
Abstract
Interleukin (IL)-17A is a pro-inflammatory cytokine in mice and humans. It is recognized as a key factor for the protection of mice against various pathogens, but it also underlies pathogenic inflammatory responses in numerous mouse models. The inborn errors of IL-17A- and IL-17F-mediated immunity identified in humans in the last decade have revealed that IL-17A and IL-17F are key players in mucocutaneous immunity to Candida albicans, and, to a lesser extent, Staphylococcus aureus. By contrast, there is currently no genetic evidence for a causal link between excess of IL-17 and autoimmunity, autoinflammation, or allergy in humans. We discuss here the physiological and pathological roles of mouse and human IL-17A and IL-17F in host defense and excessive inflammation. We highlight recent advances in our understanding of the consequences of deficient or excessive IL-17 immunity at various mucocutaneous sites, including the oral cavity, skin, intestine, lungs, and vagina.
Similar content being viewed by others

IL-17 cytokines and receptors
The interleukin (IL)-17 cytokine family has six members in mice and humans: IL-17A (also known as IL-17), IL-17B, IL-17C, IL-17D, IL-17E (also known as IL-25), and IL-17F (Figure 1). The founding member, IL-17A, was originally called cytotoxic T-lymphocyte-associated antigen 8 when it was first cloned in 1993.1 It was subsequently renamed IL-17,2 and, more recently, IL-17A.3 The IL-17F protein is the protein of this family most closely related to IL-17A, and the genes encoding these two cytokines are located on the same chromosome, in both mice and humans.4 TH17 cells, a distinct lineage of CD4+ T helper cells discovered in mice in 2005, have been identified as the principal source of IL-17A and IL-17F5, 6, 7 (Figure 2). IL-22 and IL-26 (absent in mice) are the other effector cytokines preferentially produced by TH17 cells.8, 9 CD8+ T cells have also been reported to produce IL-17A and IL-17F, and are known as Tc17 cells10, 11 (Figure 2). The other cells capable of producing IL-17A and IL-17F include γδ T cells,12, 13 invariant natural killer T cells,14, 15 natural killer cells,16, 17 and type 3 innate lymphoid cells18, 19, 20, 21 (Figure 2). It remains unclear whether neutrophils produce IL-17A.22, 23, 24, 25 IL-17A and IL-17F have a similar pattern of expression in cells, driven by signal transducer and activator of transcription 3, RAR-related orphan receptor γT (RORγT), and RORα.26, 27, 28 They also display a similar cysteine knot configuration,4 acting both as IL-17A/IL-17A and IL-17F/IL-17F homodimers and as IL-17A/IL-17F heterodimers.29, 30, 31 IL-17A homodimers have been shown to be more potent than IL-17F homodimers in fibroblasts, macrophages, and epithelial cells, in terms of their ability to induce pro-inflammatory cytokines and chemokines, such as IL-6 and growth-related oncogene-α; IL-17A/IL-17F heterodimers have intermediate activity.29, 30, 31

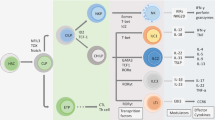
The IL-17 cytokine and receptor family. The IL-17 cytokine family has six members (IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F), whereas the IL-17 receptor family has five members (IL-17RA, IL-17RB, IL-17RC, IL-17RD, and IL-17RE). IL-17RA forms heterodimeric complexes with other subunits to induce signaling by IL-17 cytokines. The IL-17RA/IL-17RC complex binds to IL-17A/IL-17A and IL-17F/IL-17F homodimers and IL-17A/IL-17F heterodimers. The IL-17RA/IL-17RB and IL-17RA/IL-17RE receptors recognize IL-17E and IL-17C, respectively. The ligand for the IL-17RA/IL-17RD receptor complex and the receptors for IL-17B have not yet been identified. ACT1 serves as a key adaptor molecule, which is recruited to the known IL-17 receptors.

IL-17A- and IL-17F-mediated immunity. TH17, Tc17, γδ T, ILC3s, iNKT, and NK cells are the major IL-17A- and IL-17F-producing cells, whereas macrophages, fibroblasts, keratinocytes, endothelial and epithelial cells are the main IL-17RA/IL-17RC-expressing cells. The target genes of IL-17A and IL-17F include pro-inflammatory cytokines, chemokines, antimicrobial peptides, and matrix metalloproteinases (MMPs). ILC3, type 3 innate lymphoid cell; iNKT, invariant natural killer T; NK, natural killer; TNF-α, tumor necrosis factor-α; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; GRO-α, growth-related oncogene-α; ICAM-1, intercellular adhesion molecule 1; VCAM-1, vascular cell adhesion molecule 1; COX-2, cyclooxygenase 2; VEGF, vascular endothelial growth factor; PGE2, prostaglandin E2; iNOS, inducible nitric oxide synthase; NO, nitric oxide.
Soon after the discovery of IL-17A, IL-17RA (also known as IL-17R) was identified as the founding member of the IL-17 receptor family in mice and humans.2 This family has five members: IL-17RA, IL-17RB, IL-17RC, IL-17RD, and IL-17RE (Figure 1). The subunits of the IL-17 receptor, containing a conserved SEFIR (similar expression of fibroblast growth factor and IL-17R) domain, form various heterodimeric complexes with IL-17RA to induce signaling by IL-17A and IL-17F (IL-17RA/IL-17RC), IL-25 (IL-17RA/IL-17RB), and IL-17C (IL-17RA/IL-17RE)32 (Figure 1). In each case, functional mouse and human IL-17 receptors bind SEFIR domain-containing adaptor nuclear factor-κB (NF-κB) activator 1 (ACT1) to mediate the downstream NF-κB, mitogen-activated protein kinase, and CCAAT/enhancer-binding protein signaling pathways.32 The receptors for IL-17B and the ligand for the IL-17RA/IL-17RD receptor complex have not yet been identified32 (Figure 1). IL-17RA and IL-17RC are the best-characterized members of the IL-17 receptor family, largely because of their interaction with IL-17A and IL-17F. IL-17RA is ubiquitously expressed, whereas IL-17RC expression is mostly restricted to non-hematopoietic cells.33 In humans, the affinity of IL-17RA binding is much higher for IL-17A than for IL-17F.34 By contrast, IL-17RC and IL-17RA/IL-17RC bind IL-17A and IL-17F with similar affinities.34 In mice, IL-17RA binds both IL-17A and IL-17F, but IL-17RC binds strongly only to IL-17F.34 The affinity of IL-17RA/IL-17RC binding for IL-17A and IL-17F has not been tested.34 IL-17A and IL-17F, therefore, mostly activate non-hematopoietic cells (Figure 2). Genes encoding pro-inflammatory cytokines and chemokines are the major targets of IL-17A and IL-17F33 (Figure 2). For example, IL-6, granulocyte colony-stimulating factor, and granulocyte-macrophage colony-stimulating factor regulate myeloid cell functions and inflammatory responses, whereas growth-related oncogene-α, C-X-C motif chemokine ligand 2 (CXCL2), and CXCL5 act as chemoattractants for monocytes and neutrophils. Antimicrobial peptides, including β-defensins in particular, are also induced by IL-17A and IL-17F at the epithelial barrier, to protect the host against a wide range of microorganisms33 (Figure 2). C-C motif chemokine ligand 20 (CCL20) recruits IL-17A- and IL-17F-producing cells expressing C-C motif chemokine receptor 6 (CCR6)33 (Figure 2).
