
Abstract
MyD88 adapter-like (Mal)-deficient mice displayed increased susceptibility to oral but not intraperitoneal infection with Salmonella Typhimurium. Bone marrow chimeras demonstrated that mice with Mal-deficient non-hematopoietic cells were more susceptible to infection, indicating a role for Mal in non-myeloid cells. We observed perturbed barrier function in Mal−/− mice, as indicated by reduced electrical resistance and increased mucosa blood permeability following infection. Altered expression of occludin, Zonula occludens-1, and claudin-3 in intestinal epithelia from Mal−/− mice suggest that Mal regulates tight junction formation, which may in part contribute to intestinal integrity. Mal interacted with several protein kinase C (PKC) isoforms in a Caco-2 model of intestinal epithelia and inhibition of Mal or PKC increased permeability and bacterial invasion via a paracellular route, while a pan-PKC inhibitor increased susceptibility to oral infection in mice. Mal signaling is therefore beneficial to the integrity of the intestinal barrier during infection.
Similar content being viewed by others


Introduction
In recent years, there has been increasing recognition of the fundamental importance of both innate immunity and epithelial barrier dysfunction in the pathogenesis of intestinal inflammation during infection or in inflammatory bowel disease.1 Within the gastrointestinal (GI) tract, a single layer of intestinal epithelial cells (IECs) function to prevent entry of commensal bacteria and luminal antigens, whereas the gut-associated lymphoid tissue, including Peyer’s patches (PP) and lamina propria (LP) lymphocytes are at the forefront of the systemic immune response.
In order to gain access to the host, pathogens must first breach the intestinal epithelial barrier, thus, the mechanisms regulating this barrier is of fundamental importance to the prevention of many diseases. IECs express Toll-like receptors (TLRs) and NOD-like receptors that recognize microbial moieties and promote intestinal homeostasis through induction of anti-inflammatory cytokines and antimicrobial peptides, which limit bacterial invasion.2, 3, 4, 5, 6 NOD2 signaling induces cryptidins, whereas TLR signaling via MyD88 induces RegIIIγ and α-defensins, which mediate bacterial immunity within the intestine.6, 7, 8 TLR signaling also promotes barrier integrity through induction of epidermal growth factor receptor ligands, which promote tissue repair, and anti-apoptotic factors such as interleukin-6, which promote epithelial restitution and fortify intercellular tight junctions (TJs).5, 9, 10
TJs are a crucial component of the epithelial barrier, dynamic multiprotein complexes, regulated by the crosstalk of many signaling pathways that control passive diffusion of allergens and pathogens through the paracellular space.11 Zonula occludens (ZO) proteins are peripherally associated membrane proteins, which function as scaffolds, anchoring transmembrane proteins like claudins and occludin to the actin skeleton.12 Phosphorylation of TJ proteins by multiple kinases is associated with TJ regulation with ZO-1 and occludin interacting with members of the protein kinase C (PKC) family, which are associated with TJ assembly and promotion of barrier function.13, 14, 15
In the current study, we suggest a previously unidentified role for the TLR2 and more particularly TLR4 adaptor, MyD88 adapter-like (Mal), also known as TIRAP, in maintaining the intestinal epithelial barrier during infection with Salmonella enterica serovar Typhimurium UK-1 (S. Typhimurium UK-1). We have determined that Mal acts through activation of PKC to modulate TJ protein expression and is a key participant in the maintenance of intestinal barrier integrity during infection and injury.
Results
Mal signaling in epithelial cells maintains the intestinal barrier and protects against infection with S. Typhimurium UK-1
To examine the function of Mal in the intestinal epithelium and its role in defense against infection, we used an oral model of infection with the Gram-negative food-borne pathogen S. Typhimurium UK-1, recognized by both TLR2 and TLR4. Salmonella’s main route of access to the host is via the GI tract and in order to establish a successful infection, Salmonellae must first traverse the intestinal epithelium. Mice were orally infected with S. Typhimurium UK-1 and bacterial dissemination was analyzed. At 1 h post infection (p.i.), comparable levels of bacteria were detected in intestinal LP of both wild-type (WT) and Mal−/− mice, however, there was a significantly (P<0.05) greater bacterial load in the mesenteric lymph node (MLN) of Mal−/− mice compared with WT mice indicating that Salmonella transits from the GI tract to MLN at a greater rate in the absence of Mal (Figure 1a). Indeed, although no bacteria were detected in the livers and spleens of both mice at 1 h p.i., significantly a 1-2 Log increase in bacterial numbers was detected in livers and spleens of Mal−/− mice 6 days p.i., indicating that Mal−/− mice are more susceptible to dissemination of Salmonella (Figure 1b). As this may be a consequence of either increased penetration and translocation of the epithelial layer by invasive Salmonella, increased trafficking of immune cells or impaired clearance of infection, we performed intraperitoneal (i.p.) infection of WT and Mal−/− mice with S. Typhimurium to determine whether the increased bacterial load observed in livers and spleens of Mal−/− mice was due to a defect in the function of macrophages or other cell types in these organs. Interestingly, we observed similar levels of dissemination of S. Typhimurium in both WT and Mal−/− mice following i.p. infection, indicating a role for Mal in the maintenance of intestinal barrier integrity, which is independent of bacterial clearance by immune cells (Figure 1c). Indeed, to compare the role of Mal signaling in non-hematopoietic and hematopoietic cells in the maintenance of the intestinal barrier integrity, we generated WT and Mal−/− chimeras and performed oral infection with S. Typhimurium. Consistent with data in Mal−/− mice (Figure 1a), irradiated Mal−/− mice reconstituted with Mal−/−bone marrow (Mal−/−>Mal−/−) were significantly more susceptible than WT>WT mice to oral S. Typhimurium infection (Figure 1d–f). Furthermore, chimeras with non-hematopoietic cell Mal deficiency had increased susceptibility to bacterial infection relative to WT>WT mice and animals with Mal−/− hematopoietic cells, as seen by weight loss (Figure 1d) and increased bacterial burden in livers and spleens (Figure 1e and f). Collectively, these data demonstrate that Mal is involved in susceptibility to oral bacterial infection via a mechanism dependent on Mal expression in non-hematopoietic cells, which regulates bacterial translocation of gut epithelia.

MyD88 adapter-like (Mal) deficiency in mice leads to increased dissemination of Salmonella. Wild-type (WT) and Mal−/− mice were orally infected with Salmonella Typhimurium UK-1 at 5 × 107 CFU per mouse and (a) 1 h post infection (p.i.) or (b) 6 days p.i. bacterial numbers in organs and tissues were determined. Data represent means±s.e.m. (n=4; results shown are representative of three independent experiments), (c) WT and Mal−/− mice were intraperitoneally infected with Salmonella Typhimurium UK-1 at 1 × 106 colony-forming unit (CFU) per mouse and 3 days p.i. bacterial numbers in livers and spleens were determined. Data represent means±s.e.m. (n=5; results shown are representative of three independent experiments). (d–f) Mal−/− and WT bone marrow chimaeric mice were generated as described in Methods. Mice were infected orally with 3 × 108 Salmonella Typhimurium UK-1 CFU per mouse and culled after 6 days. (d) Weight loss (% change from day 0 to day 6 p.i.) and bacteria numbers in livers (e) and spleens (f) were determined. Data are means (n=7) and representative of three separate experiments. *P<0.05; **P<0.01; ***P<0.001. LP, lamina propria; MLN, mesenteric lymph node; PP, Peyer’s patches.
Mal deficiency leads to perturbed homeostatic intestinal barrier function and increased intestinal permeability following challenge
To begin deciphering the exact contribution of Mal to homeostatic intestinal barrier function, we first compared the transepithelial resistance (TER) and permeability of ex vivo colons from WT and Mal−/− mice. Although Mal-deficient animals consistently had slightly lower basal TER (Figure 2a), this effect was not significantly (P<0.073) different when compared with WT mice. Altered TER can be explained by modification of either transepithelial ion conductance and/or epithelial permeability, so to distinguish whether Mal influences homeostatic intestinal permeability and/or ion conductance, we examined ion channel flux. We found a significant (P<0.05) difference between Mal−/− and WT mice in baseline short-circuit current (Isc; Figure 2b). Subsequent analysis of Cl− secretory responses showed that stimulation with the Ca2+-dependent secretagogue carbachol led to a significant (P<0.01) difference in Isc between colons from Mal−/− and WT mice, with no differences following cyclic adenosine monophosphate-dependent forskolin stimulation (Figure 2b; data not shown).

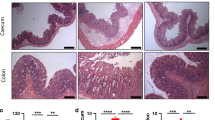
MyD88 adapter-like (Mal) deficiency leads to perturbed barrier function. (a) Intestinal resistance (transepithelial resistance (TER), Ω cm−2) was measured ex vivo on muscle stripped colons from naive C57BL/6 wild-type (WT) and Mal−/− mice mounted in Ussing chambers. (b) Apical amiloride (100 μM) was added to inhibit electrogenic Na+ absorption, and subsequent short circuit current responses (Isc) to carbachol (100 μM) and forskolin (10 μM) were measured. Values shown are means±s.d. (n=6). Intestinal mucosa-to-blood permeability measured by fluorescein isothiocyanate (FITC)-dextran 4 kDa concentration in the plasma (c) of uninfected WT and Mal−/− mice (n=4), (d) following infection of WT and Mal−/− mice with S. Typhimurium UK-1 (n=5). Data are means±s.d.. *P<0.05 (t-test). (e) Paraffin-embedded colon sections were stained using the terminal deoxinucleotidyl transferase-mediated dUTP-biotin nick end-labeling (TUNEL) assay and the numbers of TUNEL+ epithelial cells per crypt enumerated. (f) Numbers of Ki67+ epithelial cells per crypt in colon sections. Ki67+ or TUNEL+ epithelial cells were quantified in 15–20 crypts per mouse with three or four mice per group; data are represenative of three separate experiments.
We next examined in vivo intestinal paracellular permeability by luminal mucosa-to-blood flux of fluorescein isothiocyanate (FITC)-dextran in untreated WT and Mal−/− mice (Figure 2c). In normal Mal−/− and WT mice, there was no differences in serum FITC-dextran levels, consistent with a functional intestinal barrier in the steady state. There was, however, a significant difference (P<0.05) in mucosa-to-blood flux between Mal−/− and WT mice following oral challenge with Salmonella 30 min before administration of FD4 (Figure 2d).
To decipher how Mal influences intestinal permeability and barrier function, we determined the involvement of Mal in regulation of anti-apoptotic pathways by terminal deoxinucleotidyl transferase-mediated dUTP-biotin nick end-labeling staining to assess DNA fragmentation of IECs. There was no difference in apoptosis in IEC from both WT and Mal−/− (Figure 2e), indicating that increased intestinal permeability in Mal−/− is not due to loss of anti-apoptotic regulation. Furthermore, there was no difference in the proliferation of IEL in the colons of WT and Mal−/− mice (Figure 2f). Collectively, these data show that in the steady state Mal mice have a functional, but perturbed, intestinal barrier that is compromised following oral bacterial challenge.
Mal regulates expression and localization of TJ proteins within intestinal epithelium
Next, as intercellular junctions are crucial for epithelial barrier function, we analyzed the involvement of TJ dysregulation in intestinal barrier breach in Mal−/− mice by staining intestinal sections from WT and Mal−/− mice for key components of TJs, including claudin-3, ZO-1, and occludin (Figure 3a–c). Comparing naive WT and Mal−/− mice, both claudin-3 (Figure 3a top panel, Mean fluorescence intensity (MFI) WT: 14,873, Mal−/−: 8,300; P=0.008) and occludin (Figure 3a bottom panel, mean MFI WT: 9,260, Mal−/−: 6,864; P=0.020) showed significantly lower basal expression in Mal−/− mice. Although there was not a significant decrease in the basal level of ZO-1 within the epithelium (Figure 3a middle panel, mean MFI WT: 17,381, Mal−/− 12,836; P=0.067), there was a severe mislocalization of the protein in Mal−/− mice compared with WT mice (Figure 3a middle panel). All of these results are quantified in Figure 3b. In naive WT mice, ZO-1 is localized along the junction of the epithelial cells, thus forming scaffolding to which TJ proteins such as occludin and claudins can attach (Figure 3a and c). In marked contrast, in Mal−/− mice very little ZO-1 protein was localized to the TJ and instead we observed a high cytoplasmic and nuclear signal (Figure 3a and c). Thus, Mal regulates the protein expression of both claudin-3 and occludin, and the localization of ZO-1 to the TJ.

