
Abstract
Heterotrimeric G proteins are pivotal mediators of cellular signal transduction in eukaryotic cells and abnormal G-protein signaling plays an important role in numerous diseases. During the last two decades it has become evident that the activation status of heterotrimeric G proteins is both highly localized and strongly regulated by a number of factors, including a receptor-independent activation pathway of heterotrimeric G proteins that does not involve the classical GDP/GTP exchange and relies on nucleoside diphosphate kinases (NDPKs). NDPKs are NTP/NDP transphosphorylases encoded by the nme/nm23 genes that are involved in a variety of cellular events such as proliferation, migration, and apoptosis. They therefore contribute, for example, to tumor metastasis, angiogenesis, retinopathy, and heart failure. Interestingly, NDPKs are translocated and/or upregulated in human heart failure. Here we describe recent advances in the current understanding of NDPK functions and how they have an impact on local regulation of G-protein signaling.
Similar content being viewed by others

Main
The involvement of heterotrimeric G proteins in signal transduction was first described in 1971, when it was shown that G proteins have a crucial role in the activation of adenylate cyclase by glucagon.1 Since then it has been shown that G proteins have various critical and essential roles in signal transduction in every cell type and that abnormal G-protein signaling has an important role in numerous diseases.2, 3, 4 G proteins consist of three subunits, Gα, Gβ, and Gγ, and are activated by seven transmembrane G-protein-coupled receptors (GPCR).2, 5 It is generally accepted that the activation of G proteins depends on a GDP/GTP exchange in response to a conformational change induced by ligand binding to the GPCR.6, 7 However, during the last two decades it has become clear that the activation status of heterotrimeric G proteins is both highly localized and strongly regulated by a number of factors. For example, regulators of G-protein signaling (RGSs) are a large family of proteins that bind directly to activated Gα subunits and terminate their activation.8 In addition, a receptor-independent activation pathway of heterotrimeric G proteins that does not involve the classical GDP/GTP exchange, but is mediated by nucleoside diphosphate kinases (NDPKs), has been discovered.9, 10 This review describes recent insights in the localized regulation of G-protein signaling by NDPKs and caveolins.
REGULATION OF G-PROTEIN SIGNALING BY NDPKs
Background
NDPKs are NTP/NDP transphosphorylases encoded by the nme/nm23 genes, which catalyze the transfer of a γ-phosphate group from an NTP to an NDP via formation of a high-energy intermediate on the His118 residue of the NDPK.11, 12, 13, 14 The family of NDPKs consists of two groups. The first group includes four members, NDPK-A, NDPK-B, NDPK-C, and NDPK-D, also known as NME1, NME2, NME3, and NME4, respectively, which are ubiquitously expressed. The human orthologs are equally often designated Nm23H1, Nm23H2, Nm23H3, and Nm23H4, respectively. These NDPKs are highly homologous, form heterohexamers, and exert transphosphorylase activity. The second group includes a more diverse set of proteins that are mostly expressed in cilia or flagella.15 Notable differences in the protein structure within the first group of NDPKs include the N-terminal hydrophobic domain of NDPK-C, which is believed to form a transmembrane helix promoting its localization at the plasma membrane,16, 17 and the mitochondrial-targeting domain of NDPK-D.18 NDPKs are involved in a variety of complex signal processes such as tumor metastasis,19 apoptosis,20 angiogenesis,21 retinopathy,22 and development.23
Mechanisms of G-Protein Signaling Regulation by NDPKs
By catalyzing the transfer from GDP to GTP, NDPKs regulate the amount of GTP available for G-protein activation, thereby indirectly controlling G-protein signaling (Figure 1). In the early 1990s, it was proposed that this regulation takes places in multiprotein complexes in which NDPKs are involved in channeling GTP to G proteins.24, 25, 26 Subsequent work showed that NDPK-B, but not NDPK-A, forms a complex with Gβγ subunits of heterotrimeric G proteins and activates the G proteins in a receptor-independent manner.9, 10 These studies discovered that NDPK-B likely autophosphorylates at His118 in the presence of an NTP, eg, ATP, and subsequently transfers this phosphate to His266 of the Gβ subunit. This phosphate is still of high energy and thus produces a GTP, most likely from the GDP dissociating from the Gα subunit. Rebinding of this newly formed GTP causes activation of the G protein in the absence of a GPCR agonist (Figure 2). Thus, in addition to their transphosphorylase activity, NDPKs act as protein histidine kinases. In agreement, accumulating evidence suggests that NDPKs can directly regulate ion-channel function through histidine phosphorylation and corresponding specific histidine phosphatases (eg, PHP, PGAM5) have been identified.27, 28 For example, besides Gβ subunit modulation, NDPK-B-mediated histidine phosphorylation activates the small-conductance Ca2+-activated K+ channel KCa3.129 and the transient receptor potential Ca2+ channel TRPV5.30 The phosphorylation of a histidine residue involves bonding of a phosphoryl group to the imidazole ring of the histidine through a phosphoramidate bond. As this imidazole ring has two nitrogen atoms, multiple forms of phosphorylation involving the N1 (1-pHis) or N3 (3-pHis) position are possible.27, 31 Recently developed monoclonal antibodies that specifically recognize the 1-pHis and 3-pHis forms have shown that the high energetic phopshoramidate bond at the His118 of NDPKs involves the 1-pHis form, whereas the histidine phosphorylation of NDPK substrate like Gβ1 or KCa3.1 involves the 3-pHis form.28, 32 For a more detailed overview of these mechanisms, the reader is referred to previous reviews.33, 34, 35, 36 Although NDPK-C can activate G proteins similar to NDPK-B (discussed below), other potential NDPK-C phosphorylation targets have not yet been identified.

General role of nucleoside diphosphate kinase (NDPK) transphosphorylase activity in G-protein activation. Locally present NDPKs replenish the GTP required for G-protein activation. In particular, NDPK-B and NDPK-C can form GTP out of ATP and GDP using a high-energy intermediate on His118 of the NDPK.

Regulation of G-protein activation by nucