,21. Other important immunophenotypic features include lack of reactivity for KIT, and frequent absence of (or weak and focal) cytokeratin expression6,7,13,14.
After our initial descriptions of SDH-deficient RCC and the utility of SDHB IHC for the detection of SDH mutations7,8,9, the entity has now been formally recognized in the major classification systems for renal carcinoma including the World Health Organization (WHO) Classification of Tumours of the Urinary System and Male Genital Organs, as well as the International Society of Urological Pathology (ISUP) and the Genitourinary Pathology Society (GUPS) classification of renal tumors6,13,14,22. Since these publications, additional cases of SDH-deficient RCC have been reported in patients of all ages but more commonly in the young, and include rare tumors demonstrating variant histological features and more aggressive biological behavior12,23,24,25,26. Explicit recommendations, which we endorse, have therefore been made to have a very low threshold for performing SDHB IHC in any unclassified eosinophilic RCC or renal oncocytic tumor, particularly those arising in young patients23.
The key features of all previously reported SDH-deficient RCCs with variant morphology and/or aggressive biological behavior are summarized in Table 1. However, given the very small number of cases and the limited data in most instances, our understanding of their clinical features, including long-term outcomes, remains limited. We therefore sought to further study this rare entity by initiating a multi-institutional international collaboration with the following aims:
-
(1)
To identify previously unreported cases of SDH-deficient RCC to further expand knowledge and experience with these tumors.
-
(2)
To review the morphologic features of SDH-deficient RCC, with a particular focus on cases showing variant morphologies.
-
(3)
To define the unique natural history and clinical features of SDH-deficient RCC with variant morphologies in comparison with those showing the typical histologic features.
Methods
Case retrieval and review
Surgical pathologists from institutions across the Americas, Europe, Asia, Africa, and Australia with subspecialty interest in urologic pathology, or in the pathology of SDH-deficient tumors, were contacted to submit cases of previously unreported renal carcinomas occurring in the setting of a proven SDH mutation, or cases suspected to be associated with SDH deficiency based on morphology, IHC, or a personal or family history of paragangliomas or SDH-deficient GIST. Contributors were asked to provide any available clinical information including follow-up data for each case, in addition to either a representative formalin-fixed paraffin-embedded block, or 10 to 15 unstained slides for centralized pathology review and IHC. Cases from patients previously reported in any form were excluded. All submitted cases underwent centralized pathologic review by two pathologists (A.J.G. and T.L.F.). Cases without material available for centralized pathology review and repeat IHC were excluded.
Immunohistochemistry
Cases with proven SDH mutation or with compatible morphology underwent IHC analysis for SDHB and SDHA, which was performed on whole sections using mouse monoclonal antibodies against SDHB (ABCAM, Cambridge, UK: ab14714, clone 21A11, dilution 1 in 100) and SDHA (Mitosciences, Abcam, Cambridge, UK: MS204, clone 2E, dilution of 1 in 1000). Detailed IHC methods have been previously described3,7,8,9,10,11,27. Cases with definite granular cytoplasmic staining were classified as SDHB/SDHA-positive (retained). Cases with absent cytoplasmic staining in the presence of an internal positive control in the non-neoplastic cells were classified as negative (SDH deficient). If there was negative staining in the neoplastic cells but no internal positive control in the non-neoplastic cells, staining was considered indeterminate and was repeated. SDH IHC for all cases was performed and reported by one pathologist (A.J.G.) with extensive experience with interpreting these stains. Only tumors with confirmed loss of expression of SDH by IHC were included in this study. The results of IHC markers commonly used in urologic pathology (PAX8, AMACR, CD10, KIT, AE1/AE3, CK8/18, cytokeratin 7 [CK7; HUGO gene nomenclature KRT7], cytokeratin 20 [CK20], EMA [HUGO gene nomenclature MUC1], fumarate hydratase [FH], and TFE3) were also reported if available.
Statistical analysis
Statistical analyses were performed using SPSS, version 25.0 (IBM Inc., Armonk, NY). Median overall survival following diagnosis was estimated using the Kaplan–Meier method. Survival was compared across different subgroups using the log-rank test. All statistical tests were 2-sided and P-values < 0.05 were considered significant.
