
Abstract
Current directions in medication development for alcohol use disorder (AUD) emphasize the need to identify novel molecular targets and efficiently screen new compounds aimed at those targets. Ibudilast (IBUD) is a neuroimmune modulator that inhibits phosphodiesterase-4 and -10 and macrophage migration inhibitory factor and was recently found to reduce alcohol intake in rats by ∼50%. To advance medication development for AUD, the present study consists of a randomized, crossover, double-blind, placebo-controlled laboratory study of IBUD in nontreatment-seeking individuals with current (ie, past month) mild-to-severe AUD. This study tested the safety, tolerability, and initial human laboratory efficacy of IBUD (50 mg b.i.d.) on primary measures of subjective response to alcohol as well as secondary measures of cue- and stress-induced changes in craving and mood. Participants (N=24) completed two separate 7-day intensive outpatient protocols that included daily visits for medication administration and testing. Upon reaching a stable target dose of IBUD (or matched placebo), participants completed a stress-exposure session (day 5; PM), an alcohol cue-exposure session (day 6; AM), and an i.v. alcohol administration session (day 6; PM). Participants stayed overnight after the alcohol administration, and discharge occurred on day 7 of the protocol. Medication conditions were separated by a washout period that was ⩾7 days. IBUD was well tolerated; however, there were no medication effects on primary measures of subjective response to alcohol. IBUD was associated with mood improvements on the secondary measures of stress exposure and alcohol cue exposure, as well as reductions in tonic levels of craving. Exploratory analyses revealed that among individuals with higher depressive symptomatology, IBUD attenuated the stimulant and mood-altering effects of alcohol as compared with placebo. Together, these findings extend preclinical demonstrations of the potential utility of IBUD for the treatment of AUD and suggest that depressive symptomatology should be considered as a potential moderator of efficacy for pharmacotherapies with neuroimmune effects, such as IBUD.
Similar content being viewed by others

INTRODUCTION
Alcohol use disorder (AUD) is a chronic and relapsing condition marked by repeated alcohol use despite a host of negative physical and psychosocial consequences. To date, only four pharmacotherapies are approved by the Food and Drug Administration for the treatment of AUD, and these medications are only modestly effective (Johnson, 2008). Therefore, the development of efficacious medications for AUD remains a high research priority. To that end, current directions in medication development emphasize the need to identify novel molecular targets for AUD treatment and to more efficiently screen new compounds aimed at those targets (Litten et al, 2012). Novel molecular targets for the treatment of AUD include modulation of neurotrophin signaling and neuroimmune function.
Neurotrophins, including glial-derived neurotrophic factor (GDNF) and brain-derived neurotrophic factor (BDNF), are essential for synaptic plasticity (Airaksinen and Saarma, 2002), neuron survival, and basic cell signaling, including midbrain dopamine transmission (Altar et al, 1992; Lin et al, 1993). In rodent models of AUD, reductions in GDNF and BDNF expression underlie dysfunctional striatal dopamine signaling, increased motivation to consume alcohol, and heightened alcohol reward (Ahmadiantehrani et al, 2014; Barak et al, 2015; Carnicella et al, 2009a; Hensler et al, 2003). Conversely, increases in GDNF and BDNF signaling restores mesolimbic dopamine function, reduces alcohol self-administration, and attenuates relapse to alcohol seeking (Barak et al, 2011, 2015; Carnicella et al, 2008, 2009b; McGough et al, 2004). These results suggest that medications that can increase GDNF and BDNF expression may be useful treatments for AUD.
Chronic alcohol consumption produces a sustained inflammatory state such that individuals with AUD have increased neuroinflammation throughout the brain (He and Crews, 2008), and elevated peripheral levels of proinflammatory cytokines have been proposed as a biomarker for AUD (Achur et al, 2010). In turn, alcohol-induced neuroinflammation is thought to contribute to chronic drug-seeking behavior as well as the behavioral and neurotoxic effects of alcohol (Mayfield et al, 2013). In rodents, lipopolysaccharide-induced neuroinflammation produces prolonged increases in alcohol consumption (Blednov et al, 2011), and knocking out neuroimmune signaling genes attenuates alcohol preference and self-administration (Blednov et al, 2012). Alcohol-induced proinflammatory signaling also mediates acute motor impairment by alcohol (Wu et al, 2011), and chronic alcohol exposure produces long-lasting neuroinflammation that is associated with sustained cognitive and behavioral impairment, as well as brain damage (Alfonso-Loeches et al, 2010). Neuroinflammation mediated by glucocorticoid pathways is thought to increase vulnerability to stress-induced drug seeking and relapse (Frank et al, 2011). In sum, a medication that reduces proinflammatory signaling may produce antialcohol and neuroprotective effects that may be beneficial for the treatment of AUD.
Ibudilast (IBUD; also known as MN-166, previously AV411) has been used in Japan for asthma and cerebrovascular disorders (Ledeboer et al, 2007). IBUD has been recently advanced as a novel addiction pharmacotherapy that targets neurotrophin signaling and neuroimmune function. IBUD inhibits phosphodiesterase-4 (PDE4) and -10 (PDE10) and macrophage migration inhibitory factor (MMIF) (Johnson et al, 2014). As PDE4 and MMIF are critically involved in proinflammatory signaling (Calandra and Roger, 2003; Teixeira et al, 1997), and PDE10 negatively regulates neurotrophin expression (Giampà et al, 2010), the inhibition of these molecules by IBUD has been theorized to reduce neuroinflammation and promote neurotrophin expression (Johnson et al, 2014). In support, IBUD enhances neurotrophin expression, reduces proinflammatory cytokine release, and attenuates neuronal death (Mizuno et al, 2004). Most important to the present study, IBUD has been demonstrated to reduce ethanol intake by ∼50% in selectively bred alcohol-preferring (P) and high alcohol-drinking (HAD1) rats both under conditions of maintenance and relapse testing (Bell et al, 2015). These recent preclinical findings for IBUD support prior studies indicating that pharmacological inhibition of PDE4 and PDE10 decreases alcohol intake (Blednov et al, 2014; Logrip et al, 2014; Wen et al, 2012). These results suggest IBUD represents a promising treatment for AUD; however, the safety and efficacy of IBUD in combination with alcohol administration has not been tested in humans.
In summary, the available preclinical data have advanced neuroinflammatory and neurotrophin signaling pathways as plausible pharmacotherapy targets for AUD. To advance medication development for AUD, the present study consists of a randomized, crossover, double-blind, placebo-controlled human laboratory study of IBUD in nontreatment-seeking individuals with current (ie, past month) mild-to-severe AUD. Specifically, this study tests the safety, tolerability, and initial human laboratory efficacy of IBUD (50 mg b.i.d.). The a priori primary outcomes, as registered in clinicaltrials.gov (NCT02025998), were IBUD effects on subjective response to alcohol, whereas secondary outcomes were IBUD effects on cue-induced craving and on stress-induced craving and mood. IBUD has been well tolerated in opioid users (Cooper et al, 2015) and it reduced subjective response to methamphetamine administration in patients with methamphetamine use disorder (Worley et al, 2016). Therefore, it was hypothesized that IBUD would be well tolerated, alone and in combination with alcohol, and that it would reduce the rewarding subjective effects of alcohol as well as cue- and stress-induced alcohol craving and negative mood.
Given that neuroinflammation is increasingly implicated in mood disorders (Miller et al, 2009; Moieni et al, 2015; Raison and Miller, 2011), exploratory analyses were conducted to test the effects of IBUD on mood ratings during the experimental protocol as well as the moderating role of depressive symptomatology on the effects of IBUD. AUD severity was considered in these exploratory analyses given the putative overlap between severity of AUD and depressive symptomatology (Heilig et al, 2010; Sinha et al, 2009).
