
Abstract
Hypoglutamatergic function may contribute to cognitive impairment in schizophrenia (CIS). Subchronic treatment with the N-methyl-D-aspartate receptor antagonist, phencyclidine (PCP), induces enduring deficits in novel object recognition (NOR) in rodents. Acute treatment with atypical antipsychotic drugs (APDs), which are serotonin (5-HT)2A/dopamine D2 antagonists, but not typical APDs, eg, haloperidol, reverses the PCP-induced NOR deficit in rats. We have tested the ability of lurasidone, an atypical APD with potent 5-HT1A partial agonist properties, tandospirone, a selective 5-HT1A partial agonist, haloperidol, a D2 antagonist, and pimavanserin, a 5-HT2A inverse agonist, to prevent the development of the PCP-induced NOR deficit. Rats were administered lurasidone (0.1 or 1 mg/kg), tandospirone (5 mg/kg), pimavanserin (3 mg/kg), or haloperidol (1 mg/kg) b.i.d. 30 min before PCP (2 mg/kg, b.i.d.) for 7 days (day1–7), followed by a 7-day washout (day8–14). Subchronic treatment with PCP induced an enduring NOR deficit. Lurasidone (1 mg/kg) but not 0.1 mg/kg, which is effective to acutely reverse the deficit due to subchronic PCP, or tandospirone, but not pimavanserin or haloperidol, significantly prevented the PCP-induced NOR deficit on day 15. The ability of lurasidone co-treatment to prevent the PCP-induced NOR deficit was enduring and still present at day 22. The preventive effect of lurasidone was blocked by WAY100635, a selective 5-HT1A antagonists, further evidence for the importance of 5-HT1A receptor stimulation in the NOR deficit produced by subchronic PCP. Further study is needed to determine whether these results concerning mechanism and dosage can be the basis for prevention of the development of CIS in at risk populations.
Similar content being viewed by others

INTRODUCTION
Intervening pharmacologically and in other manners during the prodromal stage of schizophrenia, primarily to prevent the onset of persistent psychosis and enduring cognitive impairment is a major goal of current strategies for reducing the disability associated with schizophrenia (Bilder et al, 1992; Klosterkötter et al, 2001). Intervention during this stage is intended to arrest or attenuate the progression of the underlying pathology (McGlashan and Fenton, 1993; Larson et al, 2010). Treatment of individuals in the prodromal period with atypical antipsychotic drugs (APDs) has been reported to reduce the rate of progression to first-episode psychosis in some high-risk indiviudals (Lee et al, 2005; Lieberman and Fenton, 2000; McGlashan et al, 2006; McGorry et al, 2008; Phillips et al, 2009; Ruhrmann et al, 2005; Salokangas and McGlashan, 2008). However, there are major concerns about this strategy, which include stigmatization, false positives, and metabolic and other side effects of the atypical APDs and stigmatization (Kaur and Cadenhead, 2010).
Even at the time of first diagnosis, deficits in multiple domains of cognition, including visual learning and declarative memory, are present in most patients with schizophrenia and are known to be a key factor leading to impaired work and social function (Saykin et al, 1991; Meltzer and McGurk, 1999). There is evidence that some of the atypical APDs, which are more potent serotonin (5-HT)2A than dopamine (DA) D2 antagonists, including the novel atypical APD, lurasidone, are more effective than typical APDs to attenuate some of these deficits (Hagger et al, 1993; Meltzer and McGurk, 1999; Woodward et al, 2005; Harvey et al, 2011), although not all studies are in accord (Keefe et al, 2007). The development of novel adjunctive or stand alone treatments that can improve some domains of cognition in schizophrenia, accompanied by functional improvement, is currently a major goal of pharmacologic research (Buchanan et al, 2010).
Hypoglutamatergic activity has been postulated to be a major cause of the cognitive impairment in schizophrenia (CIS; Goldman-Rakic and Selemon, 1997; Coyle, 2006). Important evidence that a deficit in glutamatergic function may be the basis for this component of schizophrenia is that the noncompetitive N-methyl-D-aspartate (NMDA) receptor antagonists, such as phencyclidine (PCP), dizocilpine (MK-801), and ketamine, induce schizophrenia-like cognitive impairment in healthy subjects (Javitt and Zukin, 1991; Krystal et al, 1999). The effects of NMDA receptor antagonists on cognitive function in rodents and monkeys have been intensively studied as an animal model of CIS (Gunduz-Bruce, 2009). Acute or subchronic administration of PCP, MK-801, or ketamine to rodents produces cognitive impairments that model CIS, eg, novel object recognition (NOR; Neill et al, 2010, Meltzer et al, 2011). Acute administration of atypical APDs (eg, clozapine), but not the typical APD, haloperidol, has been reported to reverse cognitive deficits induced by subchronic PCP treatment in rat NOR (Grayson et al, 2007; Snigdha et al, 2010; Horiguchi et al, 2011a). We recently reported that selective 5-HT2A inverse agonists (eg, pimavanserin) can potentiate the ability of sub-effective doses of several atypical APDs, including lurasidone, to ameliorate the PCP-induced NOR deficit (Snigdha et al, 2010). However, there are no reports that show a preventive effect of typical or atypical APDs.
Lurasidone is an atypical APD that has D2, 5-HT2A, and 5-HT7 receptor antagonist properties, as well as being a potent 5-HT1A partial agonist (Meyer et al, 2009; Ishibashi et al, 2010). We have recently reported that acute treatment with lurasidone ameliorates the subchronic PCP-induced NOR deficits in a 5-HT1A- and 5-HT7-dependent manner (Horiguchi et al, 2011b; Horiguchi and Meltzer, 2012).
Stimulation of 5-HT1A receptors has been identified as a target for improving CIS, possibly by enhancing the release of cortical DA (Ichikawa et al, 2001, 2002). We have reported that the addition of tandospirone, a 5-HT1A partial agonist (Hamik et al, 1990), to the ongoing treatment with typical APDs of patients with schizophrenia, improved executive function, verbal learning, and memory (Sumiyoshi et al, 2000, 2001a, 2001b). We have also recently reported that acute administration of tandospirone or F15599, another 5-HT1A agonist, improved the NOR deficit induced by subchronic PCP and WAY100635, a 5-HT1A antagonist, blocked the acute attenuating effect of lurasidone (Horiguchi and Meltzer, 2012). These results indicate that acute stimulation of 5-HT1A receptors is adequate to ameliorate the PCP-induced impairment in NOR.
The aim of the current study was to test the preventive effect of lurasidone, tandospirone, haloperidol, a typical APD, and pimavanserin, a 5-HT2A inverse agonist, on the subchronic PCP-induced NOR deficit in rats. We also tested whether WAY100635, a 5-HT1A antagonist, blocks the preventive effect of lurasidone.
MATERIALS AND METHODS
Animals
Thirty-four female Long-Evans (LE) rats (8 or 9 weeks old; Harlan Sprague Dawley, Indianapolis, IN, USA) were used as subjects for experiments 1–2 (rat group 1). Forty-three rats (rat group 2) were used for experiment 3. Twenty-six rats (rat group 3) were used for experiment 4. LE rats were housed in groups of three or four on a 12 h light/dark cycle. Food and water were available ad libitum. All experiments were conducted during the light phase in accordance with the Vanderbilt Animal Committee Regulations.
Drugs
Lurasidone and tandospirone were provided by Dainippon Sumitomo Pharma (Osaka, Japan). Pimavanserin was provided by Acadia Pharmaceuticals (Torrence, CA, USA). Haloperidol was obtained from Sigma-Aldrich (St Louis, MO, USA). WAY100635 was a gift from Wyeth Laboratories (Philadelphia, PA). PCP was supplied as a generous gift from the National Institute of Drug Abuse (Bethesda, MD, USA).
Lurasidone was dissolved in 0.5% methylcellulose, 0.2% Tween80. The other drugs were dissolved in distilled water. All drugs or vehicle were administered intraperitoneally (i.p.) in a volume of 1 ml/kg body.
Drug Treatment
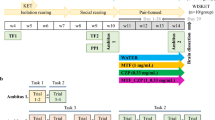
LE rats (rat group 1) were randomly assigned to four groups. For each of these groups, the first injection (A) was given 30 min before the second injection (B) twice daily for 7 days (day1–7). Group 1 (control group), A; vehicle (saline), B; vehicle (saline). Group 2 (subchronic PCP group), A; vehicle, B; PCP (2 mg/kg). Group 3 (lurasidone 0.1 mg/kg+PCP group), A; lurasidone (0.1 mg/kg), B; PCP (2 mg/kg). Group 4 (lurasidone 1 mg/kg+PCP group), A; lurasidone (1 mg/kg), B; PCP (2 mg/kg).
Rat group 2 were randomly assigned to five groups. For each of these groups, the first injection (A) was given 30 min before second injection (B) twice daily for 7 days (day1–7). Group 1 (control group), A; vehicle, B; vehicle. Group 2 (subchronic PCP group), A; vehicle, B; PCP (2 mg/kg). Group 3 (tandospirone 5 mg/kg+PCP group), A; tandospirone (5 mg/kg), B; PCP (2 mg/kg). Group 4 (haloperidol 1 mg/kg+PCP group), A; haloperidol (1 mg/kg), B; PCP (2 mg/kg). Group 5 (pimavanserin 3 mg/kg+PCP group), A; pimavanserin (3 mg/kg), B; PCP (2 mg/kg).
Rat group 3 were randomly assigned to three groups. For each of these groups, the first injection (A) and second injection (B) were given 45 min or 30 min, respectively, before the third injection (C) twice daily for 7 days (day1–7). Group 1 (control group), A; vehicle, B; vehicle, C; vehicle. Group 2 (subchronic PCP group), A; vehicle, B; vehicle, C; PCP (2 mg/kg). Group 3 (lurasidone 1 mg/kg+WAY100635 0.6 mg/kg+PCP group), A; WAY100635 (0.6 mg/kg), B; lurasidone (1 mg/kg), C; PCP (2 mg/kg).
Subsequently, animals were given a 7-day washout period (days 8–14) before NOR testing.
NOR Test
Testing was carried out according to a previously validated method (Snigdha et al, 2010). Briefly, all rats were habituated for 1 h to the NOR arena for three consecutive days (days 12–14) before the first NOR test. Rats were given a further 3-min habituation on the day of testing (days 15 or 21). After the 3-min habituation, the rats were given two 3-min trials (an acquisition trial and a retention trial) separated by a 1-min intertrial return to their home cage. During the acquisition trial, the animals were allowed to explore two identical objects (A1 and A2). During the retention trial, the animals explored a familiar object (A) from the acquisition trial and a novel object (B).
Behavior was recorded on video for blind scoring of object exploration. Object exploration is defined as an animal licking, sniffing, or touching the object with the forepaws while sniffing. The exploration time (s) of each object in each trial was recorded manually by the use of two stopwatches. The discrimination index (DI) [(time spent exploring the novel object−time spent exploring the familiar object)/total exploration time] was then calculated for retention trials.
If the exploration time in the aquisition or retention trials to either of two objects was <5 s, the data were excluded from analysis. This rarely occurred and did not affect the ability to complete the analysis using the data from the remaining animals of that group. All experimental groups consisted of six to nine rats.
Data Analysis
All data are expressed as the mean±SEM (n=6–9 per group). Exploration data were analyzed by a two-way ANOVA followed by the pairwise comparison when a significant effect was detected by the ANOVA. This analysis was used to detect the interaction of drug treatment and object exploration as well as main effects. When a significant effect was found, further analysis by a post hoc Student's t-test was performed to compare the time spent exploring the novel and familiar object. DI data were analyzed using one-way ANOVA followed by the Bonferroni test when a significant effect was detected by the ANOVA.
RESULTS
Effect of Co-Administration of Lurasidone on Subchronic PCP-Induced NOR Deficit on Day 15 (Experiment 1)
In the aquisition trial, no significant differences in time spent exploring the two identical objects were observed in any group (F7,54=0.9, p>0.05, Figure 1a). There were no significant effects of drugs on exploratory activity during the acquisition trial period (F5,42=0.64, p>0.05 for Supplementary Figure 1, F9,66=0.6, p>0.05 for Supplementary Figure 2, F5,44=0.48, p>0.05 for Supplementary Figure 3) in any of the experiments (Supplementary Figures 1–3). In the retention phase, the time spent exploring the novel vs familiar objects was significantly different among the groups (F7,54=3.8, p<0.005, Figure 1b). Using post-hoc analysis, vehicle-treated animals explored the novel object significantly longer than the familiar object (p<0.05, Figure 1b). The ability to discriminate novel and familiar objects was abolished by subchronic PCP treatment. Lurasidone (0.1 mg/kg) failed to prevent this deficit (Figure 1b). However, lurasidone (1 mg/kg) significantly attenuated the NOR deficit (p<0.005, Figure 1b). The model examining the group effect on DI was statistically significant (F3,27=5.7, p<0.005, Figure 1c). In the post-hoc analysis, the DI was significantly reduced following subchronic PCP-treatment (p<0.01). Co-administration of 1 mg/kg lurasidone significantly prevented the PCP-induced reduction in DI (p<0.01), but 0.1 mg/kg lurasidone did not (Figure 1c).

Effect of co-treatment with lurasidone (LUR, 0.1, 1 mg/kg) on PCP-induced cognitive impairment in NOR test on day 15. (a) Effect of LUR (0.1, 1 mg/kg, i.p.) on exploration of two identical objects in the aquisition trial in NOR test on day 15. Data are shown as mean±SEM (n=6–9 per group). (b) Effect of LUR (0.1, 1 mg/kg, i.p.) on exploration of a novel and a familiar object in the retention trial in NOR test on day 15. Data are shown as mean±SEM (n=6–9 per group). ***p<0.001, *p<0.05, significant difference in time spent exploring the novel compared with the familiar object. (c) Effect of LUR (0.1, 1 mg/kg, i.p.) on the DI on day 15. Data are shown as mean±SEM (n=6–9 per group). **p<0.01, significant decrease in DI compared with the vehicle. ##p<0.01, significant reversal in DI compared with PCP group.
Effect of Co-Administration of Lurasidone on Subchronic PCP-Induced NOR Deficit on Day 22 (Experiment 2)
In the retention trial, the time spent exploring the novel vs familiar objects was significantly different among the groups (F5,42=7.8, p<0.005, Figure 2a). The post-hoc analysis revealed that vehicle-treated animals showed preference for the novel object (p<0.005, Figure 2a). PCP-treated rats did not show preference for the novel object (Figure 2a). Co-treatment with lurasidone (1 mg/kg) significantly attenuated the PCP-induced deficit (p<0.005, Figure 2a) demonstrating an enduring preventive effect on the PCP-induced NOR deficit. Statistical analysis showed the model examing the group effect on DI was significant (F2,21=14.8, p<0.005, Figure 2b). Examing the post-hoc test, it was revealed that subchronic PCP-treatment significantly reduced the DI (p<0.005, Figure 2b). Co-administration of lurasidone (1 mg/kg) prevented the reduction of DI (p<0.005, Figure 2b).

Effect of co-treatment with lurasidone (LUR, 0.1, 1 mg/kg) on PCP-induced cognitive impairment in NOR test on day22. (a) Effect of LUR (0.1, 1 mg/kg, i.p.) on exploration of a novel and a familiar object in the retention trial in NOR test on day 22. Data are shown as mean±SEM (n=7–9 per group). ***p<0.001, significant difference in time spent exploring the novel compared with the familiar object. (b) Effect of LUR (0.1, 1 mg/kg, i.p.) on the DI on day 22. Data are shown as mean±SEM (n=7–9 per group). ***p<0.001, significant decrease in DI compared with the vehicle. ###p<0.001, significant reversal in DI compared with PCP group.
Effect of Co-Administration of Tandospirone, Haloperidol, or Pimavanserin on Subchronic PCP-Induced NOR Deficit on Day 15 (Experiment 3)
In the retention trial, the time spent exploring the novel vs familiar objects was significantly different among the groups (F9,66=3.8, p<0.005, Figure 3a). Using post-hoc analysis, it was found vehicle-treated rats showed exploratory preference for the novel object (p<0.005, Figure 3a). In PCP-treated rats, there was no significant difference between the time spent exploring the novel and the familiar object (Figure 3a). Tandospirone (5 mg/kg) significantly prevented the PCP-induced NOR deficit (p<0.04; Figure 3a). Neither haloperidol 1 mg/kg nor pimavanserin 3 mg/kg prevented this deficit (Figure 3a). The model examining the group effect on DI was statistically significant (F4,33=4.5, p<0.01, Figure 3b). In the post-hoc analysis, the DI was significantly reduced following subchronic PCP-treatment (p<0.005). Co-treatment with 5 mg/kg tandospirone (p<0.05), but not 1 mg/kg haloperidol or 3 mg/kg pimavanserin, significantly improved the DI reduction (Figure 3b).

Effect of co-treatment with tandospirone (TAN, 5 mg/kg), haloperidol (HAL, 1 mg/kg), and pimavanserin (PIM, 3 mg/kg) on PCP-induced cognitive impairment in NOR test on day15. (a) Effect of TAN, HAL, and PIM (5, 1, and 3 mg/kg, respectively, i.p.) on exploration of a novel and a familiar object in the retention trial in NOR test on day 15. Data are shown as mean±SEM (n=7–9 per group). ***p<0.001, *p<0.05, significant difference in time spent exploring the novel compared with the familiar object. (b) Effect of TAN, HAL, and PIM (5, 1, and 3 mg/kg, respectively, i.p.) on the DI on day 15. Data are shown as mean±SEM (n=7–9 per group). ***p<0.001, significant decrease in DI compared with the vehicle. #p<0.05, significant reversal in DI compared with PCP group.
Effect of Co-Administration of WAY100635 Plus Lurasidone on Subchronic PCP-Induced NOR Deficit on Day 15 (Experiment 4)
In the retention trial, the time spent exploring the novel vs familiar objects was significantly different among the groups (F5,44=3.0, p<0.02, Figure 4a). Using post-hoc analysis, it was found that the vehicle-treated rats showed preference for the novel object (p<0.005); this preference was abolished by subchronic PCP-treatment (Figure 4a). WAY100635 (0.6 mg/kg) plus lurasidone (1 mg/kg) did not prevent this deficit (Figure 4a). The model examing the group effect on DI was statistically significant (F2,22=4.4, p<0.05, Figure 4b). In the post-hoc analysis, the DI was significantly reduced following subchronic PCP-treatment (p<0.05). Treatment with 0.6 mg/kg WAY100635 and 1 mg/kg lurasidone did not improve the reduction in the DI (Figure 4b).

Effect of co-treatment with WAY100635 (WAY, 0.6 mg/kg) plus lurasidone (LUR, 1 mg/kg) on PCP-induced cognitive impairment in NOR test on day 15. (a) Effect of WAY (0.6 mg/kg, i.p.) plus LUR (1 mg/kg, i.p.) on exploration of a novel and a familiar object in the retention trial in NOR test on day 15. Data are shown as mean±SEM (n=7–9 per group). ***p<0.001, significant difference in time spent exploring the novel compared with the familiar object. (b) Effect of WAY (0.6 mg/kg, i.p.) plus LUR (1 mg/kg, i.p.) on the DI on day 15. Data are shown as mean±SEM (n=7–9 per group). *p<0.05, significant decrease in DI compared with the vehicle.
DISCUSSION
NOR is a possible analog of declarative memory in humans (Winters et al, 2010) and a frequently studied model of CIS. In this study, we again confirmed that subchronic administration of PCP produces a severe deficit in NOR and that the co-treatment with lurasidone, an atypical APD with 5-HT1A partial agonism, or with tandospirone, a selective 5-HT1A agonist, prevented the NOR deficit induced by subchronic PCP. On the other hand, haloperidol, a typical antipsychotic with selective D2 antagonist properties, or pimavanserin, a selective 5-HT2A inverse agonist, did not prevent the PCP-induced deficit in NOR.
This is the first demonstration of the preventive effect of an atypical APD (lurasidone), or a 5-HT1A partial agonist (tandospirone) on subchronic PCP-induced disruption in NOR in rats. We also found that the preventive effect of lurasidone lasted at least 14 days after the final PCP injection (on day 22). It has been reported that lurasidone has cognitive benefits in some animal models of CIS, in which other APDs were ineffective or even worsened the impairment in cognition (Ishiyama et al, 2007; Enomoto et al, 2008). Lurasidone was reported to be somewhat more effective than ziprasidone to improve general cognitive function in patients with schizophrenia in a double-blind study of 21 days duration (Harvey et al, 2011). In the present experiment, co-administration of lurasidone, 0.1 mg/kg, did not show the preventive effect, although acute treatment with this dose of lurasidone can reverse the subchronic PCP-induced NOR deficit (Horiguchi et al, 2011a, 2011b). Acute treatment with lurasidone, 0.1 mg/kg, is sufficient to temporarily reverse the subchronic PCP-induced NOR deficit 30 min after administration, but when co-administration with subchronic PCP, this dose is insufficient to prevent the cognitive impairment in NOR induced by the subchronic PCP regimen. These results are inconsistent with several other preclinical reports using risperidone, suggest that lower doses of risperidone might be effective in some animal models of psychosis (Piontkewitz et al, 2011; Richtand et al, 2006). Thus, risperidone, at a dose of 0.045 mg/kg per day attenuated the abnormally elevated locomotor response to amphetamine following hippocampal lesions and behavioral abnormalities in the offsprings of poly I:C mother mice (Piontkewitz et al, 2011; Richtand et al, 2006). Our results suggest that for lurasidone or other atypical APDs higher doses than those needed to prevent recurrence of psychosis might be needed to impact CIS.
The results in this study are consistent with the acute reversal studies in which atypical APDs, including lurasidone and the 5-HT1A agonist, tandospirone, but not pimavanserin or haloperidol, are effective reversing PCP-induced cognitive deficits (Grayson et al, 2007; Snigdha et al, 2010; Horiguchi et al, 2011a, 2011b; Horiguchi and Meltzer, 2012). It is of interest to compare these results from acute reversal studies with studies in mice that showed a subchronic PCP (10 mg/kg)-induced NOR deficit in mice and examined the ability of subsequent subchronic administration of APDs to reverse the impairment (Hashimoto et al, 2005; Hagiwara et al, 2008; Nagai et al, 2009; Tanibuchi et al, 2009). This deficit was recovered by subsequent subchronic (14 days) administration of clozapine (5 mg/kg, i.p.), but not haloperidol (0.1 mg/kg, i.p.; Hashimoto et al, 2005). This deficit was also subsequently improved by subchronic treatment with perospirone or aripiprazole both of which are 5-HT1A partial agonists (Hagiwara et al, 2008; Nagai et al, 2009). Tanibuchi et al (2009) reported that subsequent treatment with quetiapine, another atypical APD with 5-HT1A partial agonism, also reversed the subchronic PCP-induced deficit in mice. On the other hand, in rat NOR, McKibben et al (2010) reported that treatment with risperidone (0.5 mg/kg, i.p.) twice daily for 10 days, beginning 3 days before the start of PCP administration (2 mg/kg, i.p., b.i.d. for 7 days), did not show a protective effect against the NOR deficit induced by subchronic PCP. More studies with other atypical APDs are needed to better understand the role of atypical APDs on cognitive impairments in NOR induced by subchronic PCP. These results suggest that at least some atypical APDs (eg, lurasidone) may be effective to prevent the development of cognitive impairmant in individuals who at high risk for schizophrenia.
Stimulation of 5-HT1A receptors has been identified as a target for improving CIS (Meltzer, 1999). In this study, not only lurasidone but also the 5-HT1A agonist, tandospirone, showed the preventive effect on subchronic PCP-induced NOR deficit. Moreover, WAY100635, a selective 5-HT1A antagonist, blocked the preventive effect of lurasidone, thereby demonstrating the involvement of 5-HT1A agonism in the effect of lurasidone. As mentioned above, these results are consistent with the acute studies with 5-HT1A agonists in this model (Horiguchi and Meltzer, 2012). These data suggest that tandospirone by itself or as an add on treatment with an atypical APD might have value to prevent the development of CIS. The 5-HT1A agonists, eg, tandospirone, have a lower side effect burden than most atypical APDs, especially of the metabolic variety (Feighner and Boyer, 1989). It is noteworthy that lurasidone shares important structural similarities with tandospirone, and that lurasidone is also a 5-HT1A partial agonist (Meltzer et al, 2011).
Postmortem studies have reported that the density of 5-HT1A receptors is increased in frontal and temporal cortices in schizophrenia (Burnet et al, 1996, 1997; Gurevich and Joyce, 1997; Hashimoto et al, 1991; Simpson et al, 1996; Sumiyoshi et al, 1996). Positron emission tomography studies confirm an increase in cortical 5-HT1A receptor binding in schizophrenia (Kasper et al, 2002; Tauscher et al, 2002). Subchronic treatment with PCP has been reported to increase 5-HT1A receptor binding in the medial- and dorsolateral-frontal cortex (Choi et al, 2009). Microdialysis studies report that acute administration of PCP increases cortical 5-HT release (Etou et al, 1998; Martin et al, 1998; Millan et al, 1999; Adams and Moghaddam, 2001; Amargós-Bosch et al, 2006). This effect is blocked by clozapine and olanzapine but not haloperidol (Amargós-Bosch et al, 2006). It is possible that lurasidone and tandospirone, through their 5-HT1A agonist properties, suppress cortical 5-HT release, thereby blocking effects of PCP related to 5-HT release that lead to interference with NOR.
Haloperidol and pimavanserin did not show a preventive effect in this model. As mentioned above, these results are in agreement with the lack of effectiveness of these drugs to acutely reverse the effects of subchronic PCP on NOR (Grayson et al, 2007; Snigdha et al, 2010). Subchronic treatment with haloperidol also did not reverse the NOR deficit induced by subchronic PCP in mice (Hashimoto et al, 2005; Nagai et al, 2009). Haloperidol (1 mg/kg) has been reported to reverse PCP-induced morphologic deficits in the auditory system, but not to reverse PCP-induced decreases in prefrontal cortical GABAergic interneurons in rats (Cochran et al, 2003). Pimavanserin (3 mg/kg) has been shown to achieve essentially 100% 5-HT2A receptor occupancy (Vanover et al, 2006) and potentiated the ability of sub-effective doses of atypical APDs to reverse the NOR deficit induced by subchronic PCP (Snigdha et al, 2010). It is noteworthy that haloperidol and pimavanserin effectively block acute NMDA receptor antagonist (eg, PCP, MK-801)-induced hyperlocomotion, considered a model of psychosis (Maurel-Remy et al, 1995; Vanover et al, 2006; Gardell et al, 2007). Ritanserin, another 5-HT2A inverse agonist, was able to block the ability of PCP to increase cortical 5-HT efflux (Amargós-Bosch et al, 2006). Co-administration of haloperidol or pimavanserin with PCP was ineffective in restoring performance in the NOR test in subchronic PCP-treated rats, indicating that D2 or 5-HT2A receptor blockade alone is insufficient to reverse cognitive impairment induced by subchronic PCP treatment, unlike the blockade of the effect of acute PCP or MK-801 on locomotor activity.
It has been reported that subchronic PCP induces a reduction in the density of paravalbumin-containing GABAergic interneurons in the hippocampus in both male and female rats (Abdul-Monim et al, 2007; Jenkins et al, 2008). A number of groups have conducted studies investigating the neuroprotective effects of atypical APDs. Chronic PCP treatment decreases parvalbumin mRNA expression and chronic administration of clozapine, but not haloperidol, reversed the PCP-induced decreases in parvalbumin mRNA expression in prefrontal cortical GABAergic interneurons (Cochran et al, 2003). Chronic treatment with olanzapine, but not haloperidol, has been reported to slow volume loss in the prefrontal cortex in a prefrontal cortical dopamine denervation model of schizophrenia (Wang and Deutch, 2008). Some clinical reports also suggest some atypical APDs may have a neuroprotective effect. A study of patients with first-episode schizophrenia reported that basal ganglia volume is increased in patients treated with risperidone (Massana et al, 2005). Treatment with risperidone increased the volume of gray matter in neuroleptic-naive patients (Molina et al, 2005). Further studies with lurasidone and tandospirone on the subchronic PCP-induced GABAergic interneuron deficit could provide valuable insight into the mechanism of subchronic PCP to induce cognitive impairment in rat NOR.
Female rats were used in this study primarily because they have been found to perform significantly better than male rats in the NOR task (Sutcliffe et al, 2007). Further, no effect of estrous cycle on performance in the NOR task has been found (Sutcliffe et al, 2007). Finally, female rats have been shown to differ in pharmacokinetics of PCP metabolism rendering them more sensitive to PCP than males, because of slower metabolism and higher PCP tissue levels (Nabeshima et al, 1984).
In conclusion, these results indicate that lurasidone or tandospirone, but not haloperidol, a typical APD, nor pimavanserin, may prevent the development of cognitive impairment in individuals who are at the risk for schizophrenia or related disorders with cognitive impairment, eg, bipolar disorder.
References
Abdul-Monim Z, Neill JC, Reynolds GP (2007). Sub-chronic psychotomimetic phencyclidine induces deficits in reversal learning and alterations in parvalbumin-immunoreactive expression in the rat. J Psychopharmacol 21: 198–205.
Adams BW, Moghaddam B (2001). Effect of clozapine, haloperidol, or M100907 on phencyclidine-activated glutamate efflux in the prefrontal cortex. Biol Psychiatry 50: 750–757.
Amargós-Bosch M, López-Gil X, Artigas F, Adell A (2006). Clozapine and olanzapine, but not haloperidol, suppress serotonin efflux in the medial prefrontal cortex elicited by phencyclidine and ketamine. Int J Neuropsychopharmacol 9: 565–573.
Bilder RM, Lipschutz-Broch L, Reiter G, Geisler SH, Mayerhoff DI, Lieberman JA (1992). Intellectual deficits in first-episode schizophrenia: evidence for progressive deterioration. Schizophr Bull 18: 437–448.
Buchanan RW, Keefe RS, Umbricht D, Green MF, Laughren T, Marder SR (2010). The FDA-NIMH-MATRICS guidelines for clinical trial design of cognitive-enhancing drugs: what do we know 5 years later? Schizophr Bull 37: 1209–1217.
Burnet PW, Eastwood SL, Harrison PJ (1996). 5-HT1A and 5-HT2A receptor mRNAs and binding site densities are differentially altered in schizophrenia. Neuropsychopharmacology 15: 442–455.