IL-17 in the oral cavity and skin
In mice, IL-17A, IL-17F, and their receptors, IL-17RA/IL-17RC, have been shown to have a major role in host defense against experimental infections of the oral cavity and skin caused by C. albicans and S. aureus.35 In a mouse model of oropharyngeal candidiasis, IL-17RA-, and IL-17RC-deficient mice, like anti-IL-17A/IL-17F antibody (Ab)-treated mice, were found to have higher fungal burdens on the tongue than wild-type controls.36, 37, 38 Neutrophil recruitment and function, which have been reported to be important for host defense against oropharyngeal candidiasis in mice,39 were essentially normal in these mice.38 A recent study, using mice with a conditional deficiency of IL-17RA in superficial oral and esophageal epithelial cells, suggested that the IL-17RA-dependent antifungal response was mediated by the production of β-defensin 3.40 Moreover, innate lymphoid cells, γδ T and thymus-derived TH17 cells (natural TH17 cells) have been shown to be major sources of IL-17A and IL-17F in response to C. albicans in mice, and indispensable for IL-17-mediated antifungal immunity in the oral mucosa.41, 42, 43 IL-17A-deficient mice are more susceptible to cutaneous infection with C. albicans than wild-type mice.44 Mice lacking IL-12p40 or IL-23p19, the two subunits of IL-23, which is essential for TH17 cell development and maintenance in mice,45 also have high fungal burdens in the skin when challenged with C. albicans.44 These results suggest that the immunity mediated by IL-17A and IL-17F is the key to mucocutaneous protection against C. albicans in mice, at least in experimental conditions. However, C. albicans is a commensal in humans, but not in mice. In addition, IL-17RA- and γδ T-cell-deficient mice have larger skin lesions with higher bacterial counts upon cutaneous S. aureus infection, and these phenotypes can be rescued by the administration of recombinant IL-17A in γδ T-cell-deficient mice.46 Furthermore, mice lacking both IL-17A and IL-17F are particularly susceptible to S. aureus infection, developing mucocutaneous abscesses around the nose and mouth.47 These findings highlight the crucial importance of IL-17A and IL-17F in protective immunity to experimental infection with S. aureus, which is commensal in mice.48
Humans with inborn errors of IL-17 immunity present chronic mucocutaneous candidiasis (CMC).49, 50 CMC is characterized by recurrent or persistent lesions of the skin, nails, oral and genital mucosae caused by Candida spp., mostly C. albicans.51, 52, 53 Autosomal recessive (AR) IL-17RA and autosomal dominant (AD) IL-17F deficiencies were the first genetic etiologies identified in patients with ‘isolated’ inherited CMC, who develop CMC with no other prominent clinical signs except staphylococcal skin lesions and pulmonary bacterial diseases in some cases.54 AR IL-17RA deficiency has been reported in 23 patients from 13 unrelated kindreds, and shown to be caused by 12 different homozygous IL17RA mutations and one large homozygous deletion encompassing IL-17RA and adenosine deaminase 2.54, 55, 56 These patients lack fibroblast responses to IL-17A and IL-17F homodimers and heterodimers and leukocyte responses to IL-25.54, 56 These cellular responses are also impaired in patients with AR ACT1 deficiency (two patients from one kindred).57 By contrast, patients with AD IL-17F (five patients from one kindred) or AR IL-17RC (three patients from unrelated kindreds) deficiencies display impaired or abolished responses to IL-17A and IL-17F homodimers and heterodimers in fibroblasts, but their leukocytes respond normally to IL-25.54, 58 Interestingly, staphylococcal diseases are frequently seen in patients with AR IL-17RA or ACT1 deficiencies, but not in those with AD IL-17F or AR IL-17RC deficiencies.49, 50 It has thus been suggested that compromised responses to IL-25 or another IL-17RA-dependent cytokine may account for staphylococcal diseases in patients with inborn errors of IL-17 immunity. Taken together, these data reveal that human IL-17 immunity is indispensable for mucocutaneous immunity to C. albicans and S. aureus in natural conditions, especially in the oral cavity and skin.