MATERIALS AND METHODS
Participants
A community-based, nontreatment-seeking sample of individuals with current DSM-5 AUD was recruited via online and print advertisements in the Los Angeles area between January 2014 and June 2015. The study protocol and all procedures were approved by the Institutional Review Board of the University of California, Los Angeles. Inclusion criteria were: (1) age between 21 and 65 years; and (2) meet current (ie, past month) DSM-5 diagnostic criteria for AUD (mild, moderate, or severe). Exclusion criteria were: (1) current treatment for alcohol problems, a history of treatment in the 30 days before enrollment or treatment seeking; (2) DSM-IV diagnosis of dependence on any psychoactive substances other than alcohol and nicotine in the past 12 months; (3) a lifetime DSM-IV diagnosis of schizophrenia, bipolar disorder, or any psychotic disorder; (4) current use of psychoactive drugs, other than marijuana, verified by a toxicology screen; (5) clinically significant alcohol withdrawal symptoms as indicated by a score of ⩾10 on the Clinical Institute Withdrawal Assessment for Alcohol-Revised (CIWA-R); (6) pregnancy, nursing, or a refusal to use reliable method of birth control (if female); (7) a medical condition that may interfere with safe study participation (eg, unstable cardiac, renal, or liver disease, uncontrolled hypertension, or diabetes); and (8) AST, ALT, or GGT ⩾3 times upper normal limit.
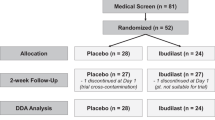
A total of 138 individuals consented to participate in the initial screening/eligibility visit. Of these, 62 were clinically eligible and invited to complete a screening physical exam and laboratory tests. Initial screen failures were primarily because of not meeting diagnostic criteria for AUD and/or meeting criteria for one of the exclusionary diagnoses. Of the 62 eligible participants from the initial screening visit, 47 completed a physical exam and 38 were deemed medically eligible. A total of 32 participants were randomized to the first study medication, 28 of whom completed the first medication condition and 24 of whom completed the entire study. Demographic characteristics and alcohol use variables for the 24 completers are summarized in Table 1. This study was registered in clinicaltrials.gov (NCT02025998).
Screening Procedures
Interested individuals called the laboratory and completed a telephone-screening interview. Eligible callers were invited to the laboratory, and after receiving a full explanation of study procedures and providing written informed consent, they completed the in-person assessment visit. At the beginning of the screening visit, participants were required to have a breath alcohol concentration (BrAC) of 0.00 g/dl (measured by Alcotest 6510; Drager Safety Diagnostics) and a urine toxicology test (CLIAWaived, 10-panel drug test; Medimpex United) negative for all drugs (excluding marijuana). The toxicology panel comprised cocaine, marijuana, opiate (heroin, morphine, and codeine), amphetamine, methamphetamine, PCP, benzodiazepine, barbiturate, methadone, and oxycodone.
During the in-person screening visit, participants completed questionnaires on demographics, drug use history, and psychological functioning. The following interviews were administered by trained master’s-level clinicians supervised by a licensed psychologist (LAR): (1) the 30-day Timeline Follow-Back (TLFB) to capture daily alcohol use over the 30 days before the visit (Sobell et al, 1988); (2) CIWA-R (Sullivan et al, 1989) to assess for exclusionary clinically significant alcohol withdrawal; and (3) the Structured Clinical Interview for DSM-IV (SCID) (First, 2005) to assess criteria for alcohol abuse and dependence and to screen for exclusionary psychiatric diagnoses. In addition, the symptom of alcohol craving was added to the SCID using an interview item from the SSAGA (Bucholz et al, 1994), thus allowing for diagnosing participants on AUD according to DSM-5 criteria (which is reported herein). The craving symptom was required to be current (ie, present within the past month). The Penn Alcohol Craving Scale (PACS) (Flannery et al, 1999), the Beck Depression Inventory II (Beck et al, 1996), and the Beck Anxiety Inventory (Beck and Steer, 1993) were administered during the screening visit.
Participants deemed eligible following the in-person screening were invited to return to the laboratory to complete a physical exam with the study physician (KM). Participants were required to provide a negative urine toxicology screen for all drugs (excluding marijuana) at the time of the physical exam that consisted of clinical laboratory testing (ie, a blood chemistry panel and liver profile) and an electrocardiogram (EKG).
Medication Administration and Intensive Outpatient Procedures
The study medication and matched placebo were provided by Medicinova and dispensed daily by the UCLA Health Pharmacy Investigational Drug Section that also managed the blind. Medically eligible participants were then randomized to receive the first study medication. Each participant completed two separate 7-day intensive outpatient protocols at the UCLA Clinical and Translational Research Center (CTRC), during which they completed daily morning visits with a research nurse to take the AM study medication under observation and assessments of vital signs and side effects (collected using an open-ended question format). At each daily visit the study nurse completed BrAC and toxicology tests. All samples were negative for alcohol and drugs (except for marijuana) during the course of the experiment. Participants were asked to take the PM dose at home and a riboflavin (50 mg) tracer was used to verify compliance and all samples fluoresced during the trial. Although a recent study has raised concerns regarding the use of riboflavin as a tracer substance for compliance testing (Herron et al, 2013), the fact that individuals knew they would be tested by the study nurse on a daily basis may have accounted for all the samples fluorescing in this trial.
To assess tonic levels of alcohol craving, depression, and anxiety, the PACS, BDI-II, and BAI were also administered at day 1 (intake) and again at day 7 (discharge) of each medication condition. See Figure 1 for study flowchart.

Flowchart of study design.
IBUD was available in 10 mg capsules and participants were titrated as follows: 20 mg (ie, two 10 mg capsules) b.i.d. during days 1 and 2, and 50 mg (ie, five 10 mg capsules) b.i.d. during days 3–6. Matched placebo was provided and medication order was randomized and counterbalanced. The AM medication dose and assessments took place at 0800 h during days 1–6. Upon reaching a stable target dose on the study medication (or placebo), participants completed a stress-exposure paradigm (on day 5; PM), an alcohol cue-exposure session (day 6; AM), and an i.v. alcohol administration session (day 6; PM), which started at 1300 h. Participants stayed overnight at the CTRC after the alcohol administration and clinical labs were repeated before discharge (day 7) from each medication condition. The protocol required a minimum 7-day washout period; however, the observed washout period was on average 16 days (mean=16.58, SD=10.44, range=7–40). For safety reasons, participants were instructed not to drink during the medication period and abstinence was verified daily by the study nurse using a breathalyzer, thus limiting the naturalistic alcohol use data for analyses. Although no positive breathalyzer readings were obtained in the study, self-report TLFB data indicated that five participants drank alcohol during the medication period (four in the IBUD condition and one in the Placebo condition).
Following completion of the entire study and immediately before discharge on day 7 of the second medication condition, participants completed a motivational interviewing (MI) session targeting alcohol use reduction and supporting treatment seeking. The intervention was delivered by a master’s-level clinician under the supervision of a licensed psychologist (LAR). Participants received $30 for completing the in-person screening visit and $10 for the physical exam. Daily medication visits with the study nurse were compensated at $20/each. Participants received $40 for completing each stress-exposure and cue-exposure session, and $100 for each alcohol administration session. Participants who completed the entire study received a $50 bonus. The total compensation for all study procedures was $590.