Results
Clinical features
Following centralized pathologic review and confirmation of absent SDHB expression in tumor cells by IHC, we identified a total of 62 previously unreported SDH-deficient RCCs from 59 patients. The clinical and IHC features are summarized in Table 2. Briefly, the mean age at presentation with a renal tumor was 42.1 years (range 19 to 80 years; median 39.0 years), with a slight male predominance (M:F = 1.6:1). Eighteen patients had documented germline pathogenic variants in the SDHB gene. None of the patients had mutations in SDHC, SDHD, or SDHA genes, and all cases were positive for SDHA IHC.
The mean duration of follow-up from initial presentation was 23.5 months (median 10 months, range 0.5 to 120 months). Of the 59 patients, 9 (15%) were known to have developed metastatic disease. Sites of metastatic spread included: lymph nodes (n = 6), bone (n = 5), liver (n = 4), lung (n = 3), and brain (n = 1). Three patients died of disease at 24, 26, and 27 months following diagnosis. One of these patients had documented hepatic and bony metastases and a second had retroperitoneal lymph node and hepatic metastases. Two deceased patients had known germline SDHB mutations. A fourth patient died of unknown cause at 3.5 months following diagnosis.
Seven patients (12%) had a relevant personal or family history of other SDH-deficient tumors. One patient had a personal history of multiple GISTs, a retroperitoneal paraganglioma, and a pulmonary chondroma; 3 had a family history of paraganglioma (2 in first-degree relatives and 1 in a second-degree relative); and 3 patients had a family history of SDH-deficient RCC in a first-degree relative. Three patients had known germline SDH mutations prior to their diagnosis of RCC, 2 of whom also had relatives with known SDH mutations. In the remaining 15 patients with subsequently proven germline SDHB mutations, the diagnosis of SDH-deficient RCC was the sentinel event for their diagnosis with a hereditary tumor syndrome.
Pathologic features
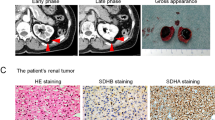
Centralized pathologic review was undertaken on all 62 SDH-deficient RCCs from 59 patients. Macroscopic descriptions were limited, but when available, tumors were described as being circumscribed with a tan or light-brown cut surface (Fig. 1). Four patients had multifocal tumors in the same kidney and 2 had bilateral tumors. Tumors ranged in size from 1 to 22 cm (mean 7.1 cm, median 6.5 cm). Tumor stage was recorded for 54 cases, of which 27 were pT1 (50%), 16 were pT2 (30%), 9 were pT3 (17%) and 2 were pT4 (4%).

The tumors were often solid, with a tan or light-brown cut surface, and frequently showed central hemorrhage and cystic degeneration.
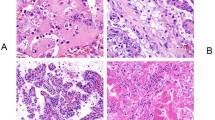
Histologically, the typical morphology was consistent with previous descriptions7,8 and was present, at least focally, in 59 tumors (95%). Briefly, the tumors were well-demarcated or coarsely lobulated, with a pushing border, sometimes associated with a pseudocapsule (Fig. 2). Cystic spaces containing pale eosinophilic material and/or blood were frequently seen. In a few tumors, the stroma showed areas of prominent myxoid change, containing scattered individual tumor cells. The tumor cells were arranged in solid sheets or variably sized nests. In some cases, nests of tumor cells surrounded cystic spaces, imparting a pseudoglandular/tubular appearance. Entrapped non-neoplastic tubules or glomeruli were a frequent finding.

They were well circumscribed, sometimes with a pseudocapsule separating them from the adjacent non-neoplastic kidney (A). Entrapped non-neoplastic tubules or glomeruli were a frequent finding (B). In some cases, the cells had dense eosinophilic cytoplasm (C), but in most instances (D, E) the cytoplasm was pale and flocculent, often with intracytoplasmic inclusions.
The neoplastic cells were cuboidal, but with indistinct cell borders, and had round to oval nuclei containing dispersed chromatin and generally inconspicuous nucleoli, equivalent to a WHO/ISUP nucleolar grade 1 to 2 in 38 (61%) cases. Grade 3 nuclei with prominent nucleoli were identified in 18 (29%) cases, and grade 4 nuclei were identified in 6 (10%) cases (all of which demonstrated at least focal sarcomatoid change). The cytoplasm was eosinophilic and flocculent, but not oncocytic. As we have previously described7, the most consistent and distinctive histologic feature was the presence of cytoplasmic vacuoles and inclusions containing pale eosinophilic or flocculent material (Fig. 2). True tumor necrosis (also termed ‘granular necrosis’ and defined as well-defined necrotic foci being sharply demarcated from adjacent viable tumor28), was present in 13 tumors (21%), 11 of which were WHO/ISUP nuclear grade 3 or 4. Psammomatous calcifications were seen in 3 cases, 2 of which showed otherwise typical morphology, whilst the third showed variant morphology, as described below. Allowing for secondary effects of the tumor, the adjacent non-neoplastic kidney was normal, and no dysplastic or precursor lesions were identified.
Variant morphologic features were present in 13 tumors (21%)—illustrated in Figs. 3, 4. These tumors showed high-grade features, including 7 cases with WHO/ISUP grade 3 nuclei and 6 cases with WHO/ISUP grade 4 nuclei. In addition to prominent nucleoli, the neoplastic cells in the higher-grade areas acquired darker and coarser chromatin and denser eosinophilic (rather than flocculent) cytoplasm. The nuclei in these areas were about 2 times larger than those in the low-grade areas, with oval to slightly elongated shape and irregular nuclear outlines. Various architectural patterns were observed in the high-grade tumors, including solid sheet-like growth, irregular anastomosing tubular structures, and papillary architecture. Stromal changes included prominent desmoplasia, hyalinization, deposition of basophilic myxoid material, and prominent peritumoral lymphocytes. In some cases, areas showing the typical low-grade features of SDH-deficient RCC were present, while in others, the tumors showed diffuse high-grade variant morphology throughout. Interestingly, 3 of the tumors with variant morphologic features arose in patients who also had separate renal tumors showing entirely low-grade morphology more typical of SDH-deficient RCC. Follow-up data was available for 2 of these patients, 1 of whom later developed metastatic disease, and the other who died of disease 27 months after diagnosis (Fig. 5).

These ncluded: (A, B) sheets of undifferentiated epithelioid or sarcomatoid cells; (C, D) tubular structures merging with areas of more conventional morphology; (E, F) papillary growth, often with necrosis (F); and (G, H) irregular tubular and microcystic structures with extracellular myxoid material.

Occasional cases with otherwise typical low-grade morphology showed areas of higher-grade nuclear features (A, B) and/or tumor necrosis (C).

The largest tumor was 10 cm and demonstrated the stereotypical low-grade features of SDH-deficient renal carcinoma (A, B). In contrast, the second tumor measured 4.5 cm and exhibited widespread high-grade morphology including occasional rhabdoid cells (C). Focal intracytoplasmic inclusions were identified in the high-grade tumor after careful inspection (D). The patient died of disease 27 months after diagnosis.
Tumors with variant morphology were more likely to present at higher tumor stage compared to those with conventional morphology. Of the 13 cases with variant morphology in this cohort, 4 (31%) were pT3 or pT4, 5 (38%) were pT1 or pT2, and 4 did not have tumor stage recorded. In contrast, of the 49 cases with conventional morphology, 7 (14%) were pT3, none were pT4, 38 (78%) were pT1 or pT2, and 4 did not have tumor stage recorded.
Six cases demonstrated frank sarcomatoid transformation and 2 showed areas of rhabdoid morphology. The sarcomatoid areas were composed of pleomorphic spindled cells essentially indistinguishable from other high-grade sarcomatoid renal carcinomas. In 5 of these cases, the sarcomatoid areas were seen in direct continuity with areas showing the stereotypical low-grade morphology (including WHO/ISUP nucleolar grade 2 nuclei), indicating true dedifferentiation, rather than existence of a different tumor type.
Only 3 of 62 tumors (5%) lacked any areas with typical morphologic features or cytoplasmic inclusions and would not have been recognizable as SDH-deficient RCC based on morphology. These 3 tumors demonstrated high-grade nuclear features (equivalent to WHO/ISUP nucleolar grade 3/4). In 1 patient, the morphology was that of an undifferentiated sarcomatoid malignancy and the patient was alive with bone and lymph nodes metastases at 103 months post-diagnosis. Genetic testing revealed a germline SDHB mutation, in addition to somatic NF2 and ARID1A aberrations in a lymph node metastasis. The 2 other tumors without typical morphologic features of SDH-deficient RCC showed diffuse papillary architecture with necrosis. One of these also contained prominent desmoplastic stroma, while the other showed focal rhabdoid differentiation. No information on germline testing was available for these 2 patients.
