
Abstract
Influenza A is a highly contagious respiratory virus that causes seasonal epidemics and occasional worldwide pandemics. The primary cause of influenza-related mortality is bacterial superinfection. There are numerous mechanisms by which preceding influenza infection attenuates host defense, allowing for increased susceptibility to bacterial pneumonia. Herein, we demonstrate that influenza inhibits Staphylococcus aureus-induced production of interleukin-33 (IL-33). Restoration of IL-33 during influenza A and methicillin-resistant S. aureus superinfection enhanced bacterial clearance and improved mortality. Innate lymphoid Type 2 cells and alternatively activated macrophages are not required for IL-33-mediated protection during superinfection. We show that IL-33 treatment resulted in neutrophil recruitment to the lung, associated with improved bacterial clearance. These findings identify a novel role for IL-33 in antibacterial host defense at the mucosal barrier.
Similar content being viewed by others

Introduction
Pneumonia is a significant cause of mortality in the United States and worldwide. Numerous organisms cause pneumonia, including bacteria and viruses. Influenza is a common respiratory virus that results in 3–5 million cases of severe illness and 250,000–500,000 worldwide deaths annually. Although most cases of influenza do not result in death, secondary bacterial superinfections are associated with increased hospitalization and mortality. Bacterial pneumonia is a severe complication of influenza infection.1 Staphylococcus aureus and Streptococcus pneumoniae typically cause bacterial pneumonia following influenza infection. The prevalence of S. aureus has increased in recent years with the emergence of methicillin-resistant S. aureus (MRSA) and is now the most prevalent cause of pneumonia in this context.2, 3, 4 Understanding the mechanisms by which influenza alters the immune system and reduces host defense against secondary bacterial infections is critical to decreasing morbidity and mortality during future influenza pandemics.
Interleukin-33 (IL-33) is an alarmin cytokine that is expressed in the nucleus of epithelial and endothelial cells and released in response to cellular stress and injury. IL-33 acts through the ST2 receptor, and its known target cells include T helper 2 cells, innate lymphoid Type 2 cells (ILC2), macrophages, CD8+ T cells, dendritic cells, basophils, and B cells.5, 6 IL-33 promotes myeloid cell migration, polarizes macrophages towards an alternatively activated macrophage (M2a) phenotype and induces Type 2 immune responses through the induction of IL-5 and IL-13.7, 8, 9, 10, 11, 12 IL-33 is known to have both pro-inflammatory and anti-inflammatory effects depending on the model being utilized. During influenza infection, endothelial cells, epithelial cells, and macrophages express IL-33 and can lead to IL-13 production by ILCs.11, 13 ILCs also produce amphiregulin in response to IL-33 stimulation, and amphiregulin expression is enhanced in the lung following influenza infection. ILCs can promote airway epithelial repair through amphiregulin in influenza-infected mice.14 IL-33 is known to promote host defense against bacterial infections through recruitment of neutrophils in sepsis and skin models and enhanced antimicrobial capacity of macrophages in the skin.7, 15, 16 Little is known about the role of IL-33 in mucosal antibacterial host defense and published studies focus on IL-33 stimulation of ILC2 and alternatively activated macrophages (AAM). The mucosal barriers of the respiratory, gastrointestinal, reproductive, and urinary tracts encompass large surface areas in which the host is exposed to the environment and serves as a potential route for acquisition of infection.
We have previously demonstrated that mice infected with influenza A and S. aureus have increased bacterial burden and higher mortality compared with mice infected with singular S. aureus infection.17 In addition, we have shown that influenza attenuates S. aureus-induced IL-1β production, increasing susceptibility to secondary bacterial pneumonia.18 Our current work explores the novel protective role of IL-33, an IL-1 family member, in a murine model of influenza A and S. aureus superinfection in the lung.
Results
Preceding influenza infection inhibits IL-33 production in response to secondary bacterial challenge
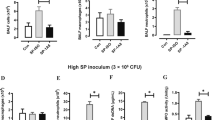
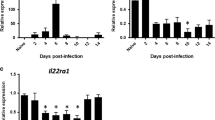
Mice infected with influenza and S. aureus have increased bacterial burden, inflammation, and mortality compared with those infected with S. aureus alone.17 We have previously demonstrated that influenza attenuates S. aureus-induced IL-1β production, increasing susceptibility to secondary bacterial pneumonia in mice.18 The role of IL-33, an IL-1 family member, during influenza A and S. aureus superinfection has yet to be explored. We challenged C57BL/6 mice with influenza A PR/8/34 H1N1 for 6 days followed by methicillin-resistant S. aureus (USA 300) and after 24 h saw a decreased IL-33 response in superinfected mice compared with those infected with either singular MRSA or influenza infection. (Figure 1a and c). Similar to previously published data, bacterial burden was significantly higher in the superinfected mice compared with those that received MRSA alone (Supplementary Figure 1A online). During influenza infection alone, IL-33 gene expression peaks at day 2 post infection and returns to baseline by day 6 post infection (Figure 1b). In addition, similar data were seen in mice that received influenza followed by secondary challenge with methicillin-sensitive S. aureus (ATCC 49775) or Pseudomonas aeruginosa (Figure 1d–f). These data represent a novel impairment of host defense during influenza and secondary bacterial pneumonia. Importantly, the attenuation of IL-33 production by preceding influenza infection occurs with both gram-positive and gram-negative bacterial challenge.