Altered epithelial tight junction (TJ) due to downregulation and mislocalization of TJ protein expression in Mal−/− mice. (a) Expression of TJ proteins claudin-3, Zonula occludens (ZO-1), and occludin was measured by immunofluorescence in intestinal samples from wild-type (WT) and Mal−/− mice under basal conditions or following infection with S. Typhimurium UK-1. Representative images are shown. (b) The mean fluorescence intensity (MFI) in Salmonella-infected and basal WT and Mal−/− mice are shown as means±s.e.m. (c) Three-dimensional images of ZO-1 staining in Mal−/− mice compared with WT controls. For all immunofluoresence analysis, three biological replicates (n=3 mice per group) were used and 20–30 images were obtained for each biological replicate, thus a total of 60–90 images were analyzed for each experimental group. Mean MFI values for each biological replicate were used to perform an unpaired two-tailed t-test. **P<0.01.
Upon assessment of S. Typhimurium-infected tissues, we determined that in WT mice S. Typhimurium infection caused a significant decrease in both ZO-1 (Figure 3a middle panel, MFI WT: 17,381, Infected: 10,149; P<0.001) and occludin (Figure 3a bottom panel, MFI WT: 9,260, Infected: 7,206; P=0.022) proteins (Figure 3a, quantified in Figure 3b). Claudin-3 levels in WT mice were upregulated, although with great variability and thus were not significant (MFI WT: 14,873, Infected: 22,407; P=0.26; Figure 3a and b). The decreases in expression of ZO-1 and occludin were comparable in intensity to levels observed in uninfected Mal−/− mice. Thus, infection of Mal−/− mice did not cause an additional decrease in TJ protein expression and exacerbate the effects of S. Typhimurium; however, this may be due to expression of these proteins already being at a minimal level.
Mal deficiency leads to disruption of TJ and increased bacterial invasion via the paracellular route in a PKC-dependent manner
The human Caco-2 cell line was employed as an in vitro tool to analyze the mechanism by which Mal regulates intestinal barrier function. Permeability through the paracellular space can be evaluated by comparing rates of bacterial invasion and translocation across monolayers grown on porous inserts, and also by measuring the TER of these monolayers. In Caco-2 monolayers pre-treated with an inhibitory Mal peptide, there was no difference in invasion of these monolayers by Salmonella compared with untreated monolayers (Figure 4a). However, we observed fivefold greater levels of bacteria translocating the epithelial barrier upon inhibition of Mal (P<0.05) (Figure 4b). There was also a significant (P<0.05) decrease in TER across monolayers after inhibition of Mal (Figure 4c). This indicates that bacteria are translocating the epithelial barrier via the paracellular route. Caco-2 monolayers pre-treated with an inhibitor to MyD88, a TLR adaptor that functions downstream of Mal on the TLR4 pathway, also displayed similar levels of invasion (Figure 4d) compared with untreated controls but increased translocation (Figure 4e) by S. Typhimurium and reduced TER (Figure 4f) across monolayers after inhibition of MyD88. As the kinase PKC has been shown to maintain TJs, we also tested a pan-PKC inhibitor (bisPKC) here. In Caco-2 monolayers pre-treated with the inhibitor bisPKC, although there was no difference in invasion by Salmonella of these monolayers compared with untreated controls (Figure 4a), there was a significant (P<0.05) increase in the number of bacteria translocating PKC-inhibited monolayers (Figure 4b). There was also a significant (P<0.05) decrease in TER across monolayers after treatment with the pan-PKC inhibitor (Figure 4c).

MyD88 adapter-like (Mal) deficiency leads to disruption of epithelial tight junctions via a protein kinase C (PKC)-dependent pathway and promotes paracellular bacterial invasion. (a) Invasion and (b) translocation of Caco-2 monolayers by S. Typhimurium UK-1 following treatment with Mal-inhibitory peptide or pan-PKC (bisPKC) inhibitor (open bars). (c) Transepithelial electrical resistance (TER) measured in Caco-2 monolayers grown on transwell inserts and treated with Mal-inhibitory peptide or pan-PKC (bisPKC) inhibitor (open bars), presented relative to untreated monolayers (solid bars). (d) Invasion and (e) translocation of Caco-2 monolayers by S. Typhimurium UK-1 following treatment with a MyD88 inhibitor (open bars). (f) TER measured in Caco-2 monolayers grown on transwell inserts and treated with a MyD88 inhibitor (open bar), presented relative to untreated monolayers (solid bars). Data are means±s.d. (n=3; results shown are representative of at least three independent experiments). *P<0.05 (Student’s t-test) compared with control monolayers (black bar, and corresponding gray bars) for a, b, c, d. CFU, colony-forming unit, DMSO, dimethyl sulfoxide.
Mal interacts with PKC
A previous report indicated that Mal could interact with PKCδ in macrophages.16 To determine whether Mal interacts with PKC in our study, Caco-2 cells were transfected with hemagglutinin (HA)-Mal and stimulated with lipopolysaccharide (LPS). Lysates were subjected to immunoprecipitation with an antibody to phosphorylated pan-PKC and immunoprecipitates were analyzed for HA-Mal. As can be seen in Figure 5a, a marked increased in association between Mal and phosphoPKC was evident following 120 min treatment with LPS (compare lanes 7 and 8). Similar levels of Mal were present in the lysates (lanes 3 and 4) and background levels of Mal isolated with a control IgG were negligible (lanes 5 and 6). The phospho-pan-PKC antibody can immunoprecipitate multiple PKC isoforms. We therefore determined that PKCs are activated following infection of Caco-2 cells with Salmonella, and the sensitivity of any isoforms to Mal inhibition. As shown in Figure 5b top panel, Salmonella infection increased phospho-pan-PKC in Caco-2 cells from 20 min, the amount waning by 120 min (top panel, left hand side). Pre-treatment with the Mal inhibitory peptide decreased this response (for example, compare lanes 9 and 10 to lanes 3 and 4, respectively). Salmonella infection activated phospho-PKCα/β from 30 min (second panel, left hand side), PKCμ/PKD (fourth panel, left hand side) from 20 min, PKCζ from 10 min (fifth panel, left hand side), but not PKCδ/θ (third panel, left hand side). The activation of PKCα/β was not inhibited by the Mal inhibitory peptide (second panel, right hand side). However, activation of both PKCμ/PKD and PKCζ were inhibited (right hand side, panels 4 and 5, respectively). As PKCζ has been found associated with TJs in Caco-2 cells,17 we next tested for an interaction with Mal. From co-immunoprecipitation assays, we identified an interaction between Mal and PKCζ, both in unstimulated cells and in cells treated with LPS (Figure 5c, lanes 7 and 8) and infected with Salmonella (Figure 5d, lanes 7 and 8). No interaction was observed between Mal and PKCδ (Figure 5e).

MyD88 adapter-like (Mal) interacts with protein kinase C (PKC) and signals via a PKC-dependent pathway to maintain tight junction integrity. (a) Lysates from Caco-2 cells treated with lipopolysaccharide (LPS) were immunoprecipitated (IP) with phosphorylated pan-PKC antibody, fractionated by SDS-polyacrylamide gel electrophoresis and blotted for HA-Mal. (b) Activation of indicated PKC isoforms (as assessed by immunoblotting (IB) for phosphorylated forms) in Caco-2 cells infected with S. Typhimurium UK-1 and treated with a Mal-inhibitory peptide or corresponding control inhibitor peptide. Loading controls of PKCζ and β-actin are shown in lower 2 panels. (c) Caco-2 cells treated with LPS, lysates prepared, and IP with PKCζ antibody and blotted for HA-Mal. (d) Lysates from Caco-2 cells infected with S. Typhimurium UK-1 were IP with PKCζ antibody and blotted for HA-Mal. (e) Lysates from Caco-2 cells treated with LPS were IP with PKCδ antibody and blotted for HA-Mal. (f) Transepithelial electrical resistance (TER) measurements across Caco-2 monolayers grown on transwell inserts and treated with PKC inhibitors to θ, Gö 6983, and ζ/ι, (5 μg ml−1), data presented relative to untreated monolayers. Data are means±s.d. (n=3; data shown is representative of three independent experiments). *P<0.05 (Student’s t-test) compared with control monolayers (black bar and gray bar). (g) Treatment of mice with a pan-PKC inhibitor (bisPKC) (0.2 mg per mouse) before oral infection with Salmonella greatly increases dissemination of bacteria to livers and spleens (n=7). *P<0.05 compared with phosphate-buffered saline (PBS) treated mice (Student’s t-test). DMSO, dimethyl sulfoxide.
We also tested selective inhibition of PKCθ and PKCζ on TER in Caco-2 cells and found that PKCζ/ι inhibition decreased this significantly (P<0.05), whereas inhibition of PKCθ had no effect (Figure 5f). The atypical PKC inhibitor Gö 6983, also significantly (P<0.05) reduced TER (Figure 5f). To confirm a role for PKC in barrier function in vivo, mice were gavaged with the bisPKC inhibitor 30 min before oral infection with S. Typhimurium UK-1 (Figure 5g). Mice in which PKC was inhibited displayed significantly (P<0.05) more bacterial dissemination to livers and spleens compared with mice given a phosphate-buffered saline (PBS) control (Figure 5g). Taken together, our data point to a critical role for Mal in maintaining the intestinal epithelial barrier, via the observed interaction with and activation of PKC, with PKCζ being a key isoform for maintenance of TJs.
Discussion
The ultrastructure of TJs and the intracellular signaling events governing their modulation is becoming better understood. However, the regulation of TJs secondary to extracellular stimuli resulting in disease or injury, remains less well characterized.18 Our present findings identify a mechanism whereby TLR signaling via Mal regulates TJs through regulation of TJ proteins ZO-1, claudin-3, and occludin, and simultaneous activation of PKC in particular PKCζ, causing occludin, ZO-1, and claudin-3 to localize at TJs.
Expression of Mal has previously been shown to be detrimental to the clearance of Salmonella following infection of mice intravenously, most likely because of impaired function of Mal-deficient macrophages.19 However, as Salmonella is primarily a food-borne pathogen whose normal route of entry into the host is via the GI tract, we performed oral infection of mice and observed increased susceptibility of Mal−/− mice to dissemination by Salmonella. The clear distinction between the intravenous (i.v.) and oral route in terms of Mal dependency is worth emphasizing. Clearly during i.v. infection, Salmonella provokes a systemic inflammatory response via induction of tumor necrosis factor-α in the macrophage. Mal-deficient mice are therefore protected from i.v.-administered Salmonella. When given orally, however, the natural route of infection, Mal-deficient mice are more susceptible because of enhanced bacterial dissemination.
Following oral infection, Salmonellae cross intestinal epithelium mainly, but not exclusively, through M-cells in follicle-associated epithelium of PPs and are then internalized by macrophages, dendritic cells, or neutrophils, which serve as vehicles for bacterial dissemination from MLN to deeper organs.20 However, while we observed increased bacterial numbers in MLN of Mal-deficient mice at 1 h p.i., there were similar levels in PPs of WT and Mal-deficient mice suggesting a route of entry independent of M cells following oral infection in the absence of Mal. Several PP-independent mechanisms of entry have been suggested for Salmonella including a recent observation that myeloid cells residing in LP provide a direct portal of entry for Salmonella, with dendritic cells transporting bacteria to MLN and spleen.21, 22 In our study, despite an increase in bacterial load in the MLN at 1 h p.i. in Mal-deficient mice, similar levels were detected in LP of both WT and Mal-deficient mice. This could have been due to enhanced trafficking of Salmonella by dendritic cells to the MLN, but this was thought unlikely given the lack of increased dissemination of Salmonella in chimeric mice reconstituted with Mal-deficient bone marrow. This result, along with the lack of difference in dissemination in Mal-deficient mice infected by the IP route, indicated that the defect in Mal-deficient mice was unlikely to be in myeloid cells. It is possible that Mal in the endothelium might limit trafficking of Salmonella to the MLN. The observation that there was no increase in bacterial load in the LP at 1 h p.i. is difficult to explain if there is a defective barrier in the Mal-deficient mice. It is possible that Mal does not have a role in preventing entry of bacteria into M cells and subsequent entry into the LP, but instead Mal in the endothelium prevents trafficking to the MLN. These aspects require further investigation but it is likely that Mal is playing a role outside the intestinal barrier in oral challenge studies described herein.
TLR2 has been shown to mediate intestinal homeostasis through induction of anti-apoptotic events mediated by the PI3K/Akt pathway, promoting cell survival and preventing stress-induced disruption and depolarization of the barrier and redistribution of ZO-1 at TJ.23, 24 Oral treatment of mice with the TLR2 ligand Pam3Cys-SK4 suppressed epithelial apoptosis and prevented inflammation highlighting the potential to develop adjuvants, which stimulate intestinal barrier function.23 TLR4−/− mice display increased epithelial damage due to reduced epithelial cell proliferation and increased bacterial translocation to MLNs, however, systemic dissemination was not detected.25, 26 The TLR adaptor MyD88 has also been shown to protect against intestinal inflammation with MyD88−/− mice developing more severe colitis in both the dextran sodium sulfate and Citrobacter rodentium models.27, 28 This susceptibility was due in part to increased bacterial burden due to impaired tumor necrosis factor-α and interleukin-6 responses.28 MyD88−/− mice also displayed reduced expression of epidermal growth factor receptor ligands, amphiregulin and epiregulin, in nonhematopoietic cells.8, 10 As these ligands are anti-apoptotic in the epithelium, this observation highlights the importance of IEC homeostasis in protection against development of colitis.
The observed increased susceptibility of Mal−/− mice to Salmonella infection following oral administration of stimuli, led us to investigate how intestinal barrier function and homeostasis was impaired in these mice. Permeability through the paracellular space can be evaluated by two methods, the measurement of TER and the flux of tracers such as FITC-dextran. Reduced TER and permeability changes in colons from Mal−/− mice ex vivo indicates an impaired barrier. As the integrity of TJs and thus paracellular permeability are dependent on proper expression and localization of TJ proteins, using immunohistochemistry we investigated the expression and distribution of occludin, claudin-3, and ZO-1 within intestinal samples from WT and Mal−/− mice, naive and following infection with S. Typhimurium. We observed reduced basal expression of claudin-3, occludin, and ZO-1 in intestinal epithelia from naive Mal−/− mice compared with naive WT mice, suggesting impaired TJ structure. Following infection, expression of claudin-3 was further reduced in Mal−/− epithelium, suggesting that TJ are more readily disrupted in the absence of Mal signaling. Interestingly, we also observed redistribution of ZO-1 in the epithelium of Mal−/− mice with ZO-1 having a focused nuclear expression in these mice. ZO-1 has been shown to be key to the formation of TJs.29, 30, 31 Localization is very important for ZO-1 as it is the adapter protein to which the “backbone” TJ proteins such as claudin and occludins bind to. It can be found throughout the cell, but needs to be localized to the cell membrane to function properly as a TJ adapter between the TJ proteins and F-actin. Epithelial cells that lack ZO-1 fail to properly assemble TJ structures and lack a TJ permeability barrier.31 It is possible that the defect in ZO-1 could also alter the actin cytoskeleton, which could contribute to the altered intestinal integrity in Mal-deficient mice. Although nuclear localization of ZO-1 has been described, its nuclear function still remains ill-defined. Current experimental evidence links nuclear ZO-1 localization as inversely related to cellular maturity and cell-to-cell contact/adhesion.29, 32, 33 In the case of ZO-2, it has been shown to concentrate at the nucleus when subjected to adverse conditions such as chemical stress or mechanical injury.34
Barrier dysfunction and increased permeability were confirmed in Caco-2 cells, in which inhibition of Mal decreases TER, suggesting a basal role for Mal in maintaining the barrier. What activates Mal in this situation is not known. It is possible that commensals are responsible for Mal activation in the intestine. Caco-2 cells may produce extracellular matrix components such as fibronectin, which act as a TLR4 ligand and therefore may be responsible for Mal activation in the absence of bacteria.35 Although there is no overt “leakiness” in the barrier in vivo as assessed using FITC-dextran permeability, this is significantly increased in Mal−/− following oral challenge with Salmonella confirming a role for Mal in the maintenance of intestinal barrier integrity. Mal is therefore likely to regulate barrier function in response to TLR4 stimuli such as LPS from Salmonella, which would lead to the tightening of the paracellular pathway, and thus reduced permeability to FITC-dextran in the gut of WT mice. However, we must not rule out the possibility that the increased paracellular permeability following Salmonella challenge is due to an enhanced ability of Salmonella to disrupt already weakened epithelial TJs in Mal−/− mice, as Salmonellae have been shown to disrupt TJs, thus promoting paracellular invasion.36 Indeed, we observed a further reduction in expression of claudin-3 in Mal−/− epithelium following infection, suggesting that TJs are more readily disrupted in the absence of Mal signaling.
The question arose as to how Mal was maintaining the intestinal barrier. Using the well-studied Caco-2 model and immunohistochemistry of intestinal samples, we could confirm that Mal maintains paracellular permeability by regulating expression of TJ proteins and further that PKC was involved. PKC is known to regulate localization of TJ proteins within epithelial cells. Indeed, TLR2 activation has been shown to enhance ZO-1-associated barrier integrity via phosphorylation of PKC, which regulates localization of TJ proteins within epithelial cells.37 Stimulation of HT29 and Caco-2 epithelial cells with TLR2 ligands leads to activation of specific PKC isoforms α and δ, which enhances barrier function through translocation of ZO-1 to the TJ.37 TLR2 activation enhances TJ-associated barrier function in epidermal keratinocytes via PKCζ/ι.38 PKCζ has been shown to regulate both ZO-1 and occludin localization at the epithelial TJ in Caco-2 and MDCK monolayers, with inhibition of PKCζ by a specific PKCζ pseudosubstrate peptide resulting in reduced phosphorylation of occludin and ZO-1 and redistribution of both TJ proteins from the intercellular junction and disruption of barrier function.13, 15 Recently, intratracheal administration of a specific PKCδ peptide inhibitor was shown to protect mice against polymicrobial sepsis-induced lung injury.39 Here, we show for the first time that oral treatment of mice with a bisPKC inhibitor as little as 30 min before infection with Salmonella significantly increases the dissemination of bacteria to organs. These findings highlight the potential for in vivo modification of the PKC-TJ pathway through the use of adjuvants that would strengthen intestinal barrier integrity against infection. Multiple PKC isoforms may be involved here. PKCα/β, ζ, and μ are activated by Salmonella in Caco-2, however, we found Mal strongly interacts with PKCζ and a selective inhibitor led to decreased TER. This indicated that PKCζ may be a key PKC isoform downstream of Mal, which executes the role of Mal in barrier function. Our observation is the first describing interaction between PKCζ and Mal. Mal has previously been shown to interact with PKCδ following LPS stimulation of rat peritoneal macrophages and THP1 cell lysates.16 Thus, in epithelial cells we have identified a TLR4-Mal-dependent pathway that requires Mal activation to drive expression of TJ proteins via induction of interleukin-10 and while simultaneously activating PKC, and specifically PKCζ, to regulate subsequent TJ assembly in the intestinal epithelium. Interestingly, in the steady state, despite a perturbed intestinal barrier, Mal−/− mice do not develop overt inflammation. However, during infection or injury, this essential role for Mal in maintaining intestinal barrier integrity is evident.
Intestinal barrier function and “leaky gut” are crucial to the pathogenesis of many disorders, not just GI disorders. Interestingly, intestinal barrier breach by non-self-antigens has been implicated in development of multiple sclerosis and autism, with patients being associated with increased intestinal permeability in both cases.40, 41 The identification of a previously uncharacterized role for Mal in the maintenance of intestinal barrier function aids our understanding of the signaling pathways which maintain barrier integrity. These new insights for a role for Mal in maintenance of GI integrity could ultimately enable the design of new therapeutic approaches for the treatment of a broad spectrum of human disorders based on targeted modulation of barrier-protective TLR signaling pathways and re-establishment of intestinal homeostasis.
Methods
Mice. Mal-deficient (−/−) mice, on a C57BL/6 background, were described previously.42 WT mice were bred in house. CD45.1+ mice were obtained from Jackson Labs (Bar Harbor, ME) and bred in house. Animal experiments were performed in compliance with Irish Department of Health and Children Regulations and approved by Trinity College Dublin’s BioResources Ethical Review Board.
In vivo infections. Mice were orally inoculated with 5 × 107 colony-forming unit (CFU) of S. Typhimurium UK-1 and 1 h or 6 days p.i. Log CFU per organ was determined. Mice were intraperitoneally infected with S. Typhimurium UK-1 at 1 × 106 CFU and 3 days p.i. Log CFU per organ was determined.
Bone marrow chimeras. Lethally irradiated CD45.1+ WT and CD45.2+ Mal−/− recipient mice (9 Gy in 2 doses, 3 h apart) were reconstituted with 1 × 107 bone marrow cells from CD45.1+ WT or CD45.2+ Mal−/− mice. The following chimeras were generated: WT>WT; WT>Mal−/−; Mal−/−>Mal−/−; Mal−/−>WT. To ensure efficient irradiation and reconstitution, after 6 weeks cell expression of CD45.1 (A20; BD Biosciences) vs. CD45.2 (104; BD Biosciences, Franklin Lakes, NJ) was assessed in the blood by flow cytometry with >95% reconstitution achieved.
Intestinal permeability. Intestinal permeability was determined by measuring the appearance in blood of FITC-dextran (molecular mass 4 kDa; Sigma, St Louis, MO) administered by gavage at 12 mg per mouse in sterile PBS as previously described.43 Mice were either uninfected or orally infected with 5 × 107 CFU of S. Typhimurium UK-1 30 min before gavage.
Electrophysiological measurements. Electrophysiological measurements were performed on the distal colon as previously reported.44 Following equilibration, TER was recorded every 5 min for 60 min, and averaged to assess the basal TER. TER data were normalized and expressed as Ω cm−2. To examine Cl- secretion, following amiloride (10 μM) treatment, the secretagogues carbachol (100 μM basolaterally) and forskolin (10 μM apically) were used to stimulate Ca2+ and cyclic adenosine monophosphate-mediated Cl− secretion, respectively. Results were normalized and expressed as ΔIsc (μA cm−2).
Cell culture. Caco-2 cells were seeded at a high density of 5 × 105 cells per ml for invasion assays and at 5 × 105 cells per transwell filter (3 μm pore size, Costar, Corning, NY) for translocation studies, and cultured postconfluence.
Inhibition of Mal, PKC, and MyD88 activity in vitro.Before in vitro assays, monolayers were pre-treated for 2 h with 5 μg ml−1 of either Mal-inhibitory peptide, inhibitor peptide control, MyD88 inhibitor, bisindolylmaleimide (bis)-PKC inhibitor, or PKC inhibitors to θ, ζ/ι, and Gö 6983 (α, β, γ, δ, ζ; all Calbiochem, Merck, Millipore, MA).
In vitro infection and translocation assays. Following pre-treatments, S. Typhimurium UK1 were added at 1 × 107 CFU per well for 1 h or 1.5 h for invasion and translocation, respectively. To determine infection, media containing gentamycin (100 μg ml−1) were added for 15 min, monolayers washed and lyzed. Rates of invasion were expressed as LogCFU per ml. For translocation, samples were taken from the basolateral chamber and rates of translocation determined, expressed as LogCFU per ml. TER, expressed as TER Ω cm−2 and as a percentage of untreated monolayers, was measured before and after pre-treatment with inhibitors.
Quantification of epithelial cell proliferation and apoptosis. Epithelial cell proliferation was analyzed by Ki67 staining. Colon sections were blocked for nonspecific background staining using 10% normal goat serum (DakoCytomation, Glostrup, Denmark), incubated overnight with rabbit anti-mouse-Ki67 (Abcam, Cambridge, UK), visualized by EnVision Detection System (DakoCytomation) and counterstained using Mayer’s hematoxylin. Epithelial cell apoptosis was analyzed by terminal deoxinucleotidyl transferase-mediated dUTP-biotin nick end-labeling assay using a commercial kit (In Situ Cell Death Detection kit, Roche, Dublin, Ireland) according to the manufacturer. Sections were imaged using an Olympus BX43 with an Olympus DP25 Camera.