Burnet PW, Eastwood SL, Harrison PJ (1997). [3H]WAY-100635 for 5-HT1A receptor autoradiography in human brain: a comparison with [3H]8-OH-DPAT and demonstration of increased binding in the frontal cortex in schizophrenia. Neurochem Int 30: 565–574.
Choi YK, Snigdha S, Shahid M, Neill JC, Tarazi FI (2009). Subchronic effects of phencyclidine on dopamine and serotonin receptors: implications for schizophrenia. J Mol Neurosci 38: 227–235.
Cochran SM, Kennedy M, McKerchar CE, Steward LJ, Pratt JA, Morris BJ (2003). Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation by antipsychotic drugs. Neuropsychopharmacology 28: 265–275.
Coyle JT (2006). Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol 26: 365–384.
Enomoto T, Ishibashi T, Tokuda K, Ishiyama T, Toma S, Ito A (2008). Lurasidone reverses MK-801-induced impairment of learning and memory in the Morris water maze and radial-arm maze tests in rats. Behav Brain Res 186: 197–207.
Etou K, Kuroki T, Kawahara T, Yonezawa Y, Tashiro N, Uchimura H (1998). Ceruletide inhibits phencyclidine-induced dopamine and serotonin release in rat prefrontal cortex. Pharmacol Biochem Behav 61: 427–434.
Feighner JP, Boyer WF (1989). Serotonin-1A anxiolytics: an overview. Psychopathology 22 (Suppl 1): 21–26.
Gardell LR, Vanover KE, Pounds L, Johnson RW, Barido R, Anderson GT et al. (2007). ACP-103, a 5-hydroxytryptamine 2A receptor inverse agonist, improves the antipsychotic efficacy and side-effect profile of haloperidol and risperidone in experimental models. J Pharmacol Exp Ther 322: 862–870.
Goldman-Rakic PS, Selemon LD (1997). Functional and anatomical aspects of prefrontal pathology in schizophrenia. Schizophr Bull 23: 437–458.
Grayson BI, Idris NF, Neill JC (2007). Atypical antipsychotics attenuate a sub-chronic PCP-induced cognitive deficit in the novel object recognition task in the rat. Behav Brain Res 184: 31–38.
Gunduz-Bruce H (2009). The acute effects of NMDA antagonism: from the rodent to the human brain. Brain Res Rev 60: 279–286.
Gurevich EV, Joyce JN (1997). Alterations in the cortical serotonergic system in schizophrenia: a postmortem study. Biol Psychiatry 42: 529–545.
Hagger C, Buckley P, Kenny JT, Friedman L, Ubogy D, Meltzer HY (1993). Improvement in cognitive functions and psychiatric symptoms in treatment-refractory schizophrenic patients receiving clozapine. Biol Psychiatry 34: 702–712.
Hagiwara H, Fujita Y, Ishima T, Kunitachi S, Shirayama Y, Iyo M et al. (2008). Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of the antipsychotic drug perospirone: role of serotonin 5-HT1A receptors. Eur Neuropsychopharmacol 18: 448–454.
Hamik A, Oksenberg D, Fischette C, Peroutka SJ (1990). Analysis of tandospirone (SM-3997) interactions with neurotransmitter receptor binding sites. Biol Psychiatry 28: 99–109.
Harvey PD, Ogasa M, Cucchiaro J, Loebel A, Keefe RS (2011). Performance and interview-based assessments of cognitive change in a randomized, double-blind comparison of lurasidone vs. ziprasidone. Schizophr Res 127: 188–194.
Hashimoto K, Fujita Y, Shimizu E, Iyo M (2005). Phencyclidine-induced cognitive deficits in mice are improved by subsequent sub-chronic administration of clozapine, but not haloperidol. Eur J Pharmacol 519: 114–117.
Hashimoto T, Nishino N, Nakai H, Tanaka C (1991). Increase in serotonin 5-HT1A receptors in prefrontal and temporal cortices of brains from patients with chronic schizophrenia. Life Sci 48: 355–363.
Horiguchi M, Huang M, Meltzer HY (2011a). Interaction of mGlu(2/3) agonism with clozapine and lurasidone to restore novel object recognition in subchronic phencyclidine-treated rats. Psychopharmacology (Berl) 217: 13–24.
Horiguchi M, Huang M, Meltzer HY (2011b). The role of 5-HT7 receptors in the phencyclidine (PCP)-induced novel object recognition (NOR) deficit in rats. J Pharmacol Exp Ther 338: 605–614.
Horiguchi M, Meltzer HY (2012). The role of 5-HT1A receptors in phencyclidine (PCP)-induced novel object recognition (NOR) deficit in rats. Psychopharmacology (Berl) 221: 205–215.
Ichikawa J, Ishii H, Bonaccorso S, Fowler WL, O′Laughlin IA, Meltzer HY (2001). 5-HT(2A) and D(2) receptor blockade increases cortical DA release via 5-HT(1A) receptor activation: a possible mechanism of atypical antipsychotic-induced cortical dopamine release. J Neurochem 76: 1521–1531.
Ichikawa J, Li Z, Dai J, Meltzer HY (2002). Atypical antipsychotic drugs, quetiapine, iloperidone, and melperone, preferentially increase dopamine and acetylcholine release in rat medial prefrontal cortex: role of 5-HT1A receptor agonism. Brain Res 956: 349–357.
Ishibashi T, Horisawa T, Tokuda K, Ishiyama T, Ogasa M, Tagashira R et al. (2010). Pharmacological profile of lurasidone, a novel antipsychotic agent with potent 5-HT7 and 5-HT1A receptor activity. J Pharmacol Exp Ther 334: 171–181.
Ishiyama T, Tokuda K, Ishibashi T, Ito A, Toma S, Ohno Y (2007). Lurasidone (SM-13496), a novel atypical antipsychotic drug, reverses MK-801-induced impairment of learning and memory in the rat passive-avoidance test. Eur J Pharmacol 572: 160–170.
Javitt DC, Zukin SR (1991). Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry 148: 1301–1308.
Jenkins TA, Harte MK, McKibben CE, Elliott JJ, Reynolds GP (2008). Disturbances in social interaction occur along with pathophysiological deficits following sub-chronic phencyclidine administration in the rat. Behav Brain Res 194: 230–235.