Influenza attenuates the IL-33 response to secondary bacterial challenge. C57BL/6 mice were infected with 100 PFU of influenza A PR/8/34 for 6 days, then challenged with 5 × 107 CFU of MRSA for 24 h. (a) IL-33 expression in lung tissue measured by RT-PCR (n=3–7). C57BL/6 mice were infected with influenza A and harvested at different time points following influenza infection. (b) IL-33 expression in lung tissue measured by RT-PCR (n=3–4). Mice were infected with influenza and MRSA. (c) IL-33 protein in lung tissue measured by western blot (n=8). C57BL/6 mice were infected with 100 PFU of influenza A PR/8/34 for 6 days, then challenged with 1 × 108 CFU of MSSA for 24 h. (d) IL-33 expression in lung tissue measured by RT-PCR (n=3–7). (e) IL-33 protein in lung tissue measured by Lincoplex (n=7). C57BL/6 mice were infected with 100 PFU of influenza A PR/8/34 for 6 days, then challenged with PA01 for 24 h. (f) IL-33 expression in lung tissue measured by RT-PCR (n=3–8). *P<0.05. Significance was tested by unpaired t-test or one-way ANOVA. Each experiment was independently performed twice and data are shown from combined experiments with the exception of Panel a data from naive mice and singular infection with influenza and Panel (b), these experiments were performed once. Panel (f) represents two independently performed experiments with representative data shown.
Restoration of IL-33 improves S. aureus clearance in the lung during superinfection
Next, we proposed that restoration of IL-33 would improve bacterial clearance during influenza and S. aureus superinfection. To test this, we challenged C57BL/6 mice with influenza A PR/8/34 H1N1 for 6 days followed by MRSA and restored IL-33 by two methods: recombinant murine IL-33 or adenoviral vector expressing IL-33. In both cases, IL-33 restoration resulted in improved clearance of S. aureus in superinfected mice (Figure 2a, Supplementary Figure 1B) with no change in viral burden (data not shown). Of note, in a murine model of influenza with complicating S. aureus pneumonia, twofold changes in colony-forming units have been associated with increased mortality.17, 19 Consistent with these data, exogenous IL-33 improved survival during superinfection (Figure 2b). Although not statistically significant, mice that received IL-33 had median survival of 39 h vs. mice that did not receive IL-33 with median survival of 24 h. These data indicate that the inhibition of IL-33 during viral infection may have a role in enhanced susceptibility to secondary bacterial pneumonia. Of note, although exogenous IL-1β induces Type 17 immune activation,18 restoration of IL-33 does not alter Type 17 immunity-related antimicrobial peptide gene expression (Lcn2, RegIIIβ, S100A8, S100A9) compared with the untreated group (Supplementary Figure 1C).

Restoration of IL-33 improves S. aureus clearance in the lung during superinfection. Mice were infected with influenza or control and MRSA and received 2 μg recombinant mouse IL-33 at 24 and 2 h prior to MRSA challenge. (a) Bacterial colony counts in the lung (n=6 in MRSA groups and n=12 in Flu/MRSA groups). (b) Survival curve (n=5–6). (c,e,f) Type 2 immune-related gene expression in lung tissue measured by RT-PCR (n=12). (d,g–i) Protein production in lung tissue measured by Lincoplex (n=12). (j) CXCR2 gene expression in lung tissue measured by RT-PCR. *P<0.05 Significance was tested by unpaired t-test or one-way ANOVA. Each experiment was independently performed twice and data are shown from combined experiments with the exception of the singular MRSA groups in panel (a) and the survival curve in panel (b), these experiments were performed once.
IL-33 induces a Type 2 immune response in the lung during superinfection
IL-33 is known to promote a Type 2 immune response through the induction of IL-5 and IL-13 in multiple established models of both infection and allergic disease. During influenza and bacterial superinfection, we found that gene expression of IL-13, clca3, and muc5b were significantly increased in mice that received recombinant IL-33 compared with control mice (Figure 2c, e and f) at 24 h post MRSA challenge. In addition, protein production of IL-5 and eotaxin were significantly increased (Figure 2d and g). This confirms that IL-33 induces a Type 2 immune response in the lung during superinfection. In our model, IL-33 did not enhance production of pro-inflammatory cytokines IL-6 or TNF-α (Figure 2h and i), or alter gene expression of the chemokine receptor CXCR2 (Figure 2j).