Immunohistochemical features
All cases showed loss of expression of SDHB by IHC in the presence of internal positive controls (an inclusion criterion for the study) (Fig. 6). All cases also showed preserved positive staining for SDHA. PAX8 was positive in all but 2 of the 23 cases stained with this marker. All 11 cases stained with antibodies to EMA were positive. Focal KIT expression was reported in 4 of 30 cases (13%), and focal/patchy CK7 expression was found in only 3 of 32 cases (9%). Of the 14 cases stained with antibodies to broad spectrum cytokeratins (CAM5.2 and/or AE1/AE3), 6 demonstrated completely negative staining for all cytokeratins, and 1 showed only focal CK20 positivity. There were no significant immunophenotypic differences between tumors showing variant and conventional morphologies.

Serial sections stained with hematoxylin and eosin (A, C) and SDHB IHC (B, D). Entrapped benign tubules were frequently seen at the edge of the tumors. SDHB IHC shows retained positive cytoplasmic staining in the internal controls (including entrapped benign tubules), but all the neoplastic cells are negative.
Morphologic predictors of metastasis
Nine patients (15%) developed metastatic disease, 2 of whom died of disease at 24 and 26 months after initial presentation. Seven patients with metastatic disease had nuclear atypia equivalent to WHO/ISUP grade 3 or 4 at presentation, and 2 had WHO/ISUP nuclear grade 2. Six had tumor necrosis, and 5 showed variant high-grade morphology in their primary tumor. Of the 5 cases with variant high-grade morphology, 2 had papillary architecture, and 3 also had a minor component of low-grade conventional morphology. Of the 9 patients who developed metastatic disease, 7 were alive at the last follow-up (follow-up ranged from 0 to 103 months), 2 had documented germline SDHB mutations, 1 had multifocal disease, and 1 had a family history of SDH-deficient RCC in a first-degree relative.
One patient who died of disease at 27 months after initial presentation had a documented germline SDHB mutation, but no metastatic disease was reported. Interestingly, this patient had 2 discrete tumors in the right kidney, one demonstrating the typical low-grade morphology, and the other showing high-grade features including rhabdoid cells and extensive perineural invasion. However, close inspection of this tumor revealed occasional intracytoplasmic inclusions typical of SDH-deficient RCC. A third patient died of unknown causes at 3.5 months following diagnosis of their 2.6-cm tumor that showed typical low-grade morphology without necrosis. Unfortunately, no further clinical information or follow-up data was available.
Survival analyses
Survival analysis was limited by incomplete follow up for many patients. With this caveat, mean overall survival was 65.0 months for cases with variant morphology, compared to 93.9 months for those with conventional morphology, however this difference did not reach statistical significance (p = 0.736). Overall survival was significantly shorter in patients with variant morphology and a tumor stage of pT3 or pT4 (n = 4), compared with the remainder of the cohort (mean 3.8 vs. 31.7 months; median 1.0 vs. 13.0 months; p = 0.024).
Discussion
In this study, we evaluated a multi-institutional cohort of 62 previously unreported SDH-deficient RCCs from 59 patients, with a particular focus on variant morphologic features and their clinical associations. This study represents the largest cohort of SDH-deficient RCC to date, and our findings confirm that the previously reported distinctive morphologic features of this entity are found, at least focally, in most cases.
When obtaining cases for this study, in addition to retrieving all tumors occurring in the setting of known germline SDH mutation or with compatible morphology, contributors were also specifically asked to submit any renal tumors with unusual or difficult to classify morphology, especially those occurring in younger patients. Furthermore, many of the co-authors in this study have large consultation practices in tertiary or quaternary referral centers, and frequently receive cases with unusual morphologies which they screen with SDHB IHC. Thus, we were able to screen a broad selection of tumors with unusual or difficult to classify morphologies, which somewhat mitigates the potential selection bias of only considering the diagnosis in cases with classical morphology.
Of the 62 tumors evaluated in this study, typical histological features consistent with the previous descriptions of SDH-deficient RCC were present, at least focally, in 59 cases (95%). Variant morphologic features were identified in 13 cases (21%). Variant morphologies were diverse but were all associated with high-grade nuclear features (equivalent to WHO/ISUP grade 3 to 4), rather than the low-grade nuclei typical of this entity. The architectural patterns ranged from solid sheets of undifferentiated sarcomatoid cells, to papillary growth, and irregular tubular structures with elongated cells and myxoid stroma.
Typically, discussions of the differential diagnosis of SDH-deficient RCC center on oncocytoma, chromophobe RCC, and clear cell RCC, and are well described elsewhere8,12,24,25. However, special mention should be made of the following four relatively rare and/or emerging entities: low-grade oncocytic tumor (LOT) of the kidney, FH-deficient RCC, ALK rearranged RCC (ALK-RCC), and thyroid-like follicular RCC. LOT of the kidney has recently emerged as a provisional entity according to the GUPS classification of renal tumors13,29. These tumors, which also have oval nuclei and eosinophilic cytoplasm, demonstrate a KIT-negative/CK7-positive immunophenotype, lack the flocculent/vacuolated cytoplasm typical of SDH-deficient RCC, and show retained expression of SDHB. Rare cases of FH-deficient RCC show low-grade morphology that closely mimics conventional SDH-deficient RCC16. Therefore, FH and 2SC IHC should also be performed on morphologically suggestive cases with retained SDHB expression16,23. Occasional tumors show prominent follicular architecture with luminal eosinophilic material that may mimic the colloid-like secretions seen in thyroid-like follicular RCC30. Similarly, ALK-RCC may also enter the differential diagnosis, although this entity is characterized by highly heterogeneous morphology, often with a distinctly mucinous background, and consistently shows diffuse ALK protein expression by IHC31.
While most cases in this series showed at least focal areas of typical morphology, it is important to note that 3 cases (5%) contained no areas with the typical morphologic features of SDH-deficient RCC and would not have been recognized as such without the use of SDHB IHC. Therefore, in addition to performing SDHB IHC on all cases with compatible morphology, regardless of age or clinical features, we also recommend performing IHC on renal tumors with unusual morphologic features that are difficult to classify, particularly in cases with suggestive clinical features (for example, multifocality, young patient age, or a personal or family history of RCC, pheochromocytoma/paraganglioma, gastric GIST, or pituitary adenoma). It is important to remember that SDHB IHC requires careful interpretation, as an internal positive control in the non-neoplastic cells is always required, and a weak cytoplasmic blush in the tumor cells that contrasts to the distinctly granular internal positive control is still considered a loss of IHC staining but may be difficult to appreciate1,2.
Sarcomatoid RCC is not considered a distinct entity, but rather (like rhabdoid change), an end-stage of dedifferentiation which can occur in any RCC14,22. According to the current classification, tumors with exclusive sarcomatoid or rhabdoid differentiation, without an evident lower grade component, are generally placed in the broad category of RCC, unclassified. In this study, we report 6 cases of SDH-deficient RCC with sarcomatoid change, and 2 with areas of rhabdoid change, which suggests that SDHB IHC should be performed on any sarcomatoid or rhabdoid RCC before the category of RCC, unclassified is assigned. Although SDH-deficient RCC with rhabdoid or sarcomatoid changes have a poor prognosis (and their outcomes may not differ from other sarcomatoid tumors with current treatments), failure to identify them as such would prevent family members from accessing the benefits of genetic counseling and potential early intervention.
Of the 13 cases with variant morphology in this study, 5 developed metastatic disease and 1 died of disease at 27 months after diagnosis. Four additional patients developed metastatic disease but did not have variant morphologies in their primary tumors. However, all but 1 of these tumors had necrosis. In total, necrosis was present in 13 tumors, 7 of which exhibited variant morphologic features, while 6 had entirely conventional morphology. Interestingly, of the 6 tumors with necrosis but with otherwise entirely conventional morphology, follow-up data was available in 3 patients, all of whom developed extensive metastatic disease and one of whom died of disease at 24 months after diagnosis. This supports our previous findings7 indicating that the presence of necrosis is a strong predictor of disease progression, even in the absence of other high-grade morphologic features.
Variant morphology was also associated with higher tumor stage, which is known to be a powerful predictor of outcome in RCC, as confirmed in this cohort (p = 0.034). Of the 13 cases with variant morphology, 4 (31%) were pT3 or pT4. In contrast, only 7 (14%) of the 49 cases with typical morphology were pT3 and none were pT4. The presence of variant morphology was also shown to add predictive power to tumor stage, with overall survival being significantly shorter in pT3/pT4 tumors with variant morphology (n = 4), compared to the remainder of the cohort (mean survival 3.8 vs. 31.7 months; median survival 1.0 vs. 13.0 months; p = 0.024).
In addition to the frankly high-grade tumors with variant morphologies, there were 11 tumors that showed entirely conventional morphology, except for a focal increase in nuclear atypia (equivalent to WHO/ISUP grade 3). Follow-up was available for 9 of these patients: 3 had no evidence of disease at last follow-up, 2 died of disease at 24 and 27 months after diagnosis, 2 were alive with extensive metastatic disease, and another 2 were alive with disease at 6 and 50 months but more specific follow-up information was not available.
Of the 38 tumors with conventional morphology and low WHO/ISUP grade, only 5 patients had recurrent or metastatic disease recorded during the follow-up period. Moreover, only 2 (5%) of these tumors had necrosis, whereas necrosis was present in 4 of the 11 tumors (36%) with conventional morphology and WHO/ISUP grade 3-equivalent nuclei. At present, WHO/ISUP grade is not routinely reported for SDH-deficient RCC, given their generally favorable prognosis. However, our findings suggest that increased nuclear atypia is an adverse prognostic factor, even in tumors with otherwise conventional morphology, and supports the inclusion of the highest WHO/ISUP grade in the pathology reports for all SDH-deficient RCCs.
A key limitation of this study is the lack of follow-up information for a significant proportion of patients. This is a common problem in large series of rare tumors that are mostly encountered in the consultation setting. We are continuing to obtain all available follow-up data for each of the patients in this cohort, with the aim of providing greater insights into the biological behavior of this rare entity in the future. In the meantime, we emphasize that the morphologic features described in this series, while appearing to be correlated with adverse outcomes, cannot be definitively described as predictive factors on the basis of this limited data. Nevertheless, it is important that pathologists are aware of the potential morphologic diversity that may be encountered in SDH-deficient RCC in order to maintain a low threshold for performing IHC in any difficult to classify case.
Although limited data on genetic testing was available for this cohort, all 18 patients (31%) who underwent testing were found to harbor germline mutations in the SDHB gene. This confirms the findings from earlier studies indicating that, like SDH-deficient paragangliomas, the great majority of SDH-deficient RCCs are associated with germline mutations in one of the SDH genes2. Therefore, the diagnosis of SDH-deficient RCC should be considered an absolute indication for genetic counseling/testing.
It is important to note that germline mutations in SDHA are relatively common incidental findings in the general population (estimated to occur in up to 0.3%)17, with an extremely low lifetime penetrance (as low as 1.7%)6,14,32. For this reason, when an SDHA mutation is identified as part of a broad sequencing panel, it may be an incidental finding unrelated to renal neoplasia17,27. Therefore, it is recommended that when either a germline or a somatic SDHA mutation is identified in a patient with RCC, confirmatory IHC should be performed, as the tumor should be negative for both SDHA and SDHB if it is truly driven by biallelic SDHA mutation/inactivation6,13,14. Failure to do this may lead to other RCCs arising in patients with an incidental SDHA germline variant being incorrectly classified as SDH-deficient RCC.
None of the tumors in this cohort showed loss of SDHA expression by IHC, and no patients had germline mutations in any SDH genes other than SDHB (although many did not have testing for all genes). We were therefore unable to evaluate the possibility of any genotype-phenotype correlations between variant morphologies and specific SDH mutations. Interestingly, 3 patients in this cohort with multifocal tumors were all found to have one tumor showing the typical morphologic features of SDH-deficient RCC, while their other tumors exhibited high-grade variant morphologies. These findings indicate that various morphologic appearances can be seen with the same underlying germline mutation, possibly reflecting different second hit somatic mutations driving tumorigenesis.
It is noteworthy that 6 patients with SDH-deficient RCC, but without documented germline mutations, had clinical features highly suggestive of syndromic disease, including 3 patients (5%) with bilateral and/or multifocal disease, and another 3 with a family history of relatively rare tumors (GIST and paraganglioma) known to be associated with SDH deficiency. No doubt many of these patients had not completed full genetic testing at the time of follow up. However, as we have previously suggested7, it is possible that some patients with SDH-deficient RCC may be syndromic, even if no germline mutations are identified by current methodologies. In a practical sense, since long-term follow-up of patients with SDH-deficient RCC is required due to the possibility of late disease progression, we would also recommend long-term follow-up for other syndromic manifestations (for example, multifocal/bilateral disease, pheochromocytoma/paraganglioma, GIST, pituitary adenoma), irrespective of whether a germline mutation has been identified.
In conclusion, despite the typical appearances seen in most cases of SDH-deficient RCC, variant morphologic features are present in a subset of cases (21% in this series) and are associated with more aggressive biological behavior. Examples of variant morphologies include sheets of undifferentiated epithelioid or sarcomatoid cells, papillary growth, and irregular tubular and microcystic structures composed of cells with high-grade nuclear features, often with increased stromal myxoid material. Occasional tumors may lack any of the typical morphologic features of SDH-deficient RCC (5% in this series). Finally, our findings indicate that the presence of necrosis or increased nuclear atypia, equivalent to WHO/ISUP grade 3, even in tumors with otherwise entirely conventional morphology, can be associated with an increased risk of metastatic disease. We therefore recommend a low threshold for performing SDHB IHC screening in any unusual or difficult to classify renal tumor, particularly if occurring at a younger age (<50 years), or in the setting of suggestive clinical features, such as multifocal/bilateral disease, or a history of other tumors known to be associated with SDH deficiency.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Gill, A. J. Succinate dehydrogenase (SDH) deficient neoplasia. Histopathology 72, 106–116 (2018).
Gill, A. J. Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia. Pathology 44, 285–292 (2012).
Gill, A. J. et al. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC and SDHD in paraganglioma-phaeochromocytoma syndromes. Hum. Pathol. 41, 805–814 (2010).
Gill, A. J. et al. Succinate dehydrogenase deficiency is rare in pituitary adenomas. Am. J. Surg. Pathol. 38, 560–566 (2014).
Turchini, J. & Gill, A. J. Morphological clues to succinate dehydrogenase (SDH) deficiency in pheochromocytomas and paragangliomas. Am. J. Surg. Pathol. 44, 422–424 (2020).
Williamson, S. R. et al. Report from the International Society of Urological Pathology (ISUP) Consultation Conference on Molecular Pathology of Urogenital Cancers: III: Molecular Pathology of Kidney Cancer. Am. J. Surg. Pathol. 44, e47–e65 (2020).
Gill, A. J. et al. Succinate dehydrogenase (SDH)-deficient renal carcinoma: a morphologically distinct entity: a clinicopathologic series of 36 tumors from 27 patients. Am. J. Surg. Pathol. 38, 1588–1602 (2014).
Gill, A. J. et al. Renal tumors associated with germline SDHB mutation show distinctive morphology. Am. J. Surg. Pathol. 35, 1578–1585 (2011).
Gill, A. J. et al. Renal tumors and hereditary pheochromocytoma-paraganglioma syndrome. N Engl. J. Med. 364, 885–886 (2011).
Gill, A. J. et al. Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types. Am. J. Surg. Pathol. 34, 636–644 (2010).
Dwight, T. et al. Familial SDHA mutation associated with pituitary adenoma and pheochromocytoma/paraganglioma. J. Clin. Endocrinol. Metab. 98, E1103–E1108 (2013).
Williamson, S. R. et al. Succinate dehydrogenase-deficient renal cell carcinoma: detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod. Pathol. 28, 80–94 (2015).
Trpkov, K. et al. Novel, emerging and provisional renal entities: The Genitourinary Pathology Society (GUPS) Update on Renal Neoplasia. Mod. Pathol. 34, 1167–1184 (2021).
Trpkov, K. et al. New developments in existing WHO entities and evolving molecular concepts: The Genitourinary Pathology Society (GUPS) Update on Renal Neoplasia. Mod. Pathol. 34, 1392–142 (2021).
Gill, A. J. et al. Germline SDHC mutation presenting as recurrent SDH deficient GIST and renal carcinoma. Pathology 45, 689–691 (2013).
Smith, S. C. et al. A distinctive, low-grade oncocytic fumarate hydratase-deficient renal cell carcinoma, morphologically reminiscent of succinate dehydrogenase-deficient renal cell carcinoma. Histopathology 71, 42–52 (2017).