Psoriasis is an autoimmune and inflammatory skin disorder characterized by red scaly patches of hyperproliferating keratinocytes and hyperkeratinosis.59, 60, 61, 62 Following the intradermal injection of IL-23, wild-type mice develop erythema, epidermal hyperplasia, and massive neutrophil infiltration.63 By contrast, IL-17RA- or IL-17A- (or even IL-22-) deficient mice are resistant to IL-23-induced psoriasis-like epidermal hyperplasia and inflammation.64, 65 Dermal γδ T cells, which constitutively express the IL-23 receptor, have been shown to be the principal producers of IL-17A in mouse skin following stimulation with IL-23.64 Ablation of the IL-17RA or ACT1 gene also protects mice from imiquimod-induced psoriasis-like skin inflammation.66, 67, 68 Furthermore, K5.Stat3C transgenic mice, which express signal transducer and activator of transcription 3 constitutively in keratinocytes, develop psoriasiform lesions following treatment with 12-O-tetradecanoylphorbol-13-acetate, whereas the Ab-mediated neutralization of IL-12p40, IL-23p19, or IL-17A, or deletion of the IL-17A gene attenuates the development of psoriasis-like lesions.69 These data suggest that the IL-23-TH17-IL-17A axis contributes to the pathogenesis of skin lesions in mouse models of psoriasis. Consistent with this hypothesis, several TH17-associated molecules, including IL-17A, IL-17F, IL-22, IL-26, and RORγT, are strongly expressed in the psoriatic skin lesions of patients, probably owing to the production of IL-23 by dendritic cells in the skin.9, 70 Genome-wide association studies in humans have also identified polymorphisms weakly associated with psoriasis close to the IL23R and ACT1 genes.71, 72 Genetic proof of the involvement of IL-17 in the pathogenesis of human psoriasis is, however, lacking, in the absence of known gain-of-function mutations. The known monogenic forms of psoriasis include AR deficiency of IL-36-receptor antagonist and AD caspase recruitment domain-containing protein 14 gain-of-function.73, 74, 75, 76, 77 Nevertheless, IL-23 and IL-17A have been identified as potential treatment targets in psoriasis. Human monoclonal Abs targeting IL-23p19 (risankizumab, guselkumab, and tildrakizumab) are currently in phase III trials, and have yielded encouraging early efficacy and safety results.78, 79, 80 Anti-IL-17A (secukinumab and ixekizumab) and anti-IL-17RA (brodalumab) Abs have recently been approved by the US Food and Drug Administration for psoriasis treatment, and have been shown to clear up skin lesions totally after 12 weeks of treatment in more than a quarter of the participants in the phase III trial.81, 82, 83
IL-17 in the intestine
Not only is the immunity mediated by IL-17A and IL-17F crucial for protection against C. albicans and S. aureus, it also protects mice against experimental infection of the intestine with Citrobacter rodentium. IL-12p40-, IL-23p19-, IL-17A-, IL-17F-, IL-17A/IL-17F double-, and IL-22-deficient mice are more susceptible to C. rodentium infection than wild-type mice.47, 84, 85 Mortality is higher in IL-12p40-, IL-23p19-, or IL-22-deficient mice challenged with C. rodentium than in wild-type mice challenged with the same bacterium.84, 85 IL-22 is induced by IL-23 in the colon and maintains colonic epithelial integrity during the early phase of C. rodentium infection in mice, by inducing the regenerating islet-derived (Reg) family of antimicrobial proteins.85 Likewise, in response to C. rodentium infection, IL-17A-, IL-17F-, and IL-17A/IL-17F double-deficient mice have higher bacterial burdens and display more severe colonic inflammation, probably owing to the impairment of β-defensin production.47 The specific disruption of IL-17RA in mouse intestinal epithelial cells is associated with segmented filamentous bacteria overgrowth and lower mRNA levels of α-defensins, NADPH oxidase 1, and polymeric immunoglobulin receptor, as well as poor fecal secretory IgA production.86 However, it remains unclear whether IL-17A and IL-17F protect against experimental infection of the intestine with Salmonella enterica serovar Typhimurium. In a mouse model of streptomycin-pretreated S. typhimurium infection, IL-17RA-deficient mice challenged with strain ATCC 14028 display higher levels of bacterial translocation from the intestine to the mesenteric lymph nodes and spleen.87 By contrast, IL-17A-, IL-17F-, or IL-17RA-deficient mice, and mice treated with anti-IL-17A plus IL-17F Abs, display levels of bacterial dissemination and cecal inflammation similar to those of wild-type mice following challenge with strain SL1344.88 Local neutralization of IL-17A in the intestinal lumen decreases bacterial clearance and exacerbates epithelial damage in mice infected with S. typhimurium strain LT2.89 Further studies are, therefore, required, to clarify the contribution of IL-17 immunity to intestinal protection against experimental S. typhimurium infection in mice. In humans, deficiencies of IL-17 immunity do not seem to cause a predisposition to pathogenic infections in the intestine, at least among the patients identified to date.49, 50
Inflammatory bowel disease, including ulcerative colitis and Crohn’s disease, is a group of autoinflammatory disorders characterized by inflammation of the colon and small intestine.59 The precise role of IL-17A- and IL-17F-mediated immunity in experimental inflammatory colitis in mice remains a matter of debate. In a mouse model of acute trinitrobenzenesulfonic acid-induced colitis, IL-17RA-deficient mice are protected from weight loss and colonic inflammation, and the administration of an IL-17RA IgG1 fusion protein improves established colonic inflammation in wild-type mice.90 Mice lacking IL-17A or IL-17F are also resistant to dextran sodium sulfate-induced colitis, displaying milder acute intestinal inflammation.91, 92 However, some studies have suggested that the neutralization or genetic ablation of IL-17A, and the epithelial-specific deletion of ACT1 result in severe dextran sodium sulfate-induced colitis in mice.92, 93, 94, 95 These conflicting findings may result from differences in experimental settings, including the genetic background and intestinal microbiota of the mice. Moreover, in a mouse model of T-cell-mediated colitis, the adoptive transfer of IL-17A- or IL-17RA-deficient T cells in recombination activating gene 1-deficient mice, which lack mature T and B cells, led to exacerbated colitis.96 Multidrug resistance 1a-deficient mice infected with Helicobacter bilis develop spontaneous colitis, and the neutralization of IL-17A or IL-17RA accelerates death, by greatly weakening the intestinal epithelial barrier and increasing colonic inflammation.97 It has been suggested that the principal function of IL-17A and IL-17RA in experimental colitis is the maintenance of intestinal barrier integrity rather than driving pathogenic inflammation. Clinical trials of anti-IL-17A (secukinumab) and anti-IL-17RA (brodalumab) Abs in patients with moderate-to-severe CD have reported no improvement, or even a worsening of disease, on treatment.98, 99 The high levels of mRNA for TH17 signature cytokines, including IL-17A, IL-17F, IL-22, and IL-26, in the intestinal mucosa of patients with inflammatory bowel disease may therefore be beneficial.100, 101, 102, 103