ILC2 and AAM are not required for IL-33-mediated protection during superinfection
IL-33 stimulates Type 2 immune responses through the secretion of IL-4, IL-5, and IL-13 from T helper 2 cells, mast, and ILC2 cells. There are numerous studies showing that IL-33 acts through ILCs to protect against or promote disease pathogenesis, depending on the model being studied.20, 21 We observed that exogenous IL-33 induced a Type 2 immune response in the lung during influenza A, S. aureus superinfection (Figure 2c–g). We hypothesized that the improved bacterial clearance and survival seen in IL-33-treated mice during influenza A/MRSA bacterial superinfection were related to the accumulation of ILC2 in the lung and induction of Type 2 immunity during influenza infection. To test this, we challenged Rag2/ll2rg double knockout mice with influenza followed by MRSA and restored IL-33 with recombinant murine IL-33. Surprisingly, restoration of IL-33 in Rag2/ll2rg double knockout mice also improved bacterial clearance of S. aureus (Figure 3a). Rag2/ll2rg double knockout mice lack T cells, B cells, NK cells, and ILCs, demonstrating that ILCs are not required for IL-33-mediated protection during influenza/MRSA superinfection. In addition, there was no difference in amphiregulin gene expression seen between WT mice in which IL-33 was restored and WT control mice (data not shown), lending support that ILC2 are not required for IL-33-mediated protection during superinfection. Notably, the Rag2/ll2rg double knockout mice did have increased bacterial burden compared with WT mice, likely as a result of lacking other cell types that enhance bacterial clearance during superinfection such as IL-17-producing γδ and CD4+ T cells or natural killer cells.17, 18, 19, 22 The restoration of IL-33 in Rag2/ll2rg double knockout mice did not induce a Type 2 immunity phenotype similar to that seen in WT mice owing to their lack of ILC2 and T helper 2 cells (Figure 3b and c).

IL-33-mediated protection during superinfection does not require ILC2 or alternatively activated macrophages. C57BL/6 WT mice and B6- Rag2/ll2rg double knockout mice were infected with influenza and MRSA. Mice received IL-33 prior to MRSA challenge. (a) Bacterial colony counts in the lung. (b) Protein production of IL-5 in lung homogenate as measured by Lincoplex (n=12). (c) Type 2 cytokine/protein-related gene expression in lung tissue measured by RT-PCR (n=12). (d) M2a macrophage-related gene expression in lung tissue measured by RT-PCR (n=12). (e) Protein production in lung homogenate as measured by Lincoplex (n=12). *P<0.05 Significance was tested by unpaired t-test or one-way ANOVA. Each experiment was independently performed twice and data are shown from combined experiments with the exception of panel a, which was performed twice with representative data presented.
In addition to recruiting ILC2 to the lung, IL-33 is known to polarize macrophages toward an AAM (M2a) phenotype. Given that Rag2/ll2rg double knockout mice still have myeloid cells, we next proposed that IL-33 polarizes macrophages toward an M2a phenotype, thus promoting bacterial clearance when IL-33 is restored in both WT and Rag2/ll2rg double knockout mice. To test this, we measured gene expression of M2a-associated genes and found significant induction in WT mice following influenza A/MRSA bacterial superinfection, but not in Rag2/ll2rg double knockout mice (Figure 3d), making it unlikely that M2a macrophages are required for IL-33-mediated protection during viral/bacterial superinfection in the lung. Interestingly, we also observed differences in IL-1β production between the mouse groups (Figure 3e). There was no difference in the pro-inflammatory cytokines IL-6 or TNF-α (Figure 3f and g).
IL-33 enhances functional neutrophil recruitment to the lung during superinfection
In addition to inducing Type 2 cytokines and polarizing macrophages to the M2a phenotype, IL-33 promotes myeloid cell migration. To determine the role of IL-33 in the recruitment of myeloid cells to the lung during influenza/MRSA superinfection, we performed flow cytometry. We observed no difference in the number of alveolar macrophages (F4/80+CD11c+SiglecF+) in the lungs of IL-33-treated mice compared with untreated controls (Figure 4a), however, there were fewer interstitial macrophages (F4/80+CD11c+SiglecF−) in the lung (Figure 4b). It is possible that IL-33 enhances macrophage uptake and binding of MRSA in the lung. To test this, we treated naive alveolar macrophages with IL-33 prior to in vitro flow cytometry and found that IL-33 did not alter uptake and binding of MRSA (data not shown). In addition, we observed no difference in the expression of S. aureus-associated macrophage scavenger receptors between IL-33-treated mice compared with controls (Figure 4c). There was a significant increase in neutrophils (CD11b+Ly6G+) in the IL-33-treated mice compared with untreated controls (Figure 4d). In addition, there was increased gene expression of cathespin G and neutrophil elastase in the IL-33-treated mice, proteins that are associated with the microbiocidal activities of neutrophils (Figure 4e and f). Next, we challenged C57BL/6 mice with influenza A PR/8/34 H1N1 for 6 days followed by MRSA, treated mice with exogenous IL-33, and administered an anti-neutrophil (anti-Ly-6G) antibody. Mice that received the anti-Ly-6G antibody did not clear MRSA as well as mice that received control antibody despite both groups receiving IL-33. Mice that were treated with anti-Ly-6G antibody, without IL-33 treatment, had the highest bacterial burden compared with all of the other treatment groups (Figure 4g). IL-33 treatment significantly decreased bacterial burden with and without neutrophil depletion, however, its effect on bacterial burden was attenuated by neutrophil depletion. We performed flow cytometry to determine if the anti-Ly-6G antibody effectively depleted neutrophils in the lung in our superinfection model and observed that this was the case (Figure 4h).

