
Abstract
Cobalt-catalysed sp2 C–H bond functionalization has attracted considerable attention in recent years because of the low cost of cobalt complexes and interesting modes of action in the process. In comparison, much less efforts have been devoted to the sp3 carbons. Here we report the cobalt-catalysed site-selective dehydrogenative cyclization of aliphatic amides via a C–H bond functionalization process on unactivated sp3 carbons with the assistance of a bidentate directing group. This method provides a straightforward synthesis of monocyclic and spiro β- or γ-lactams with good to excellent stereoselectivity and functional group tolerance. In addition, a new procedure has been developed to selectively remove the directing group, which enables the synthesis of free β- or γ-lactam compounds. Furthermore, the first cobalt-catalysed intermolecular dehydrogenative amination of unactivated sp3 carbons is also realized.
Similar content being viewed by others
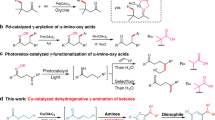

Introduction
Transition metal-catalysed direct functionalization of relatively unreactive C–H bonds has emerged as a major topic of research in organic chemistry1,2,3,4,5,6,7,8,9,10,11,12,13. This method does not require the use of prefunctionalized materials, and thus provides an attractive alternative to traditional cross-coupling reactions. Within this reaction class, cobalt-catalysed processes have received special interest due to the low cost and toxicity of cobalt complexes, and their interesting modes of action14,15,16,17. In the 1950s, Murahashi et al. demonstrated the chelation-assisted C–H functionalization process on benzaldimines and azobenzenes, and it is well accepted that the catalytic cycle is initiated by oxidative addition of a low-valent cobalt species to the aromatic C–H bonds18,19,20,21. Recently, azoles, benzamides and 2-phenylpyridines were also proven to be effective substrates through a similar reaction pathway22,23,24,25,26. Furthermore, CoII or CoIII-catalysed direct C–H functionalization of azole, 2-phenylpyridine, indole and benzamide derivatives was also demonstrated27,28,29,30. In this case, the C–H bond activation process is believed to proceed through either an electrophilic aromatic substitution or concerted metalation-deprotonation pathway. Moreover, the cobalt-catalysed hydroacylation of olefins has also been demonstrated via an sp2 C–H functionalization process in the absence of chelation assistance14,31.
In comparison with the well-established cobalt-catalysed direct functionalization on sp2 carbons, there are only a few examples of direct functionalization on sp3 C–H bonds (Fig. 1). Cenini and co-workers reported the intermolecular amination of relatively reactive sp3 carbons with moderate yields in 1999 (refs 32, 33). Recently, more efficient intramolecular version of this transformation was developed on electron-deficient sp3 carbons in Zhang’s laboratory (Fig. 1a)34,35. These reactions were proposed to proceed via the outer-sphere mechanism, in which a carbon–metal bond is not involved5,36,37. Instead, the sp3 C–H bonds were indirectly activated by an inter- or intramolecular hydrogen atom transfer of the radical intermediates. In contrast to this, Brookhart and co-workers reported intramolecular hydrogen transfer of cyclic amines in 2007 via an inner-sphere mechanism, in which a carbon–cobalt bond was formed by oxidative addition of a cobalt species to the α-sp3 C–H bond (Fig. 1b)36,37,38,39,40.

(a) Out-sphere mechanism. (b) Inner-sphere mechanism (via oxidative addition to a C–H bond). (c) Inner-sphere mechanism (via cyclometalation of an sp3 carbon).
Inspired by the reports of transition metal-catalysed bidentate ligand-directed sp3 C–H functionalization process41,42, we have explored and demonstrated here the cobalt-catalysed site-selective direct C–H functionalization on unactivated sp3 carbons with the aid of a bidentate directing group (Fig. 1c). In addition, a novel two-step procedure has been developed under oxidative conditions to remove the directing group, which enables an efficient access to β-lactam, γ-lactam or β-amino amide derivatives.