IL-17 in the lungs
The protective role of IL-17 cytokines and receptors in host defense against microorganisms was first described in a mouse model of Klebsiella pneumoniae infection. IL-17RA-deficient mice display high mortality rates, delayed neutrophil recruitment, and poor granulocyte colony-stimulating factor and CXCL2 expression in the lungs.104 This observation was supported by the findings of a recent study of mice with a conditional deficiency of IL-17RA or IL-17RC in the lung epithelium. These mice display compromised K. pneumoniae clearance, with impaired neutrophil recruitment in response to IL-17A and low levels of CXCL5 production.105 Similarly, mice lacking IL-17A owing to a genetic deficiency or Ab-mediated neutralization are more susceptible to K. pneumoniae infection than wild-type mice.106, 107, 108 It has been shown that IL-17A- and IL-17F-producing γδ T cells are essential to protect mice against K. pneumoniae,107 whereas type 3 innate lymphoid cells also have a crucial role in activating inflammatory monocytes during infection.108 In a mouse model of S. aureus pneumonia, IL-17A-, IL-17F-, and IL-17RA-deficient mice have been shown to have large bacterial burdens in the lungs.109 In addition, the genetic deletion of IL-17A or IL-17RA leads to higher bacterial counts in the lungs, associated with lower levels of granulocyte colony-stimulating factor production and an impairment of IL-12-dependent interferon-γ responses in mice infected with a live vaccine strain of Francisella tularensis.110 IL-17A induces the production of IL-12 and interferon-γ in antigen-presenting cells and enhances bacterial clearance following infection with F. tularensis live vaccine strain.110 Furthermore, upon challenge with Chlamydia muridarum, mice in which IL-17A has been neutralized display compromised Chlamydia-specific TH1 responses, with greater bacterial growth in the lungs, and lower survival.111, 112 In a dendritic cell/T-cell coculture system, dendritic cells isolated from mice in which IL-17A was neutralized induced lower levels of interferon-γ production by Chlamydia-specific T cells than those derived from wild-type mice.111 Collectively, these findings suggested that the immunity mediated by IL-17A and IL-17F makes a major contribution to the protection of mice against experimental bacterial infections of the lungs. Consistent with this hypothesis, various bacterial infections of the respiratory tract have been seen in patients with AR IL-17RA deficiency.56
Asthma is classically considered to be a TH2-mediated allergic disorder characterized by eosinophilic inflammation and airway hyperresponsiveness (AHR) that ultimately gives rise to obstructive airway and breathing problems.59 In a mouse model of ovalbumin (OVA)-induced asthma, IL-17RA-deficient mice display impaired eosinophil recruitment and compromised TH2 responses.113 Likewise, IL-17A-deficient mice have lower levels of OVA-induced AHR and pulmonary inflammation.114 The adoptive transfer of OVA-specific TH17 cells in recipient mice leads to an influx of neutrophils in the airways and AHR, whereas the transfer of OVA-specific TH2 cells results in the migration of lymphocytes and eosinophils into the lungs and AHR, following OVA challenge.115 Treatment with dexamethasone attenuates the airway inflammation and AHR induced by TH2 cells, but not those induced by TH17 cells.115 These results suggest a potential role for TH17 cells in steroid-resistant asthma in mice. In a mouse model of allergen-induced asthma, the neutralization of both IL-13 and IL-17A protects mice from eosinophilic and neutrophilic inflammation and AHR,116 suggesting that the combined targeting of TH2 and TH17 cells may maximize therapeutic efficacy. Moreover, a subset of IL-17A-producing TH2 (TH2/TH17) cells has been shown to induce higher levels of heterogeneous inflammatory leukocyte infiltration and to exacerbate asthma relative to classical TH2 and TH17 cells.117 Consistently, several studies have shown that IL-17A is upregulated in the sputum and bronchial biopsies of patients with asthma and that the production of this cytokine is correlated with disease severity.118 It has also been suggested that a higher frequency of TH2/TH17 cells in bronchoalveolar lavage fluid is associated with a more severe form of asthma in humans.119 However, a clinical trial showed that anti-IL-17RA Ab (brodalumab) alone had no effect in patients with moderate-to-severe asthma.120
Chronic obstructive pulmonary disease (COPD), encompassing chronic bronchitis and emphysema, is a progressive and largely irreversible disorder resulting in airflow limitation.59 It is caused by long-term exposure to toxic inhalants, and is most often associated with cigarette smoke.59 In mice, cigarette smoke promotes TH17 cell differentiation in vitro and in vivo via the aryl hydrocarbon receptor.121, 122 IL-17A- or IL-17RA-deficient mice are resistant to emphysema,121, 122 whereas transgenic mice displaying lung-specific IL-17A expression (Cc10-Il17a) develop severe emphysema in response to cigarette smoke.122 The adoptive transfer of lung antigen-presenting cells isolated from mice with emphysema induces emphysema and an increase in inflammation after 12 weeks in the absence of smoke in wild-type, but not in IL-17A-deficient recipient mice.122 In addition, the neutralization of IL-17A attenuates smoke-induced neutrophilic inflammation of the airways in mice.123 Large numbers of TH17 cells and the overproduction of IL-17A have also been observed in the lungs of mice treated with porcine pancreatic elastase.124 IL-17A-deficient mice display lower levels of elastase-induced pulmonary inflammation and emphysema than wild-type mice.124 Furthermore, in two mouse models of airway fibrosis triggered by COPD-relevant stimuli, exposure to an adenoviral IL-1β vector or cigarette smoke in combination with viral mimetic poly(I:C), the knockout of IL-17RA or the neutralization of IL-17A or IL-17RA results in lower levels of airway fibrosis and inflammation.125 Finally, there is growing evidence to suggest that patients with COPD have higher proportions of TH17 cells among their peripheral blood mononuclear cells and high levels of IL-17A and IL-17F on bronchial biopsies.126, 127 High levels of RORγT mRNA have also been reported in the lungs of patients with COPD.128 However, a phase II trial of anti-IL-17A Ab (CNTO 6785) did not show any significant efficacy in the treatment of patients with symptomatic moderate-to-severe COPD.129
IL-17 in the vagina
There is increasing evidence to suggest that IL-17A- and IL-17F-mediated immunity may be involved in host defense in the mouse vagina. For instance, mice in which IL-17A or IL-17RA has been neutralized are more susceptible to Neisseria gonorrhoeae infection than wild-type mice.130 They die more rapidly and have higher bacterial burdens and delayed neutrophil recruitment to the vagina during N. gonorrhoeae infection.130 By contrast, ablation of the IL-17A gene results in a lower bacterial load in the vagina upon vaginal infection with C. muridarum,131 whereas the knockout of IL-17RA does not.132 Both types of mutant mouse display lower levels of neutrophil influx into the vagina,131, 132 but IL-17A-deficient mice have high levels of Chlamydia-neutralizing Ab in the serum,131 whereas IL-17RA-deficient mice display higher levels of macrophage influx and tumor necrosis factor-α production, possibly compensating for the impaired Chlamydia-specific TH1 response and interferon-γ production.132 However, the role of IL-17 immunity in vaginal C. albicans infection in mice remains unclear. In a mouse model of estrogen-induced vulvovaginal candidiasis, wild-type mice produce larger amounts of IL-17A and display massive neutrophil influx into the vagina upon C. albicans chanllenge,133 and treatment with halofuginone, a specific inhibitor of mouse and human TH17 cell differentiation,134 results in a higher fungal load and greater β-defensin 2 production.133 By contrast, wild-type and IL-23p19-, IL-17RA-, and IL-22-deficient mice have similar vaginal fungal burdens and similar levels of neutrophil infiltration following inoculation with C. albicans.135 Humans with inborn errors of IL-17 immunity do not seem to be susceptible to pathogenic infections of the vagina.49, 50
Concluding remarks
The discovery of TH17 cells has greatly expanded and advanced our understanding of the roles of IL-17A, IL-17F, and their receptors in autoimmunity, autoinflammation, allergy, and host defense (Figure 3). However, the role of these molecules in malignancy, the fifth broad category of phenotypes associated with inborn errors of immunity, has not been studied. The development of mucocutaneous carcinomas in patients with CMC and inborn errors of IL-17 immunity suggests that IL-17 may have a role in immunosurveillance, at least indirectly, via host defense against C. albicans. Treatments with IL-17A and IL-17RA inhibitors have been shown to be beneficial in diseases characterized by excessive inflammation, such as psoriasis, but human genetic studies have shown that IL-17A and IL-17F play an essential role in mucocutaneous immunity to C. albicans and, to a lesser extent, S. aureus. A higher percentage of fungal infections, properly managed with antifungal treatments, has been reported in some trials of the blockade of IL-17-mediated immunity (e.g., anti-IL-17A or anti-IL-17RA Abs in patients with psoriasis or psoriatic arthritis).136, 137 Thus, fine control of balance between pathogenic and protective IL-17 immunity will be of crucial importance in the development of future therapeutic strategies for treating inflammatory and infectious diseases.

IL-17A and IL-17F in host defense and excessive inflammation. In mice, deficient IL-17A- and IL-17F-mediated immunity predisposes to various pathogenic infections of the oral cavity, skin, intestine, lungs, and vagina, whereas excessive IL-17A- and IL-17F-mediated immunity induces pathological inflammation in the skin, intestine, and lungs. Humans with inborn errors of IL-17 immunity present chronic mucocutaenous candidiasis (CMC) in the oral cavity, CMC and staphylococcal diseases in the skin, and various bacterial infections in the lungs. Treatments with anti-IL-17A and IL-17RA inhibitors are beneficial for patients with psoriasis characterized by excessive inflammation in the skin. SFB, segmented filamentous bacteria; TPA, 12-O-tetradecanoylphorbol-13-acetate; TNBS, trinitrobenzenesulfonic acid; DSS, dextran sodium sulfate.
References
Rouvier, E., Luciani, M. F., Mattei, M. G., Denizot, F. & Golstein, P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J. Immunol. 150, 5445–5456 (1993).
Yao, Z. et al. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity 3, 811–821 (1995).
Kolls, J. K. & Linden, A. Interleukin-17 family members and inflammation. Immunity 21, 467–476 (2004).
Hymowitz, S. G. et al. IL-17s adopt a cystine knot fold: structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. 20, 5332–5341 (2001).
Harrington, L. E. et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6, 1123–1132 (2005).
Langrish, C. L. et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201, 233–240 (2005).
Park, H. et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 6, 1133–1141 (2005).
Liang, S. C. et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 203, 2271–2279 (2006).
Wilson, N. J. et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat. Immunol. 8, 950–957 (2007).
Ciric, B., El-behi, M., Cabrera, R., Zhang, G. X. & Rostami, A. IL-23 drives pathogenic IL-17-producing CD8+ T cells. J. Immunol. 182, 5296–5305 (2009).
Guillot-Delost, M. et al. Human CD90 identifies Th17/Tc17 T cell subsets that are depleted in HIV-infected patients. J. Immunol. 188, 981–991 (2012).
Umemura, M. et al. Involvement of IL-17 in Fas ligand-induced inflammation. Int. Immunol. 16, 1099–1108 (2004).
Stark, M. A., Huo, Y., Burcin, T. L., Morris, M. A., Olson, T. S. & Ley, K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity 22, 285–294 (2005).
Michel, M. L. et al. Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J. Exp. Med. 204, 995–1001 (2007).
Michel, M. L. et al. Critical role of ROR-gammat in a new thymic pathway leading to IL-17-producing invariant NKT cell differentiation. Proc. Natl Acad. Sci. USA 105, 19845–19850 (2008).
Passos, S. T., Silver, J. S., O'Hara, A. C., Sehy, D., Stumhofer, J. S. & Hunter, C. A. IL-6 promotes NK cell production of IL-17 during toxoplasmosis. J. Immunol. 184, 1776–1783 (2010).
Pandya, A. D. et al. Identification of human NK17/NK1 cells. PLoS ONE 6, e26780 (2011).
Cupedo, T. et al. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat. Immunol. 10, 66–74 (2009).
Takatori, H. et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J. Exp. Med. 206, 35–41 (2009).
Buonocore, S. et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464, 1371–1375 (2010).
Spits, H. et al. Innate lymphoid cells—a proposal for uniform nomenclature. Nat. Rev. Immunol. 13, 145–149 (2013).
Ferretti, S., Bonneau, O., Dubois, G. R., Jones, C. E. & Trifilieff, A. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J. Immunol. 170, 2106–2112 (2003).
Lin, A. M. et al. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J. Immunol. 187, 490–500 (2011).
Taylor, P. R. et al. Activation of neutrophils by autocrine IL-17A-IL-17RC interactions during fungal infection is regulated by IL-6, IL-23, RORgammat and dectin-2. Nat. Immunol. 15, 143–151 (2014).
Huppler, A. R., Verma, A. H., Conti, H. R. & Gaffen, S. L. neutrophils do not express IL-17a in the context of acute oropharyngeal candidiasis. Pathogens 4, 559–572 (2015).