IL-33 recruits neutrophils to the lung during superinfection, which are required for MRSA clearance. C57BL/6 WT mice were infected with influenza and MRSA. Mice received IL-33 prior to MRSA challenge. (a) Total F480+CD11c+SiglecF+ cells in the lung by flow cytometry (n=6). (b) Total F480+CD11c+SiglecF− cells in the lung by flow cytometry (n=6). (c) Macrophage scavenger receptor gene expression in lung tissue measured by RT-PCR (n=12). (d) Total CD11b+Ly6G+ cells in the lung by flow cytometry (n=12). (e,f) Serine protease gene expression in lung tissue measured by RT-PCR (n=12). C57BL/6 WT mice were infected with influenza and MRSA. Mice received IL-33 prior to MRSA challenge. Mice received 250 μg anti-Ly-6G antibody at 48, 24, and 2 h prior to MRSA challenge. (g) Bacterial colony counts in the lung (n=8–16). (h) Total CD11b+Ly6G+ cells in the lung by flow cytometry (n=16). *P<0.05 vs. control group. Each experiment was independently performed twice and data are shown from combined experiments with the exception of panels (a and b), these experiments were performed once.
Discussion
The findings of this study demonstrate a novel mechanism by which influenza A predisposes to secondary bacterial pneumonia through the attenuation of bacteria-induced IL-33. The restoration of IL-33 during influenza A and MRSA superinfection in wild-type mice provides protection against the increased bacterial burden that occurs following influenza infection. Interestingly, the restoration of IL-33 during influenza A and MRSA superinfection in Rag2/ll2rg double knockout mice provides a similar protection against the increased bacterial burden following influenza infection. IL-33 does not enhance the Type 2 immune response or AAM response in Rag2/ll2rg double knockout mice during superinfection, demonstrating that ILC2s and M2a macrophages are not required for the protection that IL-33 provides. The restoration of IL-33 during influenza A and MRSA superinfection in wild-type mice results in the recruitment of neutrophils to the lung. These data establish a cellular mechanism for IL-33 protection during influenza A and MRSA superinfection through the recruitment of neutrophils to the lung and suggest a pivotal role for IL-33 in the lung and at the mucosal barrier during infection.
It has been well established that preceding influenza increases the susceptibility to secondary bacterial superinfections. Although S. pneumoniae has historically been recognized as the bacteria to most likely complicate influenza infection, S. aureus has become the most predominant pathogenic organism cultured during secondary bacterial pneumonia.23 During influenza infection, multiple cells types express IL-33 and lead to IL-13 and amphiregulin production by ILCs.11, 13, 14 In a MRSA-infection skin model, IL-33 is upregulated by MRSA challenge and exogenous IL-33 promotes wound healing and the proliferation of neutrophils and CXCR expression.16, 24 IL-33 acts on dermal macrophages to induce microbicidal nitric oxygen release. In addition to gram-positive bacteria, gram-negative bacteria can also cause bacterial superinfection following influenza. Patients with chronic respiratory disease such as chronic obstructive pulmonary disease and cystic fibrosis are frequently colonized with gram-negative bacteria and viral infections such as influenza can lead to increased bacterial burden and exacerbation of disease.25 IL-33 has been shown to polarize macrophages to an alternative activated macrophage phenotype (M2a), decrease bacterial load, and decrease neutrophil infiltrate during P. aeruginosa keratitis.8 Our data demonstrate that methicillin-sensitive S. aureus, methicillin-resistant S. aureus, and P. aeruginosa all induce IL-33 expression in the lung. Furthermore, preceding influenza attenuates the induction of IL-33 during bacterial challenge. The suppression of bacteria-induced IL-33 by preceding influenza infection could possibly represent a conserved mechanism during acute respiratory infections across gram-positive and gram-negative bacteria as it occurs during both S. aureus and P. aeruginosa pneumonia.