Results
Reaction condition optimization of intramolecular amidation
Synthesis of lactams via transition metal-catalysed C–H functionalization is of current research interest because of the biological importance of these molecules43,44. In the past two years, Pd-, Cu- or Ni-catalysed process for the formation of β- or γ-lactams has been achieved45,46,47,48,49,50,51. However, all of these approaches suffer from their own limitations on the substrate scope. To provide a complementary method and demonstrate the feasibility of cobalt catalysis on unactivated sp3 carbons, we carried out the study of cobalt-catalysed bidentate ligand-directed intramolecular cyclization of propanamides. Our investigation began with oxidative cyclization of 2-ethyl-2-methyl-N-(quinolin-8-yl)pentanamide (1a) in the presence of catalytic amount of CoCl2 by using Ag2CO3 as the oxidant (Table 1). After an initial solvent screening, chlorobenzene turned out to be optimal, producing the β-lactam compound 2a in 33% yield (entry 1). Notably, this reaction proceeded in a highly site-selective manner, favouring the C–H bond of a β-methyl group over those of β-methylene and γ-methyl groups. Next, a screening on oxidants was carried out. It was observed that the reaction could also be performed with several other oxidants with lower efficiency (entries 2–4). Further optimization of reaction conditions showed that the reaction could be significantly improved by using Co(OAc)2 as the catalyst and sodium benzoate as the base (entry 15). Considering that PhCO2Na could potentially compete with amide 1 for coordination to the cobalt complex, we further reduced the loading of this base. Delightfully, the chemical yield of this reaction was significantly improved (entry 17).
Substrate scope of intramolecular amination
With optimized conditions in hand, the substrate scope studies were carried out (Fig. 2; also see Supplementary Figs 1 and 2 for the structures of substrates). Gratifyingly, the reaction showed great generality with 2,2-disubstituted propanamides bearing either linear or cyclic chains on α-carbons with predominant selectivity for C–H bonds of β-methyl groups (2b, 2d–j). However, with α-phenyl substituted substrates, a preference of C–H bond functionalization of sp2 carbons was observed, providing indolin-2-one derivative as the major products (2k and 2l). Noticeably, the replacement of the quinolyl group with 5-methoxyquinolyl group had no apparent effect on the reaction (2c). Moreover, the removability of 5-methoxyquinolyl moiety of β- or γ-lactams has been well documented45,48,49,50,51.

Reaction conditions: 1 (0.3 mmol), Co(OAc)2 (0.03 mmol), Ag2CO3 (0.75 mmol), PhCO2Na (0.15 mmol), 0.6 ml PhCl, 150 °C, 24 h. Isolated yield based on three runs of each reaction. aRun for 48 h. bCo(OAc)2 (0.06 mmol). Q=8-quinolinyl.
Furthermore, substrates bearing a trifluoromethyl, cyano, ethoxycarbonyl, sulfonyl or phthalimidyl group on an α-carbon showed good compatibility (2m–q). In addition, although α-methoxy and acetoxy-substituted amides failed to provide the desired products (2r), substrates with an acetoxy or benzenecarboxy group on β-carbons produced β-lactams 2s and 2t in good yields. It was also noticed that the reaction favours the C–H bond of the β-methyl over that of the more reactive benzyl group (2u). Moreover, the β-benzylic sp3 C–H bonds could also be effectively functionalized (2v-ab).
Next, we carried out compatibility studies of α-monosubstituted propanamide derivatives (Fig. 3; also see Supplementary Figs 1 and 2 for the structures of substrates). To our delight, introduction of a relatively bulky group on the α-carbon could effectively initiate the process (3a–k). Furthermore, excellent diastereoselectivity was observed with β-phenyl substituted substrates (3g–k).

Reaction conditions: same as in Fig. 2. Isolated yield based on three runs of each reaction.
Interestingly, a great preference of functionalizing the C–H bonds of γ-benzylic carbons over those of β-methyl carbons was observed during the course of substrate scope studies, providing γ-lactams as the major products (Fig. 4; also see Supplementary Figs 1 and 2 for the structures of substrates). However, α-monosubstituted substrates failed to provide any γ-lactams (4d–e).

Reaction conditions: same as in Fig. 2. Isolated yield based on three runs of each reaction. aUnisolated diastereoisomers.
Mechanistic investigation
To gain some insights on the mechanism of this reaction, the deuterium-labelling experiments were carried out (Fig. 5). With the deuterium-labelled compound [D3]-1d, an apparent deuterium-proton exchange occurred with both the substrate and product (Fig. 5a). More interestingly, the product has a lower deuterium ratio compared with the recovered starting material, which is presumably due to the different reaction rates of proton- and deuterium-containing starting materials. Furthermore, a primary kinetic isotope effect was also observed for 1d based on the early relative rate of parallel reactions (see Supplementary Methods), indicating that the sp3 C–H bond cleavage of amide 1d is the rate-limiting step in the catalytic process (Fig. 5b).

(a) The deuterium–proton exchange experiment. (b) The kinetic isotope effect experiments.