Medication Dose Justification
Selecting 50 mg b.i.d. as the single target dose in this study was based on safety considerations, preclinical, and clinical data. Consistent with the primary goal of establishing safety, IBUD was tested at the highest dose that may be used clinically (50 mg b.i.d.) that in turn sets the stage for future dose-ranging studies. In addition, all in vitro (Cho et al, 2010a; Gibson et al, 2006a; Mizuno et al, 2004), in vivo animal (Beardsley et al, 2010; Hutchinson et al, 2009), and clinical neuropharmacology studies of IBUD found efficacy to be dose or concentration incremental—at least up to 80–100 mg/day doses and plasma concentrations. In essence, it appears as though ‘more is better’ up to the current clinical maximum safe and well-tolerated high dose of 50 mg b.i.d. correlates with ~100 ng/ml or ~0.5 μM steady-state plasma concentration (with daily AUC ~1500 ng × h/ml) and ~1.5 μM brain concentration. Furthermore, the half-life of IBUD is ~19 h (Rolan et al, 2008), supporting b.i.d. dosing. Relatedly, Rolan et al (2008) found that IBUD concentration was 5–10 ng/ml after 4 days of the last b.i.d. dose, suggesting that the washout period of ⩾7 days is likely sufficient to prevent carryover effects.
Experimental Procedures and Measures
The following well-validated (Bujarski and Ray, 2016; Mason and Higley, 2013) experimental paradigms were implemented to examine medication effects on subjective response to alcohol, cue reactivity, and stress reactivity:
Intravenous alcohol administration
The alcohol infusion was performed using a nomogram developed and validated in our previous work (Ray and Hutchison, 2004) (0.166 ml/min × weight in kg for males/0.126 ml/min × weight in kg for females). On testing day, the 6% alcohol solution was compounded and dispensed by the UCLA Health Pharmacy Investigational Drug Section. BrAC was monitored every 3 to 5 min and target BrACs were 0.02, 0.04, 0.06, and 0.08 g/dl. Upon reaching each of the target BrAC, infusion rates were reduced in half for testing. The duration of the ramp period where BrAC was increasing was ∼14 min to reach the 0.02 g/dl time point (SD=2), 15 min to reach the 0.04 g/dl time point (SD=7 min), 22 min to reach the 0.06 g/dl time point (SD=8 min), and 25 min to reach the final 0.08 g/dl time point (SD=8 min). Participants were held at a stable BrAC during testing for an average of 6–7 min at each target BrAC point (ie, BrAC=0.02, 0.04, 0.06, and 0.08 g/dl).
An i.v. alcohol administration method was selected given that it affords precise control over BrAC level that is a large source of between-subject variability in oral alcohol challenge studies (Ramchandani et al, 2009). This approach also eliminates alcohol cues and reduces alcohol expectancy effects that could nonpharmacologically influence the primary outcome of subjective response to alcohol.
The following measures were given at baseline and at each target BrAC: (1) The Biphasic Alcohol Effects Scale (BAES) that captures the stimulant and sedative subjective effects of alcohol (Erblich and Earleywine, 1995; Martin et al, 1993); (2) The Alcohol Urge Questionnaire (AUQ) that measures state levels of alcohol craving (Bohn et al, 1995; MacKillop, 2006); (3) The Alcohol Rating Scale that captures ‘liking’ and ‘wanting’ of alcohol; and (4) The Profile of Mood States (POMS; McNair et al, 1971) that was used to record positive and negative mood states.
Cue reactivity assessment (CR)
CR, as well as the other paradigms, were assessed at each medication condition such that participants served as their own controls. This is important as studies found that not all individuals with AUD are cue reactive (Litt et al, 1990; Rohsenow et al, 1994; Rubonis et al, 1994), and that instead between 50 (Laberg and Ellertsen, 1987) and 70% (Rubonis et al, 1994) of individuals with AUD respond to cues by showing a ⩾50% increase in subjective alcohol craving. CR followed well-established procedures (ie, Monti et al, 1987, 2001). Each session began with a 3 min relaxation period. Participants were then asked to hold and smell a glass of water for 3 min to control for the effects of simple exposure to any potable liquid. Next, they were asked to hold and smell a glass of their preferred alcoholic beverage for three 3 min trials. Order was not counterbalanced given carryover effects that are known to occur (Monti et al, 1987). Participants who were smokers were allowed a smoke break immediately before and after the CR assessment. After each exposure, participants completed the AUQ and the POMS.
Stress reactivity (SR)
Personal information collected during randomization day 1 were used to generate personalized scripts for stressful conditions following standardized procedures (Sinha, 2009; Sinha et al, 1992, 2000). Only stressful events rated ⩾8 (on 0–10 Likert scale where 10=most stressful) were used in script development and traumatic experiences were not included. The stress exposure consisted of 5 min tape-recorded scripts recounting current and unresolved stressful events in the participants’ lives, including cognitions and physical feelings. At baseline and after imagery, participants completed the POMS recording positive and negative mood, and the AUQ, assessing alcohol craving.
Statistical Analyses
To examine safety, side effects on IBUD vs PLAC were compared using Fisher’s exact test that represents a variation of the χ2 test appropriate for small cell sizes (ie, infrequent events). Repeated measures ANOVAs were used to assess for IBUD-related cardiovascular changes following administration of alcohol.
To examine human laboratory efficacy data, repeated measures analyses of variance (ANOVAs) were used to analyze the effects of IBUD on subjective response to alcohol, cue-induced craving and mood, and stress-induced craving and mood. For each test we were interested in the main effect of Medication (IBUD vs PLAC), the main effect of Trial (ie, baseline and target BrAC levels, pre–post cue exposure, and pre–post stress exposure), and the Medication × Trial interaction. To explore the moderating role of depressive symptomatology, scores on the BDI-II collected during the in-person screening visit (ie, a continuous measure, log-transformed for normality) were subsequently added to each model as a between-subjects factor with the goal of examining depressive symptoms as moderators of IBUD effects (interaction terms of interest were: Medication × BDI-II and Medication × BDI-II × Trial). An identical approach was used to probe for AUD severity as a moderator as it conceptually overlaps with depressive symptomatology such that individuals with higher AUD severity are expected to report greater depressive symptoms. Finally, a set of analyses of order effects were conducted for the purpose of probing for carryover effects given the study’s within-subjects crossover design.
RESULTS
Medication Safety and Tolerability
There were no severe adverse events in this trial, as defined by the ICH Good Clinical Practice. The complete report of adverse events, collected daily in open-ended questioning format, and coded using the Medical Dictionary for Regulatory Activities (MedDRA Version 14.0) terminology, are presented in Table 2. More participants reported at least one adverse event while on IBUD relative to placebo (Fisher’s exact p<0.001). In terms of individual adverse events, significant differences between IBUD and placebo were only observed for headaches (Fisher’s exact p<0.05), and at a trend-level effect for general gastrointestinal disorders (Fisher’s exact p=0.090), both of which were more frequent on IBUD vs PLAC. Comparisons of the results from the blood chemistry panel and liver profile, obtained at day 1 (intake) and day 7 (discharge) for each medication condition, revealed that one participant experienced a decrease in white blood cell count on the IBUD condition. All other clinical labs were within range for all participants at discharge from both IBUD and placebo conditions. Overall, IBUD was well tolerated, and there were no study dropouts directly related to IBUD as well as no dose reductions during the protocol.
To test whether IBUD would increase alcohol-induced nausea as compared with placebo, participants rated their feelings of nausea at each BrAC level during alcohol administration. Analyses revealed no significant main effect of Medication (F(1, 22)=0.38, p=0.544) or BrAC (F(4, 88)=0.91, p=0.462); however, there was a trend-level Medication × BrAC interaction (F(4, 88)=2.11, p=0.0857), suggesting that IBUD may increase feelings of nausea at rising BrAC levels. Of note, none of the participants vomited during the alcohol administration protocol.