IL-33 is known to induce Type 2 immune responses in both infection and allergic models through the induction of IL-5 and IL-13. Prior work demonstrates that influenza infection induces secretion of IL-33 from alveolar macrophages, which leads to the production of IL-13 and IL-5 from ILC2.11 ILC2s accumulate in the lung and promote tissue repair after influenza infection.14 IL-13 has shown to be protective early in the course of influenza and MRSA superinfection, although the cellular source of IL-13 has not been shown. By day 7 post influenza infection, IL-13 levels are decreased in part due to the soluble IL-13 decoy receptor and mice are more susceptible to secondary MRSA pneumonia.26, 27 In a post-influenza pneumococcal pneumonia model, ST2−/− mice have been compared with wild-type mice and showed increased bacterial burden in the lung at 48 h post bacterial challenge when receiving S. pneumoniae at day 14 of influenza infection.28 In our studies, IL-33 induced a robust Type 2 response in wild-type mice with increased expression of IL-5, IL-13, clca3, muc5b, and eotaxin at 24 h post bacterial challenge when receiving MRSA at 6 days post influenza infection, during the peak window of increased susceptibility. Owing to the timing of the IL-33 administration, 24 and 2 h prior to bacterial challenge, the Type 2 immune response we observed is likely an innate immune response with IL-33 exerting its effect on ILC2 to secrete IL-5 and IL-13. In contrast, IL-33 did not induce a Type 2 response in Rag2/ll2rg double knockout mice, likely owing to their lack of ILC2 and possibly other Type 2 cells. IL-33 rescued bacterial clearance in these mice, similar to wild-type mice, demonstrating that although ILC2 may have a protective during post influenza bacterial superinfection, they are not required for IL-33-mediated protection against influenza A and MRSA superinfection.
In addition to acting on ILC2 to promote Type 2 immune responses, IL-33 enhances the polarization of AAM, which have been shown to contribute to Type 2 immune responses and promote tissue repair.9, 29 Our data demonstrate that IL-33 polarizes macrophages to the AAM phenotype with increased expression of multiple M2a markers during post-influenza bacterial superinfection. In Rag2/ll2rg double knockout mice, IL-33 does not polarize macrophages to the AAM phenotype, possibly owing to the loss of IL-13 production by numerous cell types that are lacking in these mice as IL-33-induced AAM polarization has been shown to be IL-13-dependent. Although AAM promote tissue repair, they are not required for IL-33-mediated protection against influenza A and MRSA superinfection. Ablation of alveolar macrophages during post-influenza pneumococcus infection has been shown to impair bacterial clearance.30 However, the binding and uptake of S. aureus by alveolar macrophages from influenza-infected mice is increased compared with naive alveolar macrophages.22 Our data show no difference in the total number or effectiveness of binding and uptake of S. aureus by alveolar macrophages in IL-33-treated mice compared with controls. In addition, IL-33 leads to a decrease in the number of interstitial macrophages during post-influenza MRSA superinfection, and this is unlikely to affect bacterial clearance or killing. Influenza has also been shown to inhibit expression of the macrophage scavenger receptor MARCO, leading to decreased bacterial clearance during pneumococcal pneumonia.31 We did not observe any difference in the expression of macrophage scavenger receptors that could be attributed to IL-33. Although macrophages are highly phagocytic and alveolar macrophages secrete IL-33 during influenza infection, they do not seem to have an important role in IL-33-mediated protection during influenza and MRSA superinfection.
IL-33 also promotes host defense against bacterial infections through the recruitment of neutrophils in sepsis and skin models.7, 15, 16, 24 Prior studies have shown increased neutrophils in the lung during post-influenza bacterial pneumonia compared with singular infection,17, 32, 33 although neutrophil depletion in mice challenged with bacteria at time points mimicking human susceptibility to superinfection does not result in increased susceptibility.32 Neutrophils present during the window of increased susceptibility at 6–7 days post influenza may be dysfunctional and lead to decreased bacterial killing. Influenza suppresses NAPDH oxidase-dependent bacterial clearance during post-influenza MRSA pneumonia, increasing susceptibility to superinfection.34 Our laboratory has previously shown that influenza inhibits Type 17 immunity and Type 17-associated antimicrobial peptides, allowing for poor bacterial clearance during post-influenza staphylococcal pneumonia.17, 22 IL-33 did not cause any changes in the expression of Type 17-associated antimicrobial peptides in our model, and does not appear to be the mechanism for IL-33-dependent protection during influenza and MRSA superinfection. Interestingly, we also observed differences in IL-1β production between mouse groups: there was a significant decrease in IL-1β in super-infected WT mice treated with IL-33 compared with controls. IL-33 treatment has previously been found to downregulate IL-1β in other mouse models35 and its ability to provide protection while not inducing inflammation may be beneficial if IL-33 is considered as a therapeutic agent. We found increased IL-1β production in the super-infected Rag2/ll2rg double knockout mice compared with WT mice. IL-1β is often produced by myeloid cells during infection and the Rag2/ll2rg double knockout mice may be compensating for other immunodeficiencies by producing more IL-1β to activate monocytes, macrophages, and neutrophils. We observed an increase in the total number of neutrophils in the lung as a result of IL-33 restoration. Importantly, we also observed an increase in expression of cathepsin G and neutrophil elastase as a result of IL-33 restoration during superinfection. Neutrophil depletion with IL-33 treatment led to increased susceptibility to MRSA pneumonia, suggesting that IL-33 recruits functional phagocytic neutrophils to the lung during post-influenza MRSA pneumonia and is an important mechanism by which IL-33 leads to protection during superinfection. Importantly, neutrophil depletion without IL-33 treatment resulted in uncontrolled bacterial pneumonia compared with all other treatment groups, suggesting that neutrophils are likely not the singular cell type that responds to IL-33 during superinfection.