We then carried out a series of control experiments with 2-ethyl-2-methyl-N-(quinolin-8-yl)butanamide (1d). As shown in Table 2, the reaction failed to provide the desired product without a cobalt catalyst under the standard or modified conditions based on Shi’s study (entries 2 and 3)52. It was then noticed that the oxidant, Ag2CO3, could be replaced with Ce(SO4)2, albeit with a low yield (entries 4 and 5). Furthermore, no desired product 2d was obtained with stoichiometric amounts of commercially available Co(acac)3 or CoF3 in the absence of Ag2CO3 (entries 6 and 7). On the other hand, the reaction could be performed with a catalytic amount of Co(acac)3 or CoF3 in the presence of Ag2CO3 (entries 8 and 9). These results suggest that the C–H bond activation process could be initiated by a CoIII species28,29,30, but the product is unlikely generated from reductive elimination of a CoIII complex. It was also observed that addition of the radical inhibitor, TEMPO, had no significant effect on the reaction, indicating that a radical intermediate may not be involved in the catalytic cycle (entries 10 and 11).
Next, a series of control experiments with 1-phenethyl-N-(quinolin-8-yl)cyclohexane-1-carboxamide (1d) were carried out to explore the plausible reaction pathway for the formation of γ-lactams (Supplementary Table 1). It was found that neither a cobalt nor a silver species is required for this reaction (entries 2–5). However, the efficiency of the reaction was significantly decreased without these species. Furthermore, reaction yield was dramatically decreased by the addition of TEMPO, which indicates that an alkyl radical intermediate generated from a single electron oxidation process may be involved in the reaction (entries 6 and 7).
On the basis of the above results, a plausible catalytic cycle for the formation of β-lactams is proposed (Fig. 6)14,15,16,17,53,54. The CoIII complex A is initially generated by coordination of amide 1 to a cobalt species followed by a ligand exchange process under basic conditions. Cyclometalation of this intermediate produces the intermediate B, which is believed to be an irreversible step based on the kinetic isotope effect studies. In this process, benzoate might act as a ligand to coordinate to the CoIII complex, and subsequently facilitates the C–H bond cleavage via concerted metallation-deprotonation7,55,56. Oxidation of intermediate B with Ag2CO3 gives rise to the CoIV complex C, which produces the β-lactam compound 2 upon reductive elimination57,58. The newly generated CoII species could then be re-oxidized to the CoIII species to furnish the catalytic cycle. It is noteworthy that the catalytic CoII/CoIV cycle could not be excluded, which involves cyclometalation of amide 1 with a CoII species followed by oxidation to generate the intermediate C. It should also be mentioned that a competing side reaction, protonation of the CoIV complex C, is also possible in the process, giving the CoIV species D. Furthermore, although a radical-mediated process could not be excluded, the observed high selectivity of the β-methyl over the β-benzylic C–H bonds suggests that the catalytic cycle is unlikely performed with a radical intermediate. However, in the case of the formation of γ-lactam derivatives, a radical or cationic species is believed to be involved in the catalytic cycle because of the predominant preference of functionalization of the γ-benzylic over the β-methyl C–H bonds.

The possible mechanism involves the Co-catalysed sp3 C–H activation, oxidation and subsequent reductive elimination.
To broaden the synthetic applications of this method, we carried out studies on the selective removal of the directing group (Fig. 7). It was found that the quinolyl group could be cleaved by the introduction of a methoxy group on the C5 position of this moiety under oxidative conditions followed by oxidative cleavage of the newly generated 5-methoxyquinolyl moiety with ammonium cerium(IV) nitrate (CAN). Under these conditions, α-mono and α,α-di-substituted β-lactams, and α,α-di-substituted γ-lactams were all effective substrates, which enables the efficient synthesis of free β- or γ-lactam compounds.

Reaction conditions: (1) amide (0.15 mmol), BF3·Et2O (0.30 mmol), PhI(OAc)2 (0.45 mmol), MeOH (0.5 ml), 80 °C, 3–8 h; (2) CAN (0.45 mmol), MeCN/H2O(2.0 ml/0.4 ml), room temperature, 6 h. Isolated yield based on three runs of each reaction. aThe methoxylation step was carried out at 50 °C.