Kasper S, Tauscher J, Willeit M, Stamenkovic M, Neumeister A, Küfferle B et al. (2002). Receptor and transporter imaging studies in schizophrenia, depression, bulimia and Tourette′s disorder—implications for psychopharmacology. World J Biol Psychiatry 3: 133–146.
Kaur T, Cadenhead KS (2010). Treatment implications of the schizophrenia prodrome. Curr Top Behav Neurosci 4: 97–121.
Keefe RSE, Bilder RM, Davis SM, Harvey PD, Palmer BW, Gold JM et al. (2007). Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE trial. Arch Gen Psychiatry 64: 633–647.
Klosterkötter J, Hellmich M, Steinmeyer EM, Schultze-Lutter F (2001). Diagnosing schizophrenia in the initial prodromal phase. Arch Gen Psychiatry 58: 158–164.
Krystal JH, D′Souza DC, Petrakis IL, Belger A, Berman RM, Charney DS et al. (1999). NMDA agonists and antagonists as probes of glutamatergic dysfunction and pharmacotherapies in neuropsychiatric disorders. Harv Rev Psychiatry 7: 125–143.
Larson MK, Walker EF, Compton MT (2010). Early signs, diagnosis and therapeutics of the prodromal phase of schizophrenia and related psychotic disorders. Expert Rev Neurother 10: 1347–1359.
Lee C, McGlashan TH, Woods SW (2005). Prevention of schizophrenia: can it be achieved? CNS Drugs 19: 193–206.
Lieberman JA, Fenton WS (2000). Delayed detection of psychosis: causes, consequences, and effect on public health. Am J Psychiatry 157: 1727–1730.
Martin P, Carlsson ML, Hjorth S (1998). Systemic PCP treatment elevates brain extracellular 5-HT: a microdialysis study in awake rats. Neuroreport 9: 2985–2988.
Massana G, Salgado-Pineda P, Junqué C, Pérez M, Baeza I, Pons A et al. (2005). Volume changes in gray matter in first-episode neuroleptic-naive schizophrenic patients treated with risperidone. J Clin Psychopharmacol 25: 111–117.
Maurel-Remy S, Bervoets K, Millan MJ (1995). Blockade of phencyclidine-induced hyperlocomotion by clozapine and MDL 100,907 in rats reflects antagonism of 5-HT2A receptors. Eur J Pharmacol 280: R9–R11.
McGlashan TH, Fenton WS (1993). Subtype progression and pathophysiologic deterioration in early schizophrenia. Schizophr Bull 19: 71–84.
McGlashan TH, Zipursky RB, Perkins D, Addington J, Miller T, Woods SW et al. (2006). Randomized, double-blind trial of olanzapine versus placebo in patients prodromally symptomatic for psychosis. Am J Psychiatry 163: 790–799.
McGorry PD, Killackey E, Yung A (2008). Early intervention in psychosis: concepts, evidence and future directions. World Psychiatry 7: 148–156.
McKibben CE, Jenkins TA, Adams HN, Harte MK, Reynolds GP (2010). Effect of pretreatment with risperidone on phencyclidine-induced disruptions in object recognition memory and prefrontal cortex parvalbumin immunoreactivity in the rat. Behav Brain Res 208: 132–136.
Meltzer HY (1999). The role of serotonin in antipsychotic drug action. Neuropsychopharmacology 21 (2 Suppl): 106S–115S.
Meltzer HY, Horiguchi M, Massey BW (2011). The role of serotonin in the NMDA receptor antagonist models of psychosis and cognitive impairment. Psychopharmacology (Berl) 213: 289–305.
Meltzer HY, McGurk SR (1999). The effects of clozapine, risperidone, and olanzapine on cognitive function in schizophrenia. Schizophr Bull 25: 233–255.
Meyer JM, Loebel AD, Schweizer E (2009). Lurasidone: a new drug in development for schizophrenia. Expert Opin Investig Drugs 18: 1715–1726.
Millan MJ, Brocco M, Gobert A, Joly F, Bervoets K, Rivet J et al. (1999). Contrasting mechanisms of action and sensitivity to antipsychotics of phencyclidine versus amphetamine: importance of nucleus accumbens 5-HT2A sites for PCP-induced locomotion in the rat. Eur J Neurosci 11: 4419–4432.
Molina V, Reig S, Sanz J, Palomo T, Benito C, Sánchez J et al. (2005). Increase in gray matter and decrease in white matter volumes in the cortex during treatment with atypical neuroleptics in schizophrenia. Schizophr Res 80: 61–71.
Nabeshima T, Yamaguchi K, Yamada K, Hiramatsu M, Kuwabara Y, Furukawa H et al. (1984). Sex-dependent differences in the pharmacological actions and pharmacokinetics of phencyclidine in rats. Eur J Pharmacol 97: 217–227.
Nagai T, Murai R, Matsui K, Kamei H, Noda Y, Furukawa H et al. (2009). Aripiprazole ameliorates phencyclidine-induced impairment of recognition memory through dopamine D1 and serotonin 5-HT1A receptors. Psychopharmacology (Berl) 202: 315–328.
Neill JC, Barnes S, Cook S, Grayson B, Idris NF, McLean SL et al. (2010). Animal models of cognitive dysfunction and negative symptoms of schizophrenia: focus on NMDA receptor antagonism. Pharmacol Ther 128: 419–432.
Phillips LJ, Nelson B, Yuen HP, Francey SM, Simmons M, Stanford C et al. (2009). Randomized controlled trial of interventions for young people at ultra-high risk of psychosis: study design and baseline characteristics. Aust N Z J Psychiatry 43: 818–829.
Piontkewitz Y, Arad M, Weiner I (2012). Tracing the development of psychosis and its prevention: what can be learned from animal models. Neuropharmacology 62: 1273–1289.
Richtand NM, Taylor B, Welge JA, Ahlbrand R, Ostrander MM, Burr J et al. (2006). Risperidone pretreatment prevents elevated locomotor activity following neonatal hippocampal lesions. Neuropsychopharmacology 31: 77–89.
Ruhrmann S, Schultze-Lutter F, Maier W, Klosterkötter J (2005). Pharmacological intervention in the initial prodromal phase of psychosis. Eur Psychiatry 20: 1–6.