Ivanov, II et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133 (2006).
Yang, X. O. et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 282, 9358–9363 (2007).
Yang, X. O. et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 28, 29–39 (2008).
Chang, S. H. & Dong, C. A novel heterodimeric cytokine consisting of IL-17 and IL-17F regulates inflammatory responses. Cell Res. 17, 435–440 (2007).
Liang, S. C. et al. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J. Immunol. 179, 7791–7799 (2007).
Wright, J. F. et al. Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells. J. Biol. Chem. 282, 13447–13455 (2007).
Amatya, N., Garg, A. V. & Gaffen, S. L. IL-17 signaling: the Yin and the Yang. Trends. Immunol. 38, 310–322 (2017).
Gaffen, S. L. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 9, 556–567 (2009).
Kuestner, R. E. et al. Identification of the IL-17 receptor related molecule IL-17RC as the receptor for IL-17F. J. Immunol. 179, 5462–5473 (2007).
Cypowyj, S. et al. Immunity to infection in IL-17-deficient mice and humans. Eur. J. Immunol. 42, 2246–2254 (2012).
Conti, H. R. et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 206, 299–311 (2009).
Ho, A. W. et al. IL-17RC is required for immune signaling via an extended SEF/IL-17R signaling domain in the cytoplasmic tail. J. Immunol. 185, 1063–1070 (2010).
Trautwein-Weidner, K., Gladiator, A., Nur, S., Diethelm, P. & LeibundGut-Landmann, S. IL-17-mediated antifungal defense in the oral mucosa is independent of neutrophils. Mucosal. Immunol. 8, 221–231 (2015).
Huppler, A. R., Conti, H. R., Hernandez-Santos, N., Darville, T., Biswas, P. S. & Gaffen, S. L. Role of neutrophils in IL-17-dependent immunity to mucosal candidiasis. J. Immunol. 192, 1745–1752 (2014).
Conti, H. R. et al. IL-17 receptor signaling in oral epithelial cells is critical for protection against oropharyngeal candidiasis. Cell Host Microbe 20, 606–617 (2016).
Marks, B. R. et al. Thymic self-reactivity selects natural interleukin 17-producing T cells that can regulate peripheral inflammation. Nat. Immunol. 10, 1125–1132 (2009).
Gladiator, A., Wangler, N., Trautwein-Weidner, K. & LeibundGut-Landmann, S. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J. Immunol. 190, 521–525 (2013).
Conti, H. R. et al. Oral-resident natural Th17 cells and gammadelta T cells control opportunistic Candida albicans infections. J. Exp. Med. 211, 2075–2084 (2014).
Kagami, S., Rizzo, H. L., Kurtz, S. E., Miller, L. S. & Blauvelt, A. IL-23 and IL-17A, but not IL-12 and IL-22, are required for optimal skin host defense against Candida albicans. J. Immunol. 185, 5453–5462 (2010).
Gaffen, S. L., Jain, R., Garg, A. V. & Cua, D. J. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat. Rev. Immunol. 14, 585–600 (2014).
Cho, J. S. et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J. Clin. Invest. 120, 1762–1773 (2010).
Ishigame, H. et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30, 108–119 (2009).
Tavakkol, Z. et al. Resident bacterial flora in the skin of C57BL/6 mice housed under SPF conditions. J. Am. Assoc. Lab. Anim. Sci. 49, 588–591 (2010).
Picard, C. et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency 2015. J. Clin. Immunol. 35, 696–726 (2015).
Okada, S., Puel, A., Casanova, J. L. & Kobayashi, M. Chronic mucocutaneous candidiasis disease associated with inborn errors of IL-17 immunity. Clin. Transl. Immunol. 5, e114 (2016).
Puel, A., Picard, C., Cypowyj, S., Lilic, D., Abel, L. & Casanova, J. L. Inborn errors of mucocutaneous immunity to Candida albicans in humans: a role for IL-17 cytokines? Curr. Opin. Immunol. 22, 467–474 (2010).
Puel, A., Cypowyj, S., Marodi, L., Abel, L., Picard, C. & Casanova, J. L. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr. Opin. Allergy Clin. Immunol. 12, 616–622 (2012).
Lanternier, F. et al. Primary immunodeficiencies underlying fungal infections. Curr. Opin. Pediatr. 25, 736–747 (2013).
Puel, A. et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 332, 65–68 (2011).
Fellmann, F. et al. IL-17 receptor A and adenosine deaminase 2 deficiency in siblings with recurrent infections and chronic inflammation. J. Allergy Clin. Immunol. 137, 1189–1196 (2016) e1181-1182.
Levy, R. et al. Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc. Natl Acad. Sci. USA 113, E8277–E8285 (2016).
Boisson, B. et al. An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity 39, 676–686 (2013).
Ling, Y. et al. Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J. Exp. Med. 212, 619–631 (2015).
Weaver, C. T., Elson, C. O., Fouser, L. A. & Kolls, J. K. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu. Rev. Pathol. 8, 477–512 (2013).
Lande, R. et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat. Commun. 5, 5621 (2014).
Arakawa, A. et al. Melanocyte antigen triggers autoimmunity in human psoriasis. J. Exp. Med. 212, 2203–2212 (2015).
Cheung, K. L. et al. Psoriatic T cells recognize neolipid antigens generated by mast cell phospholipase delivered by exosomes and presented by CD1a. J. Exp. Med. 213, 2399–2412 (2016).
Chan, J. R. et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J. Exp. Med. 203, 2577–2587 (2006).
Cai, Y. et al. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity 35, 596–610 (2011).
Rizzo, H. L., Kagami, S., Phillips, K. G., Kurtz, S. E., Jacques, S. L. & Blauvelt, A. IL-23-mediated psoriasis-like epidermal hyperplasia is dependent on IL-17A. J. Immunol. 186, 1495–1502 (2011).
van der Fits, L. et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J. Immunol. 182, 5836–5845 (2009).
El Malki, K. et al. An alternative pathway of imiquimod-induced psoriasis-like skin inflammation in the absence of interleukin-17 receptor a signaling. J. Invest. Dermatol. 133, 441–451 (2013).
Ha, H. L. et al. IL-17 drives psoriatic inflammation via distinct, target cell-specific mechanisms. Proc. Natl Acad. Sci. USA 111, E3422–E3431 (2014).