To determine whether IBUD would affect cardiovascular function in the context of alcohol intake, heart rate and blood pressure were monitored during the alcohol administration at baseline and at each target BrAC level. Overall, IBUD produced a trend-level reduction in heart rate relative to placebo (Med main effect: F(1, 21)=3.55, p=0.074), but IBUD did not moderate the effect of alcohol on heart rate (Med × BrAC: F(4, 84)=0.37, p=0.83). No effects of IBUD were observed on systolic or diastolic blood pressure as either a main effect or as a moderator of alcohol’s effect across rising BrACs (p’s⩾0.35).
Analyses of Primary and Secondary Aims
Alcohol administration
The infusion of alcohol significantly increased levels of craving for alcohol (F(4, 88)=8.73, p<0.001), stimulation (F(4, 88)=8.20, p<0.001), sedation (F(4, 88)=8.20, p<0.001), positive mood (F(4, 88)=4.18, p<0.01), ‘liking’ (F(4, 88)=45.89, p<0.001), and ‘wanting’ (F(4, 88)=7.15, p<0.001), and decreased negative mood (F(4, 88)=5.01, p<0.01). Averaging across the alcohol administration (ie, across rising BrAC levels), IBUD vs PLAC did not affect subjective response to alcohol as indexed by alcohol craving, stimulation, sedation, positive mood, negative mood, ‘liking’ of the alcohol, and ‘wanting’ the alcohol (medication main effect p’s⩾0.17, medication × BrAC interaction: p’s⩾0.14).
Cue-exposure paradigm
Presentation of alcohol cues increased craving for alcohol (F(1, 22)=43.56, p<0.001) but did not affect either positive or negative mood (p’s⩾0.26). The medication main effects did not reach statistical significance for the outcomes of alcohol craving, positive mood, and negative mood (p’s⩾0.38). However, a trend-level Medication × Cue interaction was observed with respect to positive mood (F(1, 22)=3.62, p=0.07), such that IBUD vs placebo marginally increased positive mood during alcohol cue presentation vs water. The Medication × Cue interaction was not significant for craving or negative mood (p’s⩾0.14).
Stress-exposure paradigm
As expected, the stress imagery paradigm increased craving for alcohol (F(5, 115)=3.50, p<0.01), increased negative mood (F(5, 115)=6.80, p<0.001), and decreased positive mood (F(5, 115)=3.62, p<0.01). A trend-level Medication × Trial interaction was observed with respect to positive mood (F(5, 115)=2.19, p=0.06), such that IBUD produced a more rapid recovery in positive mood to baseline levels following stress exposure as compared with placebo (Figure 2). IBUD did not affect alcohol craving or negative mood during the stress imagery either as a main effect (p’s⩾0.58) or over time (p’s⩾0.93).

Positive mood during the stressful imagery paradigm (n=24). Positive mood was found to return to baseline levels following the stressful imagery more quickly while on IBUD relative to placebo (p=0.06).
Exploratory analyses
Alcohol administration. A main effect of depressive symptomatology, as measured by a log-transformed BDI-II score (to account for data nonnormality), was observed with respect to positive mood (F(1, 21)=7.86, p<0.05) and stimulation at a trend level (F(1, 21)=3.83, p=0.06). Furthermore, BDI-II scores moderated the effect of IBUD (ie, BDI-II × Medication interaction) on the outcomes of stimulation (F(1, 21)=4.78, p<0.05; Figure 3a), negative mood (F(1, 21)=4.47, p<0.05; Figure 3c), and at a trend-level positive mood (F(1, 21)=3.67, p=0.07; Figure 3b). In addition, BDI-II score moderated the effect of IBUD on alcohol-induced responses (ie, BDI-II × Medication × BrAC interaction) on positive mood (F(4, 84)=2.87, p<0.05; also in Figure 3b) as well ‘liking’ (F(4, 84)=2.35, p=0.06) and ‘wanting’ (F(4, 84)=3.17, p<0.05) of alcohol (Figure 4). Overall, these BDI-II × Medication interactive effects were such that IBUD decreased positive mood, stimulation, ‘liking,’ and ‘wanting,’ and increased negative mood responses to alcohol to a greater extent among participants with higher BDI-II scores. Log-transformed BDI-II was entered as a continuous covariate into the repeated measures ANOVA models. High and low groups were only created for the purpose of visually plotting the results, based on a median split.

Stimulation (a), positive mood (b), and negative mood (c) in response to alcohol is affected by the administration of IBUD and levels of depressive symptomatology. For ease of presentation, means and SEs are presented based on a median split of depressive symptomatology (n=12 per group); however, the analyses examined depressive symptomatology (ie, log-transformed BDI-II scores) as a continuous predictor.

Alcohol ‘Liking’ and ‘Wanting’ in response to alcohol is affected by the administration of IBUD and levels of depressive symptomatology. For ease of presentation, means and SEs are presented based on a median split of depressive symptomatology (n=12 per group); however, the analyses examined depressive symptomatology (ie, log-transformed BDI-II scores) as a continuous predictor.
Cue exposure. Depressive symptomatology, measured by the BDI-II, predicted the overall level of positive mood during the cue paradigm (F(1, 21)=6.85, p<0.05), yet BDI-II score did not significantly affect alcohol craving and mood during cue exposure (p’s⩾0.26).
Stress exposure. BDI-II score predicted levels of positive mood during the stress exposure (F(1, 22)=8.42, p<0.01), but was not a significant moderator of alcohol craving, positive mood, and negative mood during the stress paradigm (p’s⩾0.12).
Pre–post medication measures. In order to test tonic effects of IBUD on alcohol-related phenotypes, craving (PACS), depression (BDI-II), and anxiety (BAI) were measured on day 1 and day 7 of each medication. Specifically, a series of repeated measures ANOVAs were conducted on these outcomes across time (day 1 vs day 7), treatment (IBUD vs Placebo), and their interaction. Across medication conditions, decreases in tonic levels of alcohol craving (F(1, 21)=6.15, p<0.05), depression (F(1, 20)=12.76, p<0.01), and anxiety (F(1, 18)=6.73, p<0.05) were observed from day 1 to day 7, thus suggesting that IBUD was not depressogenic. Interestingly, IBUD produced a greater reduction in tonic alcohol craving over the course of the study than placebo (Figure 5; (F(1, 21)=4.55, p<0.05). Depressive symptomatology did not moderate the effect of IBUD on alcohol craving or anxiety (p’s⩾0.17).

Tonic levels of craving for alcohol on day 1 and day 7 of medication administration (n=24). Alcohol craving decreased significantly more sharply on IBUD relative to placebo (p<0.05).
Relationship between Depressive Symptomatology and AUD Severity
Because depressive symptomatology conceptually overlaps with AUD severity, we examined the correlation between the log-transformed BDI-II score and AUD severity (indexed by symptom count) and found it to be in the small-to-moderate range (r=0.34, p=0.095), suggesting that ∼11% of the variance between BDI-II and AUD severity is shared. In addition, we tested AUD severity (ie, AUD symptom count) as a moderator of IBUD effects in the laboratory paradigms (ie, primary aims) and found no significant moderator effects. Furthermore, we restricted our analyses to patients with moderate-or-severe AUD (sample size=15) and found only one significant Medication × BrAC effect on Sedation (F(4, 52)=3.74, p=0.0095), such that IBUD increased alcohol-induced sedation compared with placebo in this subsample of moderate-to-severe AUD cases. On balance, our results suggest that AUD severity did not significantly alter the results presented herein nor did it serve as a robust moderator of IBUD effects in this trial.