Immunohistochemistry. Immunohistochemical analysis of TJ proteins was performed using 4% paraformaldehyde-fixed, paraffin-embedded ileum tissues in biological and experimental triplicate. Five micrometer sections were cut and rehydrated through a graded series of ethanol and PBS. Antigen retrieval was performed with using isocitrate buffer at 95°C for 30 min. Tissues were then blocked with 1% Fc blockers (Miltenyi Biotec, Auburn, CA) and 10% donkey serum (Jackson ImmunoResearch Laboratories, West Grove, PA) for 30 min, incubated with primary antibody overnight at 4°C and then incubated with secondary antibody for 1 h at room temperature. Primary antibodies used: polyclonal rabbit anti-mouse ZO-1, occludin, and claudin-3 (Life Technologies, Carlsbad, CA). Secondary antibody: Alexa Flour 555 donkey anti-rabbit (Invitrogen,Carlsbad, CA). Nuclei were visualized using 4′-6-diamidino-2-phenylindole nucleic acid stain (Invitrogen). Images were collected using DeltaVision PersonalDV Deconvolution microscopy by Applied Precision (Issaquah, WA). The 5-μm tissue slices were imaged using Z-stack with 0.2 μm per section (25 sections total). 2 × 2 binning was utilized during image acquisition. The sum of fluorescence intensity was calculated for the stack and MFI was determined for the epithelial regions of the tissue using Image J software (National Institute of Mental Health, Bethesda, MD). For analysis, three biological replicates (n=3 mice per group) were used and 20–30 images were obtained for each biological replicate, thus a total of 60–90 images were analyzed for each experimental group. Mean MFI values for each biological replicate were used to perform an unpaired two-tailed t-test.
Co-immunoprecipitation and immunoblot. Caco-2 cells were transiently transfected with pcDNA3.1 encoding HA-tagged Mal using GeneJuice (Novagen, Madison, WI). Forty-eight hours post transfection cells were lyzed as described.45 Antibodies (Phospho-PKC (pan) (#9371, Cell Signalling Technologies, Beverly, MA), PKCζ (H-1) sc-17781, Santa Cruz Biotechnologies, Dallas, TX), anti-hemagglutinin (MMS-101R; Covance, Princeton, NJ) or relevant IgG control antibody) were pre-incubated with Protein A/G PLUS agarose beads (sc-2003; Santa Cruz Biotechnology) for 16 h at 4°C. Immune complexes/proteins were isolated, separated by SDS-polyacrylamide gel electrophoresis and detected by western blot analysis with the appropriate antibodies (as above), including relevant Phospho-PKC Antibodies from sampler kit (#9921, Cell Signalling Technologies).
In vivo administration of PKC inhibitor. bisPKC inhibitor (0.2 mg per mouse, Calbiochem) was administered to WT mice in sterile PBS by gavage 30 min before oral infection with S. Typhimurium UK-1 at 5 × 107 CFU per mouse.
Statistical analysis. Numerical results are given as arithmetic means±standard deviations of the means. Student’s t-test was performed to determine statistical significance, P⩽0.05. In vivo experiments were performed at least three separate times with three to six mice per group.
References
Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 9, 799–809 (2009).
Abraham, C. & Medzhitov, R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology 140, 1729–1737 (2011).
Rakoff-Nahoum, S., Hao, L. & Medzhitov, R. Role of Toll-like receptors in spontaneous commensal-dependent colitis. Immunity 25, 319–329 (2006).
Vora, P. et al. Beta-defensin-2 expression is regulated by TLR signaling in intestinal epithelial cells. J. Immunol. 173, 5398–5405 (2004).
Maloy, K.J. & Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474, 298–306 (2011).
Kobayashi, K.S. et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307, 731–734 (2005).
Vaishnava, S., Behrendt, C.L., Ismail, A.S., Eckmann, L. & Hooper, L.V. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. USA 105, 20858–20863 (2008).
Frantz, A.L. et al. Targeted deletion of MyD88 in intestinal epithelial cells results in compromised antibacterial immunity associated with downregulation of polymeric immunoglobulin receptor, mucin-2, and antibacterial peptides. Mucosal Immunol. 5, 501–512 (2012).
Hsu, D. et al. Toll-like receptor 4 differentially regulates epidermal growth factor-related growth factors in response to intestinal mucosal injury. Lab. Invest. 90, 1295–1305 (2010).
Brandl, K. et al. MyD88 signaling in nonhematopoietic cells protects mice against induced colitis by regulating specific EGF receptor ligands. Proc. Natl. Acad. Sci. USA 107, 19967–19972 (2010).
Balkovetz, D.F. & Katz, J. Bacterial invasion by a paracellular route: divide and conquer. Microbes Infect. 5, 613–619 (2003).
Gonzalez-Mariscal, L., Tapia, R. & Chamorro, D. Crosstalk of tight junction components with signaling pathways. Biochim. Biophys. Acta 1778, 729–756 (2008).
Stuart, R.O. & Nigam, S.K. Regulated assembly of tight junctions by protein kinase C. Proc. Natl. Acad. Sci. USA 92, 6072–6076 (1995).
Harhaj, N.S. & Antonetti, D.A. Regulation of tight junctions and loss of barrier function in pathophysiology. Int. J. Biochem. Cell Biol. 36, 1206–1237 (2004).
Jain, S., Suzuki, T., Seth, A., Samak, G. & Rao, R. Protein kinase Czeta phosphorylates occludin and promotes assembly of epithelial tight junctions. Biochem. J. 437, 289–299 (2011).
Kubo-Murai, M. et al. Protein kinase Cdelta binds TIRAP/Mal to participate in TLR signaling. Mol. Immunol. 44, 2257–2264 (2007).
Dodane, V. & Kachar, B. Identification of isoforms of G proteins and PKC that colocalize with tight junctions. J. Membr. Biol. 149, 199–209 (1996).
Fasano, A. & Shea-Donohue, T. Mechanisms of disease: the role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases. Nat. Clin. Pract. Gastroenterol. Hepatol. 2, 416–422 (2005).
Jerke, S., Srinivasan, A. & McSorley, S.J. Expression of Toll/IL-1R domain-containing adaptor protein (TIRAP) is detrimental to primary clearance of Salmonella and is not required for the generation of protective immunity. Immunol. Lett. 116, 64–71 (2008).
Santos, R.L. & Baumler, A.J. Cell tropism of Salmonella enterica. Int. J. Med. Microbiol. 294, 225–233 (2004).
Rescigno, M. Identification of a new mechanism for bacterial uptake at mucosal surfaces, which is mediated by dendritic cells. Pathol. Biol. (Paris) 51, 69–70 (2003).