Nakajima, K. et al. Distinct roles of IL-23 and IL-17 in the development of psoriasis-like lesions in a mouse model. J. Immunol. 186, 4481–4489 (2011).
Johansen, C., Usher, P. A., Kjellerup, R. B., Lundsgaard, D., Iversen, L. & Kragballe, K. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br. J. Dermatol. 160, 319–324 (2009).
Cargill, M. et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am. J. Hum. Genet. 80, 273–290 (2007).
Huffmeier, U. et al. Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat. Genet. 42, 996–999 (2010).
Marrakchi, S. et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N. Engl. J. Med. 365, 620–628 (2011).
Onoufriadis, A. et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am. J. Hum. Genet. 89, 432–437 (2011).
Fuchs-Telem, D. et al. Familial pityriasis rubra pilaris is caused by mutations in CARD14. Am. J. Hum. Genet. 91, 163–170 (2012).
Jordan, C. T. et al. Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in psoriasis. Am. J. Hum. Genet. 90, 796–808 (2012).
Jordan, C. T. et al. PSORS2 is due to mutations in CARD14. Am. J. Hum. Genet. 90, 784–795 (2012).
Papp, K. A. et al. Risankizumab versus Ustekinumab for moderate-to-severe plaque psoriasis. N. Engl. J. Med. 376, 1551–1560 (2017).
Gordon, K. B. et al. A phase 2 trial of guselkumab versus adalimumab for plaque psoriasis. N. Engl. J. Med. 373, 136–144 (2015).
Kopp, T. et al. Clinical improvement in psoriasis with specific targeting of interleukin-23. Nature 521, 222–226 (2015).
Sanford, M. & McKeage, K. Secukinumab: first global approval. Drugs 75, 329–338 (2015).
Markham, A. & Ixekizumab First global approval. Drugs 76, 901–905 (2016).
Greig, S. L. & Brodalumab First global approval. Drugs 76, 1403–1412 (2016).
Mangan, P. R. et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441, 231–234 (2006).
Zheng, Y. et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14, 282–289 (2008).
Kumar, P. et al. Intestinal interleukin-17 receptor signaling mediates reciprocal control of the gut microbiota and autoimmune inflammation. Immunity 44, 659–671 (2016).
Raffatellu, M. et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat. Med. 14, 421–428 (2008).
Songhet, P. et al. IL-17A/F-signaling does not contribute to the initial phase of mucosal inflammation triggered by S. Typhimurium. PLoS ONE 5, e13804 (2010).
Mayuzumi, H., Inagaki-Ohara, K., Uyttenhove, C., Okamoto, Y. & Matsuzaki, G. Interleukin-17A is required to suppress invasion of Salmonella enterica serovar Typhimurium to enteric mucosa. Immunology 131, 377–385 (2010).
Zhang, Z., Zheng, M., Bindas, J., Schwarzenberger, P. & Kolls, J. K. Critical role of IL-17 receptor signaling in acute TNBS-induced colitis. Inflamm. Bowel Dis. 12, 382–388 (2006).
Ito, R. et al. Involvement of IL-17A in the pathogenesis of DSS-induced colitis in mice. Biochem. Biophys. Res. Commun. 377, 12–16 (2008).
Yang, X. O. et al. Regulation of inflammatory responses by IL-17F. J. Exp. Med. 205, 1063–1075 (2008).
Ogawa, A., Andoh, A., Araki, Y., Bamba, T. & Fujiyama, Y. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin. Immunol. 110, 55–62 (2004).
Lee, J. S. et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity 43, 727–738 (2015).
Song, X. et al. Growth factor FGF2 cooperates with interleukin-17 to repair intestinal epithelial damage. Immunity 43, 488–501 (2015).
O'Connor, W. Jr. et al. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat. Immunol. 10, 603–609 (2009).
Maxwell, J. R. et al. Differential roles for interleukin-23 and interleukin-17 in intestinal immunoregulation. Immunity 43, 739–750 (2015).
Hueber, W. et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut 61, 1693–1700 (2012).
Targan, S. R. et al. A randomized, double-blind, placebo-controlled phase 2 study of brodalumab in patients with moderate-to-severe Crohn's disease. Am. J. Gastroenterol. 111, 1599–1607 (2016).
Fujino, S. et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 52, 65–70 (2003).
Rovedatti, L. et al. Differential regulation of interleukin 17 and interferon gamma production in inflammatory bowel disease. Gut 58, 1629–1636 (2009).
Sugihara, T. et al. The increased mucosal mRNA expressions of complement C3 and interleukin-17 in inflammatory bowel disease. Clin. Exp. Immunol. 160, 386–393 (2010).
Geremia, A. et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J. Exp. Med. 208, 1127–1133 (2011).
Ye, P. et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 194, 519–527 (2001).
Chen, K. et al. IL-17 receptor signaling in the lung epithelium is required for mucosal chemokine gradients and pulmonary host defense against K. pneumoniae. Cell Host Microbe 20, 596–605 (2016).
Aujla, S. J. et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 14, 275–281 (2008).
Murakami, T., Hatano, S., Yamada, H., Iwakura, Y. & Yoshikai, Y. Two types of interleukin 17A-producing gammadelta T cells in protection against pulmonary infection with Klebsiella pneumoniae. J. Infect. Dis. 214, 1752–1761 (2016).
Xiong, H., Keith, J. W., Samilo, D. W., Carter, R. A., Leiner, I. M. & Pamer, E. G. Innate lymphocyte/Ly6C(hi) monocyte crosstalk promotes klebsiella pneumoniae clearance. Cell 165, 679–689 (2016).
Kudva, A. et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J. Immunol. 186, 1666–1674 (2011).
Lin, Y. et al. Interleukin-17 is required for T helper 1 cell immunity and host resistance to the intracellular pathogen Francisella tularensis. Immunity 31, 799–810 (2009).
Bai, H. et al. IL-17/Th17 promotes type 1T cell immunity against pulmonary intracellular bacterial infection through modulating dendritic cell function. J. Immunol. 183, 5886–5895 (2009).
Zhang, X. et al. A MyD88-dependent early IL-17 production protects mice against airway infection with the obligate intracellular pathogen Chlamydia muridarum. J. Immunol. 183, 1291–1300 (2009).
Schnyder-Candrian, S. et al. Interleukin-17 is a negative regulator of established allergic asthma. J. Exp. Med. 203, 2715–2725 (2006).