Korpershoek, E. et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J. Clin. Endocrinol. Metab. 96, E1472–E1476 (2011).
Yakirevich, E. et al. A novel SDHA-deficient renal cell carcinoma revealed by comprehensive genomic profiling. Am. J. Surg. Pathol. 39, 858–863 (2015).
Ozluk, Y. et al. Renal carcinoma associated with a novel succinate dehydrogenase A mutation: a case report and review of literature of a rare subtype of renal carcinoma. Hum. Pathol. 46, 1951–1955 (2015).
Jiang, Q. et al. A novel germline mutation in SDHA identified in a rare case of gastrointestinal stromal tumor complicated with renal cell carcinoma. Int. J. Clin. Exp. Pathol. 8, 12188–12197 (2015).
McEvoy, C. R. et al. SDH-deficient renal cell carcinoma associated with biallelic mutation in succinate dehydrogenase A: comprehensive genetic profiling and its relation to therapy response. NPJ Precis. Oncol. 2, 9 (2018).
Moch, H., Humphrey, P. A., Ulbright, T. M. & Reuter, V. E. WHO Classification of Tumours of the Urinary System and Male Genital Organs 4th edn. (IARC Press, 2017).
Li, Y. et al. Re-evaluation of 33 ‘unclassified’ eosinophilic renal cell carcinomas in young patients. Histopathology 72, 588–600 (2018).
Ricketts, C. et al. Succinate dehydrogenase kidney cancer: an aggressive example of the warburg effect in cancer. J. Urol. 188, 2063–2071 (2012).
Kennedy, J. M. et al. Clinical and morphologic review of 60 hereditary renal tumors from 30 hereditary renal cell carcinoma syndrome patients: lessons from a contemporary single institution series. Med. Oncol. 36, 74 (2019).
Casey, R. T. et al. Clinical and molecular features of renal and pheochromocytoma/paraganglioma tumor association syndrome (RAPTAS): case series and literature review. J. Clin. Endocrinol. Metab. 102, 4013–4022 (2017).
Dwight, T. et al. Loss of SDHA expression identifies SDHA mutations in succinate dehydrogenase-deficient gastrointestinal stromal tumors. Am. J. Surg. Pathol. 37, 226–233 (2013).
Samaratunga, H. et al. Granular necrosis: a distinctive form of cell death in malignant tumours. Pathology 52, 507–514 (2020).
Trpkov, K. et al. Low-grade oncocytic tumour of kidney (CD117-negative, cytokeratin 7-positive): a distinct entity? Histopathology 75, 174–184 (2019).
Al-Obaidy, K. I. et al. EWSR1-PATZ1 fusion renal cell carcinoma: a recurrent gene fusion characterizing thyroid-like follicular renal cell carcinoma. Mod. Pathol. 34, 1921–1934 (2021).
Kuroda, N. et al. ALK rearranged renal cell carcinoma (ALK-RCC): a multi-institutional study of twelve cases with identification of novel partner genes CLIP1, KIF5B and KIAA1217. Mod. Pathol. 33, 2564–2579 (2020).
Benn, D. E. et al. Bayesian approach to determining penetrance of pathogenic SDH variants. J. Med. Genet. 55, 729–734 (2018).
Author information
Authors and Affiliations
Contributions
A.J.G. was responsible for study conception and design. T.L.F., F.M., and A.J.G. reviewed cases and wrote the first draft of the manuscript. A.C. was responsible for performance of IHC. The remaining authors contributed cases and reviewed the manuscript prior to submission.
Corresponding author
Ethics declarations
Ethics approval
The study was approved by the Northern Sydney Local Health District Human Research Ethics Committee.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Fuchs, T.L., Maclean, F., Turchini, J. et al. Expanding the clinicopathological spectrum of succinate dehydrogenase-deficient renal cell carcinoma with a focus on variant morphologies: a study of 62 new tumors in 59 patients. Mod Pathol 35, 836–849 (2022). https://doi.org/10.1038/s41379-021-00998-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00998-1
This article is cited by
-
Low grade oncocytic tumors of the kidney: a clinically relevant approach for the workup and accurate diagnosis
Modern Pathology (2022)
-
Overview of the 2022 WHO Classification of Familial Endocrine Tumor Syndromes
Endocrine Pathology (2022)