Although IL-33 has been shown to recruit neutrophils to the peritoneal cavity during sepsis, the novel finding that IL-33 recruits neutrophils to a mucosal barrier during infection provides a critical advancement to an evolving field. Despite the accumulation of ILC2s in the lung during influenza infection, they are not required for IL-33-mediated protection against influenza A and MRSA superinfection. Prior research regarding IL-33 in the field of pulmonary medicine focused mainly on allergic diseases and ILC2. Other groups have shown that IL-33 acts through AAM to protect against infection. Even with this ongoing work, the relationship between IL-33 and neutrophils in the lung had not previously been established. Our study provides the groundwork for future research regarding IL-33 and neutrophils in the lung and at other mucosal barrier sites. The data provide increased understanding of mucosal host defense and future studies will be needed to further define molecular mechanisms and the therapeutic potential of IL-33 during acute and chronic respiratory infections.
Methods
Mice. A total of 6–8-week-old male wild-type, C57BL/6 mice and B6-Rag2tm1Fwa Il2rgtm1Wjl were purchased from Taconic Farms (Germantown, NY, USA). Mice were maintained under pathogen-free conditions at the Children’s Hospital of Pittsburgh of University of Pittsburgh Medical. All of the studies used age- and sex-matched mice.
Bacterial and viral infections. Methicillin-resistant S. (USA300) and methicillin-sensitive S. (ATCC 49775) were cultured as detailed by American Type Culture Collection instructions in casein hydrolysate yeast extract-containing modified medium for 18 h to stationary growth phase. Mice were inoculated with 5 × 107 colony-forming units in 50 μl sterile phosphate-buffered saline (PBS) by oropharyngeal aspiration. P. aeruginosa strain PAO1 (ATCC BAA-47) was grown in 100 ml Luria-Bertani broth for 12 h. After 12 h, 1 ml of culture was added to fresh Luria-Bertani broth and grown for an additional 12 h. Bacteria were then pelleted, washed, and diluted to give a target inoculum in 50 sterile μl PBS. Serial dilutions of bacteria were grown on Luria-Bertani plates and back calculated to confirm the inocula administered. In two separate experiments, mice received inocula of 4.5 × 05 and 2.7 × 106 CFU. Influenza A PR/8/34 H1N1 was used to inoculate mice with 100 plaque-forming unit of influenza (in 40 μl sterile PBS) by oropharyngeal aspiration. Viral burden was determined by quantitative real-time RT-PCR on lung RNA for viral matrix protein as previously described.36 Mice were sequentially challenged with influenza or vehicle (PBS) for 6 days followed by infection with bacteria or vehicle (PBS) for an additional 24 h.
Recombinant IL-33. Mice received 2 μg recombinant mouse IL-33 (R&D Systems, Minneapolis, MN, 3626-ML-010) in 200 μl of sterile PBS by intraperitoneal injection 24 and 2 h prior to receiving S. aureus.
Adenoviral IL-33 infection. E1- and E3-deleted adenoviral vector encoding EGFP was constructed as previously described by Cre-lox recombination.37, 38 In brief, a SnaBI-HpaI fragment containing part of the CMV promoter, the EGFP cDNA, and part of the SV40 poly(A) sequence was inserted in the pAdlox shuttle plasmid. E1-substituted recombinant adenovirus was generated by co-transfection of SfiI-digested pAdlox-EGFP and c5 helper virus DNA into the adenoviral packaging cell line CRE8, propagated, and purified as described. The E1- and E3-deleted adenovirus encoding mouse IL-33 was constructed by cloning the mIL33 cDNA as a SalI-NotI fragment in the pAdlox shuttle plasmid. E1-substituted recombinant adenovirus was generated by co-transfection, as described above. Mice were infected with IL-33 adenovirus or EGFP control adenovirus (2.5 × 108 plaque-forming unit in 50 μl sterile PBS) by oropharyngeal aspiration 3 days after influenza A infection to ensure viral replication prior to bacterial challenge at 6 days post influenza infection.