Nakae, S. et al. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 17, 375–387 (2002).
McKinley, L. et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J. Immunol. 181, 4089–4097 (2008).
Choy, D. F. et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci. Transl. Med. 7, 301ra129 (2015).
Wang, Y. H. et al. A novel subset of CD4(+) T(H)2 memory/effector cells that produce inflammatory IL-17 cytokine and promote the exacerbation of chronic allergic asthma. J. Exp. Med. 207, 2479–2491 (2010).
Chesne, J., Braza, F., Mahay, G., Brouard, S., Aronica, M. & Magnan, A. IL-17 in severe asthma. Where do we stand? Am. J. Respir. Crit. Care Med. 190, 1094–1101 (2014).
Irvin, C. et al. Increased frequency of dual-positive TH2/TH17 cells in bronchoalveolar lavage fluid characterizes a population of patients with severe asthma. J. Allergy Clin. Immunol. 134, 1175–1186 e1177 (2014).
Busse, W. W. et al. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am. J. Respir. Crit. Care Med. 188, 1294–1302 (2013).
Chen, K. et al. IL-17RA is required for CCL2 expression, macrophage recruitment, and emphysema in response to cigarette smoke. PLoS ONE 6, e20333 (2011).
Shan, M. et al. Cigarette smoke induction of osteopontin (SPP1) mediates T(H)17 inflammation in human and experimental emphysema. Sci. Transl. Med. 4, 117ra119 (2012).
Shen, N., Wang, J., Zhao, M., Pei, F. & He, B. Anti-interleukin-17 antibodies attenuate airway inflammation in tobacco-smoke-exposed mice. Inhal. Toxicol. 23, 212–218 (2011).
Kurimoto, E. et al. IL-17A is essential to the development of elastase-induced pulmonary inflammation and emphysema in mice. Respir. Res. 14, 5 (2013).
Yanagisawa, H. et al. Role of IL-17A in murine models of COPD airway disease. Am. J. Physiol. Lung Cell Mol. Physiol. 312, L122–L130 (2017).
Vargas-Rojas, M. I., Ramirez-Venegas, A., Limon-Camacho, L., Ochoa, L., Hernandez-Zenteno, R. & Sansores, R. H. Increase of Th17 cells in peripheral blood of patients with chronic obstructive pulmonary disease. Respir. Med. 105, 1648–1654 (2011).
Chang, Y. et al. CD8 positive T cells express IL-17 in patients with chronic obstructive pulmonary disease. Respir. Res. 12, 43 (2011).
Chu, S., Zhong, X., Zhang, J., Lao, Q., He, Z. & Bai, J. The expression of Foxp3 and ROR gamma t in lung tissues from normal smokers and chronic obstructive pulmonary disease patients. Int. Immunopharmacol. 11, 1780–1788 (2011).
Eich, A. et al. A randomized, placebo-controlled phase 2 trial of cnto 6785 in chronic obstructive pulmonary disease. COPD 14, 476–483 (2017).
Feinen, B., Jerse, A. E., Gaffen, S. L. & Russell, M. W. Critical role of Th17 responses in a murine model of Neisseria gonorrhoeae genital infection. Mucosal. Immunol. 3, 312–321 (2010).
Andrew, D. W. et al. The duration of Chlamydia muridarum genital tract infection and associated chronic pathological changes are reduced in IL-17 knockout mice but protection is not increased further by immunization. PLoS ONE 8, e76664 (2013).
Scurlock, A. M. et al. Interleukin-17 contributes to generation of Th1 immunity and neutrophil recruitment during Chlamydia muridarum genital tract infection but is not required for macrophage influx or normal resolution of infection. Infect. Immun. 79, 1349–1362 (2011).
Pietrella, D. et al. Th17 cells and IL-17 in protective immunity to vaginal candidiasis. PLoS ONE 6, e22770 (2011).
Sundrud, M. S. et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science 324, 1334–1338 (2009).
Yano, J., Kolls, J. K., Happel, K. I., Wormley, F., Wozniak, K. L. & Fidel, P. L. Jr. The acute neutrophil response mediated by S100 alarmins during vaginal Candida infections is independent of the Th17-pathway. PLoS ONE 7, e46311 (2012).
Saunte, D. M., Mrowietz, U., Puig, L. & Zachariae, C. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br. J. Dermatol. 177, 47–62 (2017).
Papp, K. A. et al. Infections from 7 clinical trials of ixekizumab, an anti-interleukin-17a monoclonal antibody, in patients with moderate-to-severe psoriasis. Br. J. Dermatol. doi: 10.1111/bjd.15723 (2017).
Acknowledgements
We thank the members of the Laboratory of Human Genetics of Infectious Diseases for helpful discussions. We also thank Yelena Nemirovskaya and Cécile Patissier for their assistance. This work was supported in part by the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID); the French National Research Agency (ANR) under the “Investments for the future” program (grant number ANR-10-IAHU-01); ANR HGDIFD (ANR-14-CE15-0006-01); eRARE EURO-CMC (ANR-14-RARE-0005-02); the National Institute of Allergy and Infectious Diseases (R01AI127564); the Jeffrey Modell Foundation Translational Research Program; the St. Giles Foundation, the Rockefeller University; Institut National de la Santé et de la Recherche Médicale (INSERM); University Paris Descartes.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Rights and permissions
About this article

Cite this article
Li, J., Casanova, JL. & Puel, A. Mucocutaneous IL-17 immunity in mice and humans: host defense vs. excessive inflammation. Mucosal Immunol 11, 581–589 (2018). https://doi.org/10.1038/mi.2017.97
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2017.97
This article is cited by
-
Isolated Chronic Mucocutaneous Candidiasis due to a Novel Duplication Variant of IL17RC
Journal of Clinical Immunology (2024)
-
TH17 cell heterogeneity and its role in tissue inflammation
Nature Immunology (2023)
-
The IL-17 family in diseases: from bench to bedside
Signal Transduction and Targeted Therapy (2023)
-
The Airway Microbiome-IL-17 Axis: a Critical Regulator of Chronic Inflammatory Disease
Clinical Reviews in Allergy & Immunology (2022)
-
Safety of Ixekizumab in Adult Patients with Moderate-to-Severe Psoriasis: Data from 17 Clinical Trials with Over 18,000 Patient-Years of Exposure
Dermatology and Therapy (2022)