Intermolecular amination
Direct intermolecular amination of sp3 carbons is an important research topic because of the importance of the products in pharmaceutical industry5,59,60,61. As one of the most efficient synthetic methods, the transition metal-catalysed ligand-directed approach has attracted considerable attention and significant progress has been achieved in recent years62,63,64,65,66,67. However, within this reaction category, reports of dehydrogenative aminations are rare62,63 Encouraged by the above results, we carried out the study of a cobalt-catalysed intermolecular dehydrogenative amination of N-(quinolin-8-yl)propanamide derivatives (Fig. 8; also see Supplementary Figs 1 and 2 for the structures of substrates). After an extensive screening, trifluoroacetamide was proved to be an effective coupling partner (5a), whereas many other nitrogen sources such as acetamide, benzamide, phathalimide, sulfonamide, morpholine and aniline failed to produce the desired products (for optimization of reaction conditions, see Supplementary Table 2). Furthermore, replacement of trifluoroacetamide with heptafluorobutanamide significantly improved the reaction (5b). As expected, 2,2-disubstituted propanamides bearing either linear or cyclic chains on α-carbons proceeded smoothly to give the corresponding products 5c–j with a predominant selectivity for C–H bonds of β-methyl groups. It was also noticed that with α-phenyl-substituted substrate 1k, C–H bond functionalization of sp2 carbons was favoured, providing indolin-2-one derivative as the major product (5k1 and 2k2).

Reaction conditions: 1 (0.15 mmol), amide (0.45 mmol), Co(acac)3 (0.03 mmol), Ag2CO3 (0.45 mmol), K2HPO4 (0.23 mmol), B(OH)3 (0.075 mmol), 2.0 ml PhCF3, 160 °C, 24 h. Isolated yield based on three runs of each reaction.
We then carried out a series of control experiments with 2-ethyl-2-methyl-N-(quinolin-8-yl)butanamide (1d) to gain some insights on the reaction mechanism. As shown in Supplementary Table 3, the reaction failed to provide the desired product in the absence of a cobalt or silver species (entries 2–4). Furthermore, the addition of TEMPO had no apparent effect on the reaction, indicating that an alkyl radical intermediate may not be involved in this process (entries 5 and 6).
Discussion
As described, a highly regioselective intramolecular amination of propionamide and butyramide derivatives with an 8-aminoquinolinyl group as the bidentate directing group was developed via a cobalt-catalysed sp3 C–H bond functionalization process. The reaction favours the C–H bonds of β-methyl groups over those of the β-methylene and γ- or δ-methyl groups, providing the β-lactam derivatives in a highly site- and diastereo-selective manner. Interestingly, a predominant preference for the functionalization of γ-benzylic over β-methyl C–H bonds was observed, producing γ-lactams as the major products. On the basis of these results, it is believed that two distinct reaction pathways are involved in the formation of these four- and five-membered ring products. As mentioned earlier, synthesis of lactams has been demonstrated recently via a Pd-, Cu- or Ni-catalysed C–H functionalization process. However, the Cu- or Ni-catalysed process is restricted to substrates with an α-quaternary carbon and the formation of β-lactams. On the other hand, Pd-catalysed synthesis of β-lactams either is restricted to α-unsubstituted or α,β-cyclic substrates, or suffers from the irremovability of the directing group, whereas synthesis of γ-lactams is limited to β-substituted substrates. Therefore, this method provides a complementary approach to access monocyclic and spiro β- or γ-lactams. Furthermore, the cobalt-catalysed ligand-directed intermolecular amination of unactivated sp3 carbons was realized for the first time. The detailed mechanistic investigations of these transformations are currently undergoing in our laboratory. In the meanwhile, N-phosphonyl and phosphinyl groups will be investigated for the present intra- and intermolecular dehydrogenative amination reactions for achieving the GAP work-up68,69.
Methods
General methods
For 1H and 13C NMR spectra of compounds in this manuscript and details of the synthetic procedures, see Supplementary Figs 3–162 and Supplementary Methods.
General procedure for intramolecular amination
A 20-ml tube was charged with α,α,α-trisubstituted N-(quinolin-8-yl)acetamides (1, 0.30 mmol), Co(OAc)2 (5.3 mg, 0.030 mmol), Ag2CO3 (207 mg, 0.75 mmol), PhCO2Na (21.6 mg, 0.15 mmol) and 0.60 ml of PhCl. The reaction mixture was stirred rigorously open to the air at 150 °C for 24 h. Then, the mixture was cooled to room temperature, diluted with EtOAc (2 ml), filtered through a celite pad and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (gradient eluent of 2–5% EtOAc in hexanes, v/v) to give the desired product.