Analysis of lung inflammation. At the indicated time points, mouse lungs were lavaged with 1 ml sterile PBS for inflammatory cell differential counts. The cranial lobe of the right lung was homogenized in sterile PBS by mechanical grinding. The resulting lung homogenate was used for bacterial colony counting and cytokine analysis by Bio-plex Multiplex immunoassay (BioRad, Hercules, CA). Middle and caudal lobes of the right lung were snap-frozen and homogenized under liquid nitrogen for RNA extraction by an RNA isolation kit (Agilent Technologies, Santa Clara, CA). RNA analysis was performed by standard RT-PCR using Assay on Demand TaqMan probes and primers (Applied Biosystems, Carlsbad, CA).
Depletion antibodies. Mice received 250 μg of 1A8 (anti-Ly6G) from BioXcel (West Lebanon, NH, USA) in 200 μl of sterile PBS by intraperitoneal injection at 48, 24, and 0 h prior to S. aureus challenge.
Flow cytometry. Whole mouse lungs were digested with collagenase as previously described.18 Cells were stained with fluorescent conjugated antibodies for F480, CD11b, CD11c, Ly6G, and Siglec-F (BD Biosciences, San Jose, CA).
Statistics. All of the data are presented as the mean±s.e.m. Significance was tested by the unpaired t-test (for two means) or one-way ANOVA (for multiple data groups), followed by the Tukey post hoc test. Significance of survival data was assessed by the Gehan–Brislow–Wilcoxon test. Data were analyzed using the GraphPad Prism software.
Study approval. All animal experiments were conducted with approval from the University of Pittsburgh Institutional Animal Care and Use Committee.
References
Morens, D.M., Taubenberger, J.K. & Fauci, A.S. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J. Infect. Dis. 198, 962–970 (2008).
Rice, T.W. et al. Critical illness from 2009 pandemic influenza A virus and bacterial coinfection in the United States. Crit. Care Med. 40, 1487–1498 (2012).
Williams, D.J. et al. Influenza coinfection and outcomes in children with complicated pneumonia. Arch. Pediatr. Adolesc. Med. 165, 506–512 (2011).
Finelli, L. et al. Influenza-associated pediatric mortality in the United States: increase of Staphylococcus aureus coinfection. Pediatrics 122, 805–811 (2008).
Martin, N.T. & Martin, M.U. Interleukin 33 is a guardian of barriers and a local alarmin. Nat. Immunol. 17, 122–131 (2016).
Sims, J.E. & Smith, D.E. The IL-1 family: regulators of immunity. Nat. Rev. Immunol. 10, 89–102 (2010).
Alves-Filho, J.C. et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat. Med. 16, 708–712 (2010).
Hazlett, L.D. et al. IL-33 shifts macrophage polarization, promoting resistance against Pseudomonas aeruginosa keratitis. Investig. Ophthalmol. Vis. Sci 51, 1524–1532 (2010).
Kurowska-Stolarska, M. et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J. Immunol. 183, 6469–6477 (2009).
Moro, K. et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–544 (2010).
Chang, Y.-J. et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat. Immunol. 12, 631–638 (2011).
Li, D. et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J. Allergy Clin. Immunol. 134, 1422–1432 (2014).
Goffic, R.L.e. et al. Infection with influenza virus induces IL-33 in murine lungs. Am. J. Respir. Cell Mol. Biol. 45, 1125–1132 (2011).
Monticelli, L. et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 12, 1045–1054 (2011).
Lan, F. et al. Interleukin-33 facilitates neutrophil recruitment and bacterial clearance in S. aureus-caused peritonitis. Mol. Immunol. 72, 74–80 (2016).
Yin, H. et al. IL-33 promotes Staphylococcus aureus-infected wound healing in mice. Int. Immunopharmacol. 17, 432–438 (2013).
Kudva, A. et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J. Immunol. 186, 1666–1674 (2011).
Robinson, K.M. et al. Influenza A exacerbates Staphylococcus aureus pneumonia by attenuating IL-1β production in mice. J. Immunol. 191, 5153–5159 (2013).
Small, C.-L. et al. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. J. Immunol. 184, 2048–2056 (2010).
Rostan, O. et al. Crucial and diverse role of the interleukin-33/ST2 axis in infectious diseases. Infect. Immun. 83, 1738–1748 (2015).
Palomo, J., Dietrich, D., Martin, P., Palmer, G. & Gabay, C. The interleukin (IL)-1 cytokine family—balance between agonists and antagonists in inflammatory diseases. Cytokine 76, 25–37 (2015).