Probing for Order Effects
Given that carryover effects may be an issue with neuroimmune modulators, such as IBUD, several analyses were conducted to rule out medication order effects. First, medication order was entered as a covariate in all laboratory early efficacy analyses reported above and the results remained unchanged. Similarly, medication order did not alter the results of pre–post medication effects on alcohol-related measures as well as stress- and cue-exposure paradigms. Furthermore, Medication × Order interactions were tested and there were significant effects on Stimulation and Sedation during the alcohol administration (p=0.0095 and 0.0226, respectively). However, the direction of these effects was counter what would be expected from a carryover effect. Specifically, for stimulation, the difference between Placebo and IBUD was greater when IBUD was given first (F(1, 10)=11.43, p=0.007) than when it was given second (F(1, 11)=1.18, p=0.30). This pattern of results was also observed with respect to Sedation (IBUD first: F(1, 10)=5.08, p=0.048; Placebo first: F(1, 11)=0.88, p=0.37). Second, between-subjects analyses were conducted comparing mood variables on day 1 of the second medication based on medication order and no significant differences were found (p’s >0.10). Third, repeated measures ANOVAs were conducted to compare mood and craving variables on day 1 of the first and second medication to determine whether baseline levels at the beginning of each medication condition were comparable. Results indicated a significant decrease in BAI from the first to the second medication condition (p=0.009). This effect, however, was not found to be significantly different between the two randomization orders (ie, IBUD first vs Placebo first) (F(1, 18)=2.46, p=0.14), such that anxiety was lower to the same degree regardless of which medication was given first. No other effects were statistically significant. Taken together, these analyses provide no evidence of carryover effects in this trial.
DISCUSSION
The present study tested the effects of IBUD, a novel neuroimmune modulator, on subjective response to alcohol, cue-induced and stress-induced craving, and mood among nontreatment seekers with current (ie, past month) mild-to-severe AUD. This approach is consistent with efforts to screen novel compounds for AUD (Litten et al, 2012) as well as with the use of human laboratory paradigms to advance medication development by testing initial efficacy for alcohol (Ray et al, 2010; Yardley and Ray, 2016) as well as drug (Bigelow et al, 2012; Comer et al, 2013; Lofwall et al, 2014) use disorders.
Results of the safety aim suggest that IBUD was well tolerated in this study. Specifically, there were no study dropouts directly related to IBUD as well as no dose reductions over the course of the protocol. Analyses of specific side effects revealed significant differences between IBUD and placebo for headaches and a trend-level effect for general gastrointestinal disorders. Furthermore, the coadministration of IBUD with alcohol was not associated with significant changes in cardiovascular parameters, such as blood pressure and heart rate. A trend-level effect for nausea during alcohol administration suggested that IBUD may increase feelings of nausea at rising BrAC levels as compared with placebo. Together, these findings support the safety of IBUD, at the 50 mg b.i.d. dose, in samples with mild-to-severe AUD. This is particularly noteworthy given that PDE-4 inhibitors, such as rolipram, have yielded promising preclinical findings for AUD (Franklin et al, 2015) while raising safety concerns during testing in humans. As reviewed elsewhere (Palfreyman and Souness, 1996), PDE-4 inhibitors, including rolipram, are often associated with gastrointestinal distress, vertigo, nausea, and vomiting within their therapeutic window (Brunnee et al, 1992). These effects are believed to be due to increases in gastric acid secretion (Barnette et al, 1995) as well as central effects in brain areas regulating emesis and the vestibular system (Duplantier et al, 1996). Although the effects of IBUD are not limited to PDE-4, and include inhibition of PDE-10 and macrophage migration inhibitory factor (Cho et al, 2010b; Gibson et al, 2006b; Mizuno et al, 2004; Suzumura et al, 1999), these results support the potential application of IBUD to AUD treatment in human clinical samples and are consistent with recent safety studies for opiate (Cooper et al, 2015) and methamphetamine (Worley et al, 2016) dependence.
In addition to safety testing, this study examined initial efficacy markers using well-established human laboratory paradigms of alcohol administration, cue exposure, and stress exposure. Results for the alcohol administration paradigm, which were the primary aims of the study, revealed that IBUD did not alter the subjective responses to alcohol in this sample. Nevertheless, when exploratory analyses were conducted accounting for depressive symptomatology as a moderator of medication effects, results indicated that IBUD decreased positive mood, stimulation, ‘liking,’ and ‘wanting’ responses, and increased negative mood responses to alcohol to a greater extent among participants reporting greater levels of depressive symptoms on the BDI-II. Depressive symptomatology was advanced as a moderator in this study in light of the suppressant effects of IBUD on proinflammatory cytokines (IL-1β, IL-6, and TNF-α) that in turn have been convincingly implicated in major depression (Miller et al, 2009; Moieni et al, 2015; Raison and Miller, 2011). Therefore, it is reasonable to hypothesize that some of the beneficial effects of IBUD on efficacy markers for AUD, such as alcohol craving and subjective responses, may be, at least in part, due to its effects on mood. Furthermore, it may be that greater levels of neuroinflammation, putatively associated with mood dysregulation in participants with high BDI-II scores, may be required for IBUD to exert a stronger effect on both mood- and alcohol-related end points. These results, however, should be interpreted with caution as they represent exploratory analyses as compared with the a priori analyses for this study registered in clinicaltrials.gov.
Regarding medication effects on cue reactivity, which was a secondary aim of this study, there was a trend-level effect such that positive mood was greater when presented with the alcohol cue while on IBUD relative to alcohol cue presentation on placebo. This finding, although trend level, is generally consistent with results from the stress-reactivity paradigm wherein positive mood recovered more quickly to baseline levels following exposure to the stressful imagery on IBUD as compared with placebo. Although there was no effect of IBUD on the secondary outcomes of stress- or cue-induced craving, analyses of the tonic alcohol craving, measured by the PACS over 7 days of medication, indicated that craving decreased significantly more sharply on IBUD vs placebo.
Taken together, results from human laboratory paradigms examining early efficacy markers revealed a pattern whereby IBUD improved mood outcomes during stress and cue exposure as well as lower tonic craving during the medication period. Results for the effects of IBUD on the alcohol administration measures were largely negative in this study. A pattern of significant effects emerged only during exploratory analyses whereby depressive symptomatology was considered as a moderator. In that case, IBUD was found to produce greater decreases in the stimulant and mood-altering effects of alcohol only among individuals with higher BDI-II scores. Importantly, these findings must be interpreted in the context of the overall mean for the BDI-II scale at study entry (M=8.04, SD=9.03) that fell below the clinical depression cutoffs. This pattern suggests that the BDI-II score may be capturing subclinical mood dysphoria in this sample that in turn is thought to be associated with chronic alcohol use and AUD. As argued by the allostatic model of addiction, dysphoric mood and stress dysregulation may be particularly prominent at later stages of addiction (Koob, 2009, 2014). This is also consistent with a recent proposal to characterize addiction in terms of three domains, namely executive function, incentive salience, and negative emotionality, each domain tied to a distinct phase in the cycle of addiction (Kwako et al, 2016). This approach emphasizes the role of negative emotionality, possibly captured in this study through the BDI-II scores, in addiction that is distinct from requiring a comorbidity between AUD and depressive disorders. According to this approach, medications such as IBUD may be useful in ameliorating mood-related AUD symptomatology, and this may be particularly useful at later stages of the disorder when ‘curing the blues’ represents a clinical priority (Heilig et al, 2010). Notably, the effects of IBUD were not moderated by AUD severity, as captured by DSM-5 symptoms, suggesting that mood symptomatology may capture unique variance in IBUD response above and beyond AUD severity.