General procedure for intermolecular amination
A 20-ml tube was charged with α,α,α-trisubstituted N-(quinolin-8-yl)acetamides (1, 0.15 mmol), heptafluorobutyramide (95.9 mg, 0.45 mmol), Co(acac)3 (10.7 mg, 0.030 mmol), Ag2CO3 (75.1 mg, 0.45 mmol), K2HPO4 (39.2 mg, 0.23 mmol), B(OH)3 (4.6 mg, 0.075 mmol), 3 Å MS (200 mg) and 2.0 ml of PhCF3. Then the vial was sealed, and stirred rigorously at 160 °C for 24 h. The mixture was cooled to room temperature, diluted with CH2Cl2 (5 ml), filtered through a celite pad and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (gradient eluent of 2–5% EtOAc in hexanes, v/v) to give the desired product.
Additional information
How to cite this article: Wu, X. et al. Cobalt-catalysed site-selective intra- and intermolecular dehydrogenative amination of unactivated sp3 carbons. Nat. Commun. 6:6462 doi: 10.1038/ncomms7462 (2015).
References
Daugulis, O., Do, H.-Q. & Shabashov, D. Palladium- and copper-catalyzed arylation of carbon-hydrogen bonds. Acc. Chem. Res. 42, 1074–1086 (2009) .
Chen, X., Engle, K. M., Wang, D.-H. & Yu, J.-Q. Palladium(II)-catalyzed C-H activation/C-C cross-coupling reactions: versatility and practicality. Angew. Chem. Int. Ed. 48, 5094–5115 (2009) .
Colby, D. A., Bergman, R. G. & Ellman, J. A. Rhodium-catalyzed C−C bond formation via heteroatom-directed C–H bond activation. Chem. Rev. 110, 624–655 (2010) .
Lyons, T. W. & Sanford, M. S. Palladium-catalyzed ligand-directed C-H functionalization reactions. Chem. Rev. 110, 1147–1169 (2010) .
Jazzar, R., Hitce, J., Renaudat, A., Sofack-Kreutzer, J. & Baudoin, O. Functionalization of organic molecules by transition-metal-catalyzed C(sp3)-H activation. Chem. Eur. J. 16, 2654–2672 (2010) .
Yeung, C. S. & Dong, V. M. Catalytic dehydrogenative cross-coupling: forming carbon−carbon bonds by oxidizing two carbon−hydrogen bonds. Chem. Rev. 111, 1215–1292 (2011) .
Ackermann, L. Carboxylate-assisted transition-metal-catalyzed C–H bond functionalizations: mechanism and scope. Chem. Rev. 111, 1315–1345 (2011) .
Davis, H. M. L., Du Bois, J. & Yu, J.-Q. C-H functionalization in organic synthesis. Chem. Soc. Rev. 40, 1855–1856 (2011) .
Gutekunst, W. R. & Baran, P. S. C-H functionalization logic in total synthesis. Chem. Soc. Rev. 40, 1976–1991 (2011) .
Hartwig, J. F. Regioselectivity of the borylation of alkanes and arenes. Chem. Soc. Rev. 40, 1992–2002 (2011) .
White, M. C. Adding aliphatic C-H bond oxidations to synthesis. Science 335, 807–809 (2012) .
Yamaguchi, J., Yamaguchi, A. D. & Itami, K. C-H bond functionalization: emerging synthetic tools for natural products and pharmaceuticals. Angew. Chem. Int. Ed. 51, 8960–9009 (2012) .
Collins, K. & Glorius, F. A robustness screen for the rapid assessment of chemical reactions. Nat. Chem 5, 597–601 (2013) .
Hess, W., Treutwein, J. & Hilt, G. Cobalt-catalysed carbon-carbon bond-formation reactions. Synthesis 3537–3562 (2008) .
Kulkarni, A. A. & Daugulis, O. Direct conversion of carbon-hydrogen into carbon-carbon bonds by first-row transition-metal catalysis. Synthesis 4087–4109 (2009) .
Yoshikai, N. Chiral-auxiliary-controlled diastereoselective epoxidations. Synlett 1047–1051 (2011) .
Gao, K. & Yoshikai, N. Low-valent cobalt catalysis: new opportunities for C-H functionalization. Acc. Chem. Res. 47, 1208–1219 (2014) .
Murahashi, S. Synthesis of phthalimidines from schiff bases and carbon monoxide. J. Am. Chem. Soc. 77, 6403–6404 (1955) .
Murahashi, S. & Horiie, S. The reaction of azobenzene and carbon monoxide. J. Am. Chem. Soc. 78, 4816–4817 (1956) .