Salokangas RK, McGlashan TH (2008). Early detection and intervention of psychosis. A review. Nord J Psychiatry 62: 92–105.
Saykin AJ, Gur RC, Gur RE, Mozley PD, Mozley LH, Resnick SM et al. (1991). Neuropsychological function in schizophrenia. Selective impairment in memory and learning. Arch Gen Psychiatry 48: 618–624.
Simpson MD, Lubman DI, Slater P, Deakin JF (1996). Autoradiography with [3H]8-OH-DPAT reveals increases in 5-HT(1A) receptors in ventral prefrontal cortex in schizophrenia. Biol Psychiatry 39: 919–928.
Snigdha S, Horiguchi M, Huang M, Li Z, Shahid M, Neill JC et al. (2010). Attenuation of phencyclidine-induced object recognition deficits by the combination of atypical antipsychotic drugs and pimavanserin (ACP 103), a 5-hydroxytryptamine(2A) receptor inverse agonist. J Pharmacol Exp Ther 332: 622–631.
Sumiyoshi T, Matsui M, Yamashita I, Nohara S, Uehara T, Kurachi M et al. (2000). Effect of adjunctive treatment with serotonin-1A agonist tandospirone on memory functions in schizophrenia. J Clin Psychopharmacol 20: 386–388.
Sumiyoshi T, Matsui M, Nohara S, Yamashita I, Kurachi M, Sumiyoshi C et al. (2001a). Enhancement of cognitive performance in schizophrenia by addition of tandospirone to neuroleptic treatment. Am J Psychiatry 158: 1722–1725.
Sumiyoshi T, Matsui M, Yamashita I, Nohara S, Kurachi M, Uehara T et al. (2001b). The effect of tandospirone, a serotonin(1A) agonist, on memory function in schizophrenia. Biol Psychiatry 49: 861–868.
Sumiyoshi T, Stockmeier CA, Overholser JC, Dilley GE, Meltzer HY (1996). Serotonin1A receptors are increased in postmortem prefrontal cortex in schizophrenia. Brain Res 708: 209–214.
Sutcliffe JS, Marshall KM, Neill JC (2007). Influence of gender on working and spatial memory in the novel object recognition task in the rat. Behav Brain Res 177: 117–125.
Tanibuchi Y, Fujita Y, Kohno M, Ishima T, Takatsu Y, Iyo M et al. (2009). Effects of quetiapine on phencyclidine-induced cognitive deficits in mice: a possible role of alpha1-adrenoceptors. Eur Neuropsychopharmacol 19: 861–867.
Tauscher J, Kapur S, Verhoeff NP, Hussey DF, Daskalakis ZJ, Tauscher-Wisniewski S et al. (2002). Brain serotonin 5-HT(1A) receptor binding in schizophrenia measured by positron emission tomography and [11C]WAY-100635. Arch Gen Psychiatry 59: 514–520.
Vanover KE, Weiner DM, Makhay M, Veinbergs I, Gardell LR, Lameh J et al. (2006). Pharmacological and behavioral profile of N-(4-fluorophenylmethyl)-N-(1-methylpiperidin-4-yl)-N′-(4-(2-methylpropyloxy)phenylmethyl) carbamide (2R,3R)-dihydroxybutanedioate (2:1) (ACP-103), a novel 5-hydroxytryptamine(2A) receptor inverse agonist. J Pharmacol Exp Ther 317: 910–918.
Wang HD, Deutch AY (2008). Dopamine depletion of the prefrontal cortex induces dendritic spine loss: reversal by atypical antipsychotic drug treatment. Neuropsychopharmacology 33: 1276–1286.
Winters BD, Saksida LM, Bussey TJ (2010). Implications of animal object memory research for human amnesia. Neuropsychologia 48: 2251–2261.
Woodward ND, Purdon SE, Meltzer HY, Zald DH (2005). A meta-analysis of neuropsychological change to clozapine, olanzapine, quetiapine, and risperidone in schizophrenia. Int J Neuropsychopharmacol 8: 457–472.
Acknowledgements
This research was supported, in part, by a grant from Dainippon Sumitomo Pharma Co., Ltd. We are grateful to National Institute of Drug Abuse for supplying some of the PCP used in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Herbert Y Meltzer is a stockholder of ACADIA and SureGene. He has received grant support in the last 3 years from BioLine Rx, Cephalon, Dainippon Sumitomo, Eli Lilly, EnVivo, Janssen, Otsuka, Pfizer, and Sunovion. He is, or has been, a consultant to ACADIA, Alkemes, Astellas, Boehringer Mannheim, Bristol Myers Squibb, Cypress, Janssen, Lundbeck, Ovation, Merck, Novartis, Pfizer, Teva, and Valeant (BioVail). The research effort of HYM is supported, in part, by donations from the Hintz, Peterson and Weisman families. Masakuni Horiguchi: employed as a research scientist by Dainippon Sumitomo Pharma. The remaining authors have no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Rights and permissions
About this article
Cite this article
Horiguchi, M., Hannaway, K., Adelekun, A. et al. Prevention of the Phencyclidine-Induced Impairment in Novel Object Recognition in Female Rats by Co-Administration of Lurasidone or Tandospirone, a 5-HT1A Partial Agonist. Neuropsychopharmacol 37, 2175–2183 (2012). https://doi.org/10.1038/npp.2012.64
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2012.64
Keywords
This article is cited by
-
MEF2C transcription factor is associated with the genetic and epigenetic risk architecture of schizophrenia and improves cognition in mice
Molecular Psychiatry (2018)
-
TPA-023 attenuates subchronic phencyclidine-induced declarative and reversal learning deficits via GABAA receptor agonist mechanism: possible therapeutic target for cognitive deficit in schizophrenia
Neuropsychopharmacology (2018)
-
Effects of 2-bromoterguride, a dopamine D2 receptor partial agonist, on cognitive dysfunction and social aversion in rats
Psychopharmacology (2018)
-
The antipsychotic-like effects in rodents of the positive allosteric modulator Lu AF21934 involve 5-HT1A receptor signaling: mechanistic studies
Psychopharmacology (2015)
-
The subchronic phencyclidine rat model: relevance for the assessment of novel therapeutics for cognitive impairment associated with schizophrenia
Psychopharmacology (2015)