Although this study represents a step forward in developing IBUD for AUD, its mechanisms of action and associated clinical implications remain unclear. Based on the preclinical evidence, we expect IBUD to help individuals reduce drinking and maintain abstinence in the short or long term. Preclinical data suggest that restoration of GDNF and BDNF signaling helps normalize mesolimbic and nigrostriatal dopamine transmission, subsequently reducing motivation to consume alcohol and relapse to alcohol-seeking behavior (Barak et al, 2011, 2015). This should theoretically reduce drinking and prevent relapse, consistent with the recent preclinical findings by Bell et al (2015). In addition, blockade of proinflammatory signaling reduces motivation to consume alcohol (Blednov et al, 2011, 2012) and negative mood states (Wright et al, 2005). The latter function should theoretically promote a reduction in alcohol consumption before abstinence as well as continued abstinence after the cessation or attenuation of drinking. These hypothesized clinical effects, however, are decidedly speculative as clinical studies of IBUD for AUD have not yet been conducted (outside of the present report).
The present results should be interpreted in the context of the study strengths and limitations. Study strengths include the novel study medication with strong preclinical data, novel molecular targets, and compelling neurobiological rationale. The crossover design allowed us to reduce error variance by having individuals serve as their own controls, and the combination of three well-established human laboratory paradigms afforded an efficient screening of medication effects on initial efficacy markers. The intensive outpatient design consisted of daily visits including observed medication intake that addresses medication compliance issues and allows for careful medical monitoring of medication safety and side effects. Study limitations include the moderate sample size, although adequate for an early-phase laboratory study, and the subchronic level of IBUD dosing. This study recruited a sample of nontreatment seekers that may not generalize to treatment-seeking individuals with AUD (Rohn et al, in press). Furthermore, the inclusion of AUD of mild severity in this study may not be representative of clinical samples (ie, requiring DSM-IV alcohol dependence criteria or DSM-5 AUD moderate and severe only), particularly when the pathology associated with alcoholism is required for medications to exert clinical effects. Therefore, future studies may be better served by recruiting samples with more severe AUD than what was represented in this human laboratory trial. Laboratory measures, such as those employed in this study, may not reliably predict clinical efficacy (Yardley and Ray, 2016) and the i.v. alcohol administration method has limited external validity compared with oral alcohol intake. Regarding the monitoring of adverse events in this trial, an open-ended questioning approach was selected. Although this approach is relatively common in psychotropic medication trials (Hughes et al, 2016), it also carries the potential for underreporting of adverse events compared with scale-based adverse event monitoring (Berlin et al, 2008). In addition, multiple outcomes were examined without type I correction. However, given that IBUD is a novel pharmacotherapy with unknown mechanisms of action for AUD, a thorough screening of its potential effects was warranted. Furthermore, the sample size only allowed us to detect medium-to-large effect sizes, and a p-value correction may have resulted in an overly stringent threshold that obscures medication effects.
In conclusion, this is the first study of IBUD (50 mg b.i.d.), a neuroimmune and neurotrophin modulator, for AUD in humans. Results suggested that IBUD was well tolerated; however, there were no medication effects on primary measures of subjective response to alcohol. IBUD was associated with mood improvements on the secondary measures of stress- and alcohol-cue exposure, as well as reductions in tonic levels of craving. Exploratory analyses, however, found that among individuals with higher levels of depressive symptoms, IBUD was superior to placebo in attenuating the stimulant, rewarding, and mood-altering effects of alcohol during alcohol administration. These exploratory analyses, although interesting, should be considered with caution and future studies are needed to more clearly ascertain the biobehavioral mechanisms of action of IBUD for AUD as well as its clinical efficacy. On balance, these findings extend recent preclinical demonstrations of the potential utility of IBUD for the treatment of AUD (Bell et al, 2015) and suggest that depressive symptomatology should be considered as a potential moderator of efficacy for pharmacotherapies with neuroimmune effects, such as IBUD.
FUNDING AND DISCLOSURE
LAR has received study medication from Pfizer and consulted for GSK. Steve Shoptaw and Keith Heinzerling have received study medication from Medicinova. The authors declare no conflict of interest.
References
Achur RN, Freeman WM, Vrana KE (2010). Circulating cytokines as biomarkers of alcohol abuse and alcoholism. J Neuroimmune Pharmacol 5: 83–91.
Ahmadiantehrani S, Barak S, Ron D (2014). GDNF is a novel ethanol-responsive gene in the VTA: implications for the development and persistence of excessive drinking. Addict Biol 19: 623–633.
Airaksinen MS, Saarma M (2002). The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci 3: 383–394.
Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I, Guerri C (2010). Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J Neurosci 30: 8285–8295.
Altar CA, Boylan CB, Jackson C, Hershenson S, Miller J, Wiegand SJ et al (1992). Brain-derived neurotrophic factor augments rotational behavior and nigrostriatal dopamine turnover in vivo. Proc Natl Acad Sci USA 89: 11347–11351.
Barak S, Carnicella S, Yowell QV, Ron D (2011). Glial cell line-derived neurotrophic factor reverses alcohol-induced allostasis of the mesolimbic dopaminergic system: implications for alcohol reward and seeking. J Neurosci 31: 9885–9894.
Barak S, Wang J, Ahmadiantehrani S, Ben Hamida S, Kells AP, Forsayeth J et al (2015). Glial cell line-derived neurotrophic factor (GDNF) is an endogenous protector in the mesolimbic system against excessive alcohol consumption and relapse. Addict Biol 20: 629–642.
Barnette MS, Grous M, Cieslinski LB, Burman M, Christensen SB, Torphy TJ (1995). Inhibitors of phosphodiesterase IV (PDE IV) increase acid secretion in rabbit isolated gastric glands: correlation between function and interaction with a high-affinity rolipram binding site. J Pharmacol Exp Ther 273: 1396–1402.
Beardsley PM, Shelton KL, Hendrick E, Johnson KW (2010). The glial cell modulator and phosphodiesterase inhibitor, AV411 (ibudilast), attenuates prime- and stress-induced methamphetamine relapse. Eur J Pharmacol 637: 102–108.
Beck AT, Steer RA Beck Anxiety Inventory Manual. Psychological Corporation: San Antonio, TX; (1993).
Beck AT, Steer RA, Brown GK Manual for the Beck Depression Inventory-II. Psychological Corporation: San Antonio, TX; (1996).
Bell RL, Lopez MF, Cui C, Egli M, Johnson KW, Franklin KM et al (2015). Ibudilast reduces alcohol drinking in multiple animal models of alcohol dependence. Addict Biol 20: 38–42.
Berlin JA, Glasser SC, Ellenberg SS (2008). Adverse event detection in drug development: recommendations and obligations beyond phase 3. Am J Public Health 98: 1366–1371.
Bigelow GE, Preston KL, Schmittner J, Dong Q, Gastfriend DR (2012). Opioid challenge evaluation of blockade by extended-release naltrexone in opioid-abusing adults: dose-effects and time-course. Drug Alcohol Depend 123: 57–65.
Blednov YA, Benavidez JM, Black M, Harris RA (2014). Inhibition of phosphodiesterase 4 reduces ethanol intake and preference in C57BL/6J mice. Front Neurosci 8: 129.
Blednov YA, Benavidez JM, Geil C, Perra S, Morikawa H, Harris RA (2011). Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain Behav Immun 25: S92–S105.
Blednov YA, Ponomarev I, Geil C, Bergeson S, Koob GF, Harris RA (2012). Neuroimmune regulation of alcohol consumption: behavioral validation of genes obtained from genomic studies. Addict Biol 17: 108–120.
Bohn MJ, Krahn DD, Staehler BA (1995). Development and initial validation of a measure of drinking urges in abstinent alcoholics. Alcohol Clin Exp Res 19: 600–606.
Brunnee T, Engelstatter R, Steinijans VW, Kunkel G (1992). Bronchodilatory effect of inhaled zardaverine, a phosphodiesterase III and IV inhibitor, in patients with asthma. Eur Respir J 5: 982–985.