Le Bourhis, L. et al. Role of Nod1 in mucosal dendritic cells during Salmonella pathogenicity island 1-independent Salmonella enterica serovar Typhimurium infection. Infect. Immun. 77, 4480–4486 (2009).
Cario, E., Gerken, G. & Podolsky, D.K. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology 132, 1359–1374 (2007).
Gibson, D.L. et al. Toll-like receptor 2 plays a critical role in maintaining mucosal integrity during Citrobacter rodentium-induced colitis. Cell Microbiol. 10, 388–403 (2008).
Rakoff-Nahoum, S., Paglino, J., Eslami-Varzaneh, F., Edberg, S. & Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118, 229–241 (2004).
Fukata, M. et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G1055–G1065 (2005).
Araki, A. et al. MyD88-deficient mice develop severe intestinal inflammation in dextran sodium sulfate colitis. J. Gastroenterol. 40, 16–23 (2005).
Gibson, D.L. et al. MyD88 signalling plays a critical role in host defence by controlling pathogen burden and promoting epithelial cell homeostasis during Citrobacter rodentium-induced colitis. Cell Microbiol. 10, 618–631 (2008).
Guillemot, L., Paschoud, S., Pulimeno, P., Foglia, A. & Citi, S. The cytoplasmic plaque of tight junctions: a scaffolding and signalling center. Biochim. Biophys. Acta 1778, 601–613 (2008).
Ikenouchi, J., Umeda, K., Tsukita, S. & Furuse, M. Requirement of ZO-1 for the formation of belt-like adherens junctions during epithelial cell polarization. J. Cell Biol. 176, 779–786 (2007).
Umeda, K. et al. ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell 126, 741–754 (2006).
Bauer, H., Zweimueller-Mayer, J., Steinbacher, P., Lametschwandtner, A. & Bauer, H.C. The dual role of zonula occludens (ZO) proteins. J. Biomed. Biotechnol. 2010, 402593 (2010).
Gottardi, C.J., Arpin, M., Fanning, A.S. & Louvard, D. The junction-associated protein, zonula occludens-1, localizes to the nucleus before the maturation and during the remodeling of cell-cell contacts. Proc. Natl. Acad. Sci. USA 93, 10779–10784 (1996).
Islas, S., Vega, J., Ponce, L. & Gonzalez-Mariscal, L. Nuclear localization of the tight junction protein ZO-2 in epithelial cells. Exp. Cell Res. 274, 138–148 (2002).
Okamura, Y. et al. The extra domain A of fibronectin activates Toll-like receptor 4. J. Biol. Chem. 276, 10229–10233 (2001).
Boyle, E.C., Brown, N.F. & Finlay, B.B. Salmonella enterica serovar Typhimurium effectors SopB, SopE, SopE2 and SipA disrupt tight junction structure and function. Cell Microbiol. 8, 1946–1957 (2006).
Cario, E., Gerken, G. & Podolsky, D.K. Toll-like receptor 2 enhances ZO-1-associated intestinal epithelial barrier integrity via protein kinase C. Gastroenterology 127, 224–238 (2004).
Yuki, T. et al. Activation of TLR2 enhances tight junction barrier in epidermal keratinocytes. J. Immunol. 187, 3230–3237 (2011).
Kilpatrick, L.E. et al. Protection against sepsis-induced lung injury by selective inhibition of protein kinase C-delta (delta-PKC). J. Leukoc. Biol. 89, 3–10 (2011).
White, J.F. Intestinal pathophysiology in autism. Exp. Biol. Med. (Maywood) 228, 639–649 (2003).
Yacyshyn, B., Meddings, J., Sadowski, D. & Bowen-Yacyshyn, M.B. Multiple sclerosis patients have peripheral blood CD45RO+ B cells and increased intestinal permeability. Dig. Dis. Sci. 41, 2493–2498 (1996).
Yamamoto, M. et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature 420, 324–329 (2002).
Wang, Q., Pantzar, N., Jeppsson, B., Westrom, B.R. & Karlsson, B.W. Increased intestinal marker absorption due to regional permeability changes and decreased intestinal transit during sepsis in the rat. Scand. J. Gastroenterol. 29, 1001–1008 (1994).
Ward, J.B. et al. Hydroxylase inhibition attenuates colonic epithelial secretory function and ameliorates experimental diarrhea. FASEB J. 25, 535–543 (2011).
Bowie, A. et al. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc. Natl. Acad .Sci. USA 97, 10162–10167 (2000).
Acknowledgements
L.A.J.O’N. was supported by Science Foundation Ireland (07/IN1/B942). P.G.F. was supported by Science Foundation Ireland (07/IN1/B90; 10/IN.1/B3004) and National Children’s Research Centre.
Author Contributions
S.C.C conceived ideas, designed and performed experiments, analyzed data, and wrote the paper. L.A.J.O’N. and P.G.F. oversaw research. E.M.P-McD. contributed to Figure 5; N.J.B. and P.C. contributed to Figure 1. S.P.B., G.A., J.B.J.W., S.J.K., and P.G.F. contributed to Figures1 and 2. I.G. and S.D. contributed to Figure 3.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Rights and permissions
About this article
Cite this article
Corr, S., Palsson-McDermott, E., Grishina, I. et al. MyD88 adaptor-like (Mal) functions in the epithelial barrier and contributes to intestinal integrity via protein kinase C. Mucosal Immunol 7, 57–67 (2014). https://doi.org/10.1038/mi.2013.24
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2013.24
This article is cited by
-
Intestinal serotonergic system is modulated by Toll-like receptor 9
Journal of Physiology and Biochemistry (2022)
-
Akkermansia muciniphila-derived extracellular vesicles influence gut permeability through the regulation of tight junctions
Experimental & Molecular Medicine (2018)
-
Intestinal Epithelial Cell-Specific Deletion of PLD2 Alleviates DSS-Induced Colitis by Regulating Occludin
Scientific Reports (2017)
-
Barrier protection via Toll-like receptor 2 signaling in porcine intestinal epithelial cells damaged by deoxynivalnol
Veterinary Research (2016)
-
AIM2 contributes to the maintenance of intestinal integrity via Akt and protects against Salmonella mucosal infection
Mucosal Immunology (2016)