Funk, J., Yennawar, H. & Sen, A. Cobalt-catalyzed carbonylation of N-alkylbenzaldimines to 'N-alkylphthalimidines' (2,3-dihydro-1H-isoindol-1-ones) via tandem C-H activation and cyclocarbonylation. Helv. Chim. Acta 89, 1687–1695 (2006) .
Lee, P.-S., Fujita, T. & YoshikaI, N. Cobalt-catalyzed, room-temperature addition of aromatic imines to alkynes via directed C-H bond activation. J. Am. Chem. Soc. 133, 17283–17295 (2011) .
Gao, K., Lee, P.-S., Fujita, T. & Yoshikai, N. Cobalt-catalyzed hydroarylation of alkynes through chelation-assisted C–H bond activation. J. Am. Chem. Soc. 132, 12249–12251 (2010) .
Ding, Z.-H. & Yoshikai, N. Cobalt-catalyzed addition of azoles to alkynes. Org. Lett. 12, 4180–1283 (2010) .
Chen, Q., Ilies, L. & Nakamura, E.-I. Cobalt-catalyzed ortho-alkylation of secondary benzamide with alkyl chloride through directed C–H bond activation. J. Am. Chem. Soc. 133, 428–429 (2011) .
Li, B. et al. Direct cross-coupling of C-H bonds with Grignard reagents through cobalt catalysis. Angew. Chem. Int. Ed. 50, 1109–1113 (2011) .
Gao, K. & YoshikaI, N. Cobalt-catalyzed ortho-alkylation of aromatic imines with primary and secondary alkyl halides. J. Am. Chem. Soc. 135, 9279–9282 (2013) .
Yao, T., Hirano, K., Satoh, T. & Miura, M. Nickel- and cobalt-catalyzed direct alkylation of azoles with N-tosylhydrazones bearing unactivated alkyl groups. Angew. Chem. Int. Ed. 51, 775–779 (2012) .
Yoshino, T., Ikemoto, H., Matsunaga, S. & Kanai, M. A cationic high-valent Cp*CoIII complex for the catalytic generation of nucleophilic organometallic species: directed C-H bond activation. Angew. Chem. Int. Ed. 52, 2207–2211 (2013) .
Ikemoto, H., Yoshino, T., Sakata, K., Matsunaga, S. & Kanai, M. Pyrroloindolone synthesis via a Cp*CoIII-catalyzed redox-neutral directed C-H alkenylation/annulation sequence. J. Am. Chem. Soc. 136, 5424–5431 (2014) .
Grigorjeva, L. & Daudulis, O. Cobalt-catalyzed, aminoquinoline-directed C(sp2)-H bond alkenylation by alkynes. Angew. Chem. Int. Ed. 53, 10209–10212 (2014) .
Lenges, C. P., White, P. S. & Brookhart, M. Mechanistic and synthetic studies of the addition of alkyl aldehydes to vinylsilanes catalyzed by Co(I) complexes. J. Am. Chem. Soc. 120, 6965–6979 (1998) .
Cenini, S., Tollari, S., Penoni, A. & Cereda, C. Catalytic amination of unsaturated hydrocarbons: reactions of p-nitrophenyl azide with alkenes catalyzed by metalloporphyrins. J. Mol. Catal 137, 135–146 (1999) .
Ragaini, F. et al. Amination of benzylic C-H bonds by arylazides catalyzed by CoII-porphyrin complexes: a synthetic and mechanistic study. Chem. Eur. J. 9, 249–259 (2003) .
Lu, H.-J. & Zhang, X. P. Catalytic C-H functionalization by metalloporphyrins: recent developments and future directions. Chem. Soc. Rev. 40, 1899–1909 (2011) .
Lu, H.-J., Li, C.-Q., Jiang, H.-L., Lizardi, C. L. & Zhang, X. P. Chemoselective amination of propargylic C(sp3)-H bonds by cobalt(II)-based metalloradical catalysis. Angew. Chem. Int. Ed. 53, 7028–7032 (2014) .
Dick, A. R. & Sanford, M. S. Transition metal catalyzed oxidative functionalization of carbon-hydrogen bonds. Tetrahedron 62, 2439–2463 (2006) .
Boutadla, Y., Davies, D. L., Macgregor, S. A. & Poblador-Bahamonde, A. I. Mechanisms of C-H bond activation: rich synergy between computation and experiment. Dalton Trans. 5820–5831 (2009) .
Bolig, A. D. & Brookhart, M. Activation of sp3 C-H bonds with cobalt(I): catalytic synthesis of enamines. J. Am. Chem. Soc. 129, 14544–14545 (2007) .
Hung-Low, F., Krogman, J. P., Tye, J. W. & Bradley, C. A. Development of more labile low electron count Co(I) sources: mild, catalytic functionalization of activated alkanes using a [(Cp*Co)2-μ-(η4:η4-arene)] complex. Chem. Commun. 48, 368–370 (2012) .
Lyons, T. W. & Brookhart, M. Cobalt-catalyzed hydrosilation/hydrogen-transfer cascade reaction: a new route to silyl enol ethers. Chem. Eur. J. 19, 10124–10127 (2013) .
Corbet, M. & De Campo, F. 8-Aminoquinoline: a powerful directing group in metal-catalyzed direct functionalization of C-H bonds. Angew. Chem. Int. Ed. 52, 9896–9898 (2013) .
Rouquet, G. & Chatani, N. Catalytic functionalization of C(sp2)-H and C(sp3)-H bonds by using bidentate directing groups. Angew. Chem. Int. Ed. 52, 11726–11743 (2013) .
Tahlan, K. & Jensen, S. E. J. Origins of the β-lactam rings in natural products. Antibiotic 66, 401–410 (2013) .
Ye, L.-W., Shu, C. & Gagosz, F. Recent progress towards transition metal-catalyzed synthesis of γ-lactams. Org. Biomol. Chem. 12, 1833–1845 (2014) .
He, G., Zhang, S.-Y., Nack, W. A., Li, Q. & Chen, G. Use of a readily removable auxiliary group for the synthesis of pyrrolidones by the palladium-catalyzed intramolecular amination of unactivated γ C(sp3)-H bonds. Angew. Chem. Int. Ed. 52, 11124–11128 (2013) .
Zhang, Q. et al. Stereoselective synthesis of chiral α-amino-β-lactams through palladium(II)-catalyzed sequential monoarylation/amidation of C(sp3)-H bonds. Angew. Chem. Int. Ed. 52, 13588–13592 (2013) .
McNally, A., Haffemayer, B., Collins, B. S. & Gaunt, M. J. Palladium-catalysed C-H activation of aliphatic amines to give strained nitrogen heterocycles. Nature 510, 129–133 (2014) .
Sun, W.-W. et al. Palladium-catalyzed unactivated C(sp3)-H bond activation and intramolecular amination of carboxamides: a new approach to β-lactams. Org. Lett. 16, 480–483 (2014) .
Wang, Z., Ni, J.-Z., Kuninobu, Y. & Kanai, M. Copper-catalyzed intramolecular C(sp3)-H and C(sp2)-H amidation by oxidative cyclization. Angew. Chem. Int. Ed. 53, 3496–3499 (2014) .
Wu, X.-S., Zhao, Y., Zhang, G.-W. & Ge, H.-B. Copper-catalyzed site-selective intramolecular amidation of unactivated C(sp3)-H bonds. Angew. Chem. Int. Ed. 53, 3706–3710 (2014) .
Wu, X.-S., Zhao, Y. & Ge, H.-B. Nickel-catalyzed site-selective amidation of unactivated C(sp3)-H bonds. Chem. Eur. J. 20, 9530–9533 (2014) .
Yang, M. et al. Silver-catalysed direct amination of unactivated C-H bonds of functionalized molecules. Nat. Commun 5, 4707–4712 (2014) .
Lewis, R. A. et al. Synthesis of a cobalt(IV) ketimide with a squashed tetrahedral geometry. Chem. Commun. 49, 2888–2890 (2013) .
Nguyen, A. I., Hadt, R. G., Solomon, E. I. & Tilley, T. D. Efficient C-H bond activations via O2 cleavage by a dianionic cobalt(II) complex. Chem. Sci. 5, 2874–2878 (2014) .
Théveau, L. et al. Mechanism selection for regiocontrol in base-assisted, palladium-catalysed direct C-H coupling with halides: first approach for oxazole- and thiazole-4-carboxylates. Chem. Eur. J. 17, 14450–14463 (2011) .
Aihara, Y. & Chatani, N. Nickel-catalyzed direct arylation of C(sp3)-H bonds in aliphatic amides via bidentate-chelation assistance. J. Am. Chem. Soc. 136, 898–901 (2014) .
Brunschwig, B. S., Chou, M.-H., Creutz, C., Ghosh, P. & Sutin, N. Mechanisms of water oxidation to oxygen: cobalt(IV) as an intermediate in the aquocobalt(II)-catalyzed reaction. J. Am. Chem. Soc. 105, 4832–4833 (1983) .