Bucholz KK, Cadoret R, Cloninger CR, Dinwiddie SH, Hesselbrock VM, Nurnberger JI Jr et al (1994). A new, semi-structured psychiatric interview for use in genetic linkage studies: a report on the reliability of the SSAGA. J Stud Alcohol 55: 149–158.
Bujarski S, Ray LA (2016). Experimental psychopathology paradigms for alcohol use disorders: applications for translational research. Behav Res Ther 86: 11–22.
Calandra T, Roger T (2003). Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol 3: 791–800.
Carnicella S, Ahmadiantehrani S, Janak PH, Ron D (2009a). GDNF is an endogenous negative regulator of ethanol-mediated reward and of ethanol consumption after a period of abstinence. Alcohol Clin Exp Res 33: 1012–1024.
Carnicella S, Amamoto R, Ron D (2009b). Excessive alcohol consumption is blocked by glial cell line-derived neurotrophic factor. Alcohol 43: 35–43.
Carnicella S, Kharazia V, Jeanblanc J, Janak PH, Ron D (2008). GDNF is a fast-acting potent inhibitor of alcohol consumption and relapse. Proc Natl Acad Sci USA 105: 8114–8119.
Cho Y, Crichlow GV, Vermeire JJ, Leng L, Du X, Hodsdon ME et al (2010a). Allosteric inhibition of macrophage migration inhibitory factor revealed by ibudilast. Proc Natl Acad Sci USA 107: 11313–11318.
Cho Y, Crichlow GV, Vermeire JJ, Leng L, Du X, Hodsdon ME et al (2010b). Allosteric inhibition of macrophage migration inhibitory factor revealed by ibudilast. Proc Natl Acad Sci USA 107: 11313–11318.
Comer SD, Mogali S, Saccone PA, Askalsky P, Martinez D, Walker EA et al (2013). Effects of acute oral naltrexone on the subjective and physiological effects of oral D-amphetamine and smoked cocaine in cocaine abusers. Neuropsychopharmacology 38: 2427–2438.
Cooper ZD, Johnson KW, Pavlicova M, Glass A, Vosburg SK, Sullivan MA et al (2015). The effects of ibudilast, a glial activation inhibitor, on opioid withdrawal symptoms in opioid-dependent volunteers. Addict Biol 21: 895–903.
Duplantier AJ, Biggers MS, Chambers RJ, Cheng JB, Cooper K, Damon DB et al (1996). Biarylcarboxylic acids and -amides: inhibition of phosphodiesterase type IV versus [3H]rolipram binding activity and their relationship to emetic behavior in the ferret. J Med Chem 39: 120–125.
Erblich J, Earleywine M (1995). Distraction does not impair memory during intoxication: support for the attention-allocation model. J Stud Alcohol 56: 444–448.
Flannery BA, Volpicelli JR, Pettinati HM (1999). Psychometric properties of the Penn Alcohol Craving Scale. Alcohol Clin Exp Res 23: 1289–1295.
Frank MG, Watkins LR, Maier SF (2011). Stress- and glucocorticoid-induced priming of neuroinflammatory responses: potential mechanisms of stress-induced vulnerability to drugs of abuse. Brain Behav Immun 25 (Suppl 1): S21–S28.
Franklin KM, Hauser SR, Lasek AW, McClintick J, Ding ZM, McBride WJ et al (2015). Reduction of alcohol drinking of alcohol-preferring (P) and high-alcohol drinking (HAD1) rats by targeting phosphodiesterase-4 (PDE4). Psychopharmacology (Berl) 232: 2251–2262.
Giampà C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR (2010). Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington's disease. PLoS ONE 5: e13417.
Gibson LC, Hastings SF, McPhee I, Clayton RA, Darroch CE, Mackenzie A et al (2006a). The inhibitory profile of Ibudilast against the human phosphodiesterase enzyme family. Eur J Pharmacol 538: 39–42.
Gibson LCD, Hastings SF, McPhee I, Clayton RA, Darroch CE, Mackenzie A et al (2006b). The inhibitory profile of Ibudilast against the human phosphodiesterase enzyme family. Eur J Pharmacol 538: 39–42.
He J, Crews FT (2008). Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp Neurol 210: 349–358.
Heilig M, Thorsell A, Sommer WH, Hansson AC, Ramchandani VA, George DT et al (2010). Translating the neuroscience of alcoholism into clinical treatments: from blocking the buzz to curing the blues. Neurosci Biobehav Rev 35: 334–344.
Hensler JG, Ladenheim EE, Lyons WE (2003). Ethanol consumption and serotonin-1A (5-HT1A) receptor function in heterozygous BDNF (+/-) mice. J Neurochem 85: 1139–1147.
Herron AJ, Mariani JJ, Pavlicova M, Parrinello CM, Bold KW, Levin FR et al (2013). Assessment of riboflavin as a tracer substance: comparison of a qualitative to a quantitative method of riboflavin measurement. Drug Alcohol Depend 128: 77–82.
Hughes S, Cohen D, Johnson R (2016). Adverse event assessment methods in published trials of psychotropic drugs: poor reporting and neglect of emerging safety concerns. Int J Risk Saf Med 28: 101–114.
Hutchinson MR, Lewis SS, Coats BD, Skyba DA, Crysdale NY, Berkelhammer DL et al (2009). Reduction of opioid withdrawal and potentiation of acute opioid analgesia by systemic AV411 (ibudilast). Brain Behav Immun 23: 240–250.
Johnson BA (2008). Update on neuropharmacological treatments for alcoholism: scientific basis and clinical findings. Biochem Pharmacol 75: 34–56.
Johnson KW, Matsuda K, Iwaki Y (2014). Ibudilast for the treatment of drug addiction and other neurological conditions. Clin Invest 4: 269–279.
Koob GF (2009). Neurobiological substrates for the dark side of compulsivity in addiction. Neuropharmacology 56 (Suppl 1): 18–31.
Koob GF (2014). Neurocircuitry of alcohol addiction: synthesis from animal models. Handb Clin Neurol 125: 33–54.
Kwako LE, Momenan R, Litten RZ, Koob GF, Goldman D (2016). Addictions Neuroclinical Assessment: a neuroscience-based framework for addictive disorders. Biol Psychiatry 80: 179–189.
Laberg JC, Ellertsen B (1987). Psychophysiological indicators of craving in alcoholics: effects of cue exposure. Br J Addict 82: 1341–1348.
Ledeboer A, Hutchinson MR, Watkins LR, Johnson KW (2007). Ibudilast (AV-411). A new class therapeutic candidate for neuropathic pain and opioid withdrawal syndromes. Expert Opin Investig Drugs 16: 935–950.
Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F (1993). GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 260: 1130–1132.
Litt MD, Cooney NL, Kadden RM, Gaupp L (1990). Reactivity to alcohol cues and induced moods in alcoholics. Addict Behav 15: 137–146.
Litten RZ, Egli M, Heilig M, Cui C, Fertig JB, Ryan ML et al (2012). Medications development to treat alcohol dependence: a vision for the next decade. Addict Biol 17: 513–527.
Lofwall MR, Nuzzo PA, Campbell C, Walsh SL (2014). Aripiprazole effects on self-administration and pharmacodynamics of intravenous cocaine and cigarette smoking in humans. Exp Clin Psychopharmacol 22: 238–247.
Logrip ML, Vendruscolo LF, Schlosburg JE, Koob GF, Zorrilla EP (2014). Phosphodiesterase 10A regulates alcohol and saccharin self-administration in rats. Neuropsychopharmacology 39: 1722–1731.
MacKillop J (2006). Factor structure of the alcohol urge questionnaire under neutral conditions and during a cue-elicited urge state. Alcohol Clin Exp Res 30: 1315–1321.
Martin CS, Earleywine M, Musty RE, Perrine MW, Swift RM (1993). Development and validation of the Biphasic Alcohol Effects Scale. Alcohol Clin Exp Res 17: 140–146.
Mason BJ, Higley AE (2013). A translational approach to novel medication development for protracted abstinence. Curr Top Behav Neurosci 13: 647–670.
Mayfield J, Ferguson L, Harris RA (2013). Neuroimmune signaling: a key component of alcohol abuse. Curr Opin Neurobiol 23: 513–520.
McGough NN, He DY, Logrip ML, Jeanblanc J, Phamluong K, Luong K et al (2004). RACK1 and brain-derived neurotrophic factor: a homeostatic pathway that regulates alcohol addiction. J Neurosci 24: 10542–10552.
McNair DM, Lorr M, Droppleman LF (1971) Manual for the Profile of Mood States. Educational & Industrial Testing Service: San Diego.
Miller AH, Maletic V, Raison CL (2009). Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65: 732–741.
Mizuno T, Kurotani T, Komatsu Y, Kawanokuchi J, Kato H, Mitsuma N et al (2004). Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology 46: 404–411.
Moieni M, Irwin MR, Jevtic I, Olmstead R, Breen EC, Eisenberger NI (2015). Sex differences in depressive and socioemotional responses to an inflammatory challenge: implications for sex differences in depression. Neuropsychopharmacology 40: 1709–1716.
Monti PM, Binkoff JA, Abrams DB, Zwick WR, Nirenberg TD, Liepman MR (1987). Reactivity of alcoholics and nonalcoholics to drinking cues. J Abnorm Psychol 96: 122–126.
Monti PM, Rohsenow DJ, Swift RM, Gulliver SB, Colby SM, Mueller TI et al (2001). Naltrexone and cue exposure with coping and communication skills training for alcoholics: treatment process and 1-year outcomes. Alcohol Clin Exp Res 25: 1634–1647.
Palfreyman MN, Souness JE (1996). Phosphodiesterase type IV inhibitors. Prog Med Chem 33: 1–52.
Raison CL, Miller AH (2011). Is depression an inflammatory disorder? Curr Psychiatry Rep 13: 467–475.
Ramchandani VA, Plawecki M, Li TK, O'Connor S (2009). Intravenous ethanol infusions can mimic the time course of breath alcohol concentrations following oral alcohol administration in healthy volunteers. Alcohol Clin Exp Res 33: 938–944.
Ray LA, Hutchison KE (2004). A polymorphism of the mu-opioid receptor gene (OPRM1) and sensitivity to the effects of alcohol in humans. Alcohol Clin Exp Res 28: 1789–1795.
Ray LA, Hutchison KE, Tartter M (2010). Application of human laboratory models to pharmacotherapy development for alcohol dependence. Curr Pharm Des 16: 2149–2158.
Rohn MCH, Lee MR, Kleuter SB, Schwandt ML, Falk DE, Leggio L (in press). Differences between Treatment-Seeking and Nontreatment-Seeking Alcohol Dependent Research Participants: an Exploratory Analysis. Alcohol Clin Exp Res.
Rohsenow DJ, Monti PM, Rubonis AV, Sirota AD, Niaura RS, Colby SM et al (1994). Cue reactivity as a predictor of drinking among male alcoholics. J Consult Clin Psychol 62: 620–626.
Rolan P, Gibbons JA, He L, Chang E, Jones D, Gross MI et al (2008). Ibudilast in healthy volunteers: safety, tolerability and pharmacokinetics with single and multiple doses. Br J Clin Pharmacol 66: 792–801.
Rubonis AV, Colby SM, Monti PM, Rohsenow DJ, Gulliver SB, Sirota AD (1994). Alcohol cue reactivity and mood induction in male and female alcoholics. J Stud Alcohol 55: 487–494.
Sinha R (2009). Modeling stress and drug craving in the laboratory: implications for addiction treatment development. Addict Biol 14: 84–98.
Sinha R, Fox HC, Hong KA, Bergquist K, Bhagwagar Z, Siedlarz KM (2009). Enhanced negative emotion and alcohol craving, and altered physiological responses following stress and cue exposure in alcohol dependent individuals. Neuropsychopharmacology 34: 1198–1208.
Sinha R, Fuse T, Aubin LR, O'Malley SS (2000). Psychological stress, drug-related cues and cocaine craving. Psychopharmacology (Berl) 152: 140–148.
Sinha R, Lovallo WR, Parsons OA (1992). Cardiovascular differentiation of emotions. Psychosom Med 54: 422–435.
Sullivan JT, Sykora K, Schneiderman J, Naranjo CA, Sellers EM (1989). Assessment of alcohol withdrawal: the revised clinical institute withdrawal assessment for alcohol scale (CIWA-Ar). Br J Addict 84: 1353–1357.
Suzumura A, Ito A, Yoshikawa M, Sawada M (1999). Ibudilast suppresses TNFa production by glial cells functioning mainly as type III phosphodiesterase inhibitor in the CNS. Brain Res 837: 203–212.
Teixeira MM, Gristwood RW, Cooper N, Hellewell PG (1997). Phosphodiesterase (PDE)4 inhibitors: anti-inflammatory drugs of the future? Trends Pharmacol Sci 18: 164–171.
Wen RT, Zhang M, Qin WJ, Liu Q, Wang WP, Lawrence AJ et al (2012). The phosphodiesterase-4 (PDE4) inhibitor rolipram decreases ethanol seeking and consumption in alcohol-preferring fawn-hooded rats. Alcohol Clin Exp Res 36: 2157–2167.
Worley MJ, Heinzerling KG, Roche DJ, Shoptaw S (2016). Ibudilast attenuates subjective effects of methamphetamine in a placebo-controlled inpatient study. Drug Alcohol Depend 162: 245–250.
Wu Y, Lousberg EL, Moldenhauer LM, Hayball JD, Robertson SA, Coller JK et al (2011). Attenuation of microglial and IL-1 signaling protects mice from acute alcohol-induced sedation and/or motor impairment. Brain Behav Immun 25: S155–S164.
Yardley MM, Ray LA (2016). Medications development for the treatment of alcohol use disorder: insights into the predictive value of animal and human laboratory models. Addict Biol.
Acknowledgements
This research was supported by a grant from the National Institute on Alcohol Abuse and Alcoholism (NIAAA) to LAR (R21AA022214). SB was supported by a training grant (F31AA022569). Support for this study was also provided by a grant from the UCLA Clinical and Translational Science Institute (CTSI), grants UL1RR033176 and UL1TR000124. Study medication was provided by Medicinova.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Rights and permissions
About this article

Cite this article
Ray, L., Bujarski, S., Shoptaw, S. et al. Development of the Neuroimmune Modulator Ibudilast for the Treatment of Alcoholism: A Randomized, Placebo-Controlled, Human Laboratory Trial. Neuropsychopharmacol 42, 1776–1788 (2017). https://doi.org/10.1038/npp.2017.10
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2017.10
This article is cited by
-
Molecular mechanisms involved in alcohol craving, IRF3, and endoplasmic reticulum stress: a multi-omics study
Translational Psychiatry (2024)
-
Are medication effects on subjective response to alcohol and cue-induced craving associated? A meta regression study
Psychopharmacology (2023)
-
Additive roles of tobacco and cannabis co-use in relation to delay discounting in a sample of heavy drinkers
Psychopharmacology (2022)
-
Novel Agents for the Pharmacological Treatment of Alcohol Use Disorder
Drugs (2022)
-
The Role of Glia in Addiction: Dopamine as a Modulator of Glial Responses in Addiction
Cellular and Molecular Neurobiology (2022)

{kind=link}