Robinson, K.M. et al. Influenza a virus exacerbates staphylococcus aureus pneumonia in mice by attenuating antimicrobial peptide production. J. Infect. Dis. 209, 865–875 (2014).
Rynda-Apple, A., Robinson, K.M. & Alcorn, J.F. Influenza and bacterial superinfection: illuminating the immunologic mechanisms of disease. Infect. Immun. 83, 3764–3770 (2015).
Li, C. et al. Interleukin-33 increases antibacterial defense by activation of inducible nitric oxide synthase in skin. PLoS Pathog. 10, e1003918 (2014).
Hendricks, M.R. & Bomberger, J.M. digging through the obstruction: insight into the epithelial cell response to respiratory viral infection in cystic fibrosis. J. Virol. 90, 4258–4261 (2016).
Rynda-Apple, A. et al. Virus-like particle-induced protection against MRSA pneumonia is dependent on IL-13 and enhancement of phagocyte function. Am. J. Pathol. 181, 196–210 (2012).
Rynda-Apple, A. et al. Regulation of IFN-?? by IL-13 dictates susceptibility to secondary postinfluenza MRSA pneumonia. Eur. J. Immunol. 44, 3263–3272 (2014).
Blok, D.C. et al. Limited anti-inflammatory role for interleukin-1 receptor like 1 (ST2) in the host response to murine postinfluenza pneumococcal pneumonia. PLoS ONE 8, e58191 (2013).
Yang, Z. et al. Macrophages as IL-25/IL-33-responsive cells play an important role in the induction of Type 2 immunity. PLoS ONE 8, e59441 (2013).
Ghoneim, H.E., Thomas, P.G. & McCullers, J.A. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J. Immunol. 191, 1250–1259 (2013).
Sun, K. & Metzger, D.W. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat. Med. 14, 558–564 (2008).
Damjanovic, D., Lai, R., Jeyanathan, M., Hogaboam, C.M. & Xing, Z. Marked improvement of severe lung immunopathology by influenza-associated pneumococcal superinfection requires the control of both bacterial replication and host immune responses. Am. J. Pathol. 183, 868–880 (2013).
McCullers, J.A. & Rehg, J.E. Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J. Infect. Dis. 186, 341–350 (2002).
Sun, K. & Metzger, D.W. Influenza infection suppresses NADPH oxidase-dependent phagocytic bacterial clearance and enhances susceptibility to secondary methicillin-resistant Staphylococcus aureus infection. J. Immunol. 192, 3301–3307 (2014).
Gao, Y. et al. IL-33 exerts neuroprotective effect in mice intracerebral hemorrhage model through suppressing inflammation/apoptotic/autophagic pathway. Mol. Neurobiol. advance online publication, 12 July 2016 (2016).
Crowe, C.R. et al. Critical role of IL-17RA in immunopathology of influenza infection. J. Immunol. 183, 5301–5310 (2009).
Gambotto, A. et al. Immunogenicity of enhanced green fluorescent protein (EGFP) in BALB/c mice: identification of an H2-Kd-restricted CTL epitope. Gene. Ther. 7, 2036–2040 (2000).
Hardy, S., Kitamura, M., Harris-Stansil, T., Dai, Y. & Phipps, M.L. Construction of adenovirus vectors through Cre-lox recombination. J. Virol. 71, 1842–1849 (1997).
Acknowledgements
Funding sources include the Parker B Francis Foundation (KMR) and the NIH NHLBI (K08—HL133445-01 to KMR and R01—HL107380 to JFA)
Author contributions
K.M.R. designed research studies, conducted experiments, acquired data, analyzed data, and drafted the manuscript. K.R. conducted experiments (Figures 1, 2, 3, 4). MEC conducted experiments (Figures 1 and 2). K.J.M. conducted experiments (Figures 1, 2, 3). H.E.R. conducted experiments (Figure 1a). J.F.A. provided materials and reagents, and provided oversight in designing research studies, analyzing data, and drafting the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
Robinson, K., Ramanan, K., Clay, M. et al. Novel protective mechanism for interleukin-33 at the mucosal barrier during influenza-associated bacterial superinfection. Mucosal Immunol 11, 199–208 (2018). https://doi.org/10.1038/mi.2017.32
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2017.32
This article is cited by
-
The chemerin-CMKLR1 axis in keratinocytes impairs innate host defense against cutaneous Staphylococcus aureus infection
Cellular & Molecular Immunology (2024)
-
IFNγ-induction of TH1-like regulatory T cells controls antiviral responses
Nature Immunology (2023)
-
GRK2 regulates group 2 innate lymphoid cell mobilization in sepsis
Molecular Medicine (2022)
-
The Analyses of Cetacean Virus-Responsive Genes Reveal Evolutionary Marks in Mucosal Immunity-Associated Genes
Biochemical Genetics (2022)
-
A network map of IL-33 signaling pathway
Journal of Cell Communication and Signaling (2018)