Anson, F. C., Collins, T. J., Coots, R. J., Gipson, S. L. & Richmond, T. G. Synthesis and characterization of stable cobalt(IV) coordination complexes: molecular structure of trans-[η4-1,2-bis(3,5-dichloro-2-hydroxybenzamido)-4,5-dichlorobenzene]bis(4-tert-butylpyridine)cobalt(IV). J. Am. Chem. Soc. 106, 5037–5038 (1984) .
Collet, F., Dodd, R. H. & Dauban, P. Catalytic C-H amination: recent progress and future directions. Chem. Commun. 5061–5074 (2009) .
Ramirez, T. A., Zhao, B. & Shi, Y. Recent advances in transition metal-catalyzed sp3 C-H amination adjacent to double bonds and carbonyl groups. Chem. Soc. Rev. 41, 931–942 (2012) .
Jeffrey, J. L. & Sarpong, R. Intramolecular C(sp3)-H amination. Chem. Sci 4, 4092–4106 (2013) .
Thu, H. Y., Yu, W. Y. & Che, C. M. Intermolecular amidation of unactivated sp2 and sp2 C-H bonds via palladium-catalyzed cascade C-H activation/nitrene insertion. J. Am. Chem. Soc. 128, 9048–9049 (2006) .
Pan, J., Su, M. & Buchwald, S. L. Palladium(0)-catalyzed intermolecular amination of unactivated C(sp3)-H bonds. Angew. Chem. Int. Ed. 50, 8647–8651 (2011) .
Iglesias, Á., Álvarez, R., de Lera, Á. R. & Muñiz, K. Palladium-catalyzed intermolecular C(sp3)-H amidation. Angew. Chem. Int. Ed. 51, 2225–2228 (2012) .
Kang, T., Kim, Y., Lee, D., Wang, Z. & Chang, S. Iridium-catalyzed intermolecular amidation of sp3 C-H bonds: late-stage functionalization of an unactivated methyl group. J. Am. Chem. Soc. 136, 4141–4144 (2014) .
Wang, N. et al. Rhodium(III)-catalyzed intermolecular amidation with azides via C(sp3)-H functionalization. J. Org. Chem. 79, 5379–5385 (2014) .
Kang, T., Kim, H., Kim, J. G. & Chang, S. Facile one step-synthesis and characterisation of high aspect ratio core-shell copper-polyaniline nanowires. Chem. Commun. 50, 1207–1212 (2014) .
Sun, H., Zhang, H.-W., Han, J.-L., Pan, Y. & Li, G. Asymmetric C-C Bond Formation between Chiral N-Phosphonyl Imines and Ni(II)-Complex of Glycine Schiff Base Provides a GAP Synthesis of α,β-syn-diamino acid derivatives. Eur. J. Org. Chem. 22, 4744–4747 (2013) .
Pindi, S., Wu, J.-B. & Li, G. Design, synthesis and applications of new chiral N-2-phenyl-2-propyl sulfinylimines for Group-Assisted Purification (GAP) asymmetric synthesis. J. Org. Chem. 78, 4006–4012 (2013) .
Acknowledgements
We gratefully acknowledge Indiana University-Purdue University Indianapolis and NSF CHE-1350541 for financial support. We are also grateful for financial support from the Robert A. Welch Foundation (D-1361, USA), NSFC (No. 21332005), and the Jiangsu Innovation Programs (China)
Author information
Authors and Affiliations
Contributions
X.W. and K.Y. performed the experiments and analysed the data. All authors conceived and designed the experiments, contributed to discussions and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-162, Supplementary Table 1-3, Supplementary Methods and Supplementary References (PDF 6358 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wu, X., Yang, K., Zhao, Y. et al. Cobalt-catalysed site-selective intra- and intermolecular dehydrogenative amination of unactivated sp3 carbons. Nat Commun 6, 6462 (2015). https://doi.org/10.1038/ncomms7462
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms7462
This article is cited by
-
Transition metal-catalysed directed C–H functionalization with nucleophiles
Nature Synthesis (2022)
-
Cobalt-catalyzed electrooxidative C-H/N-H [4+2] annulation with ethylene or ethyne
Nature Communications (2018)
-
Metal-Free C–H Alkyliminylation and Acylation of Alkenes with Secondary Amides
Scientific Reports (2016)
-
Carbon-hydrogen activation in China
Science China Chemistry (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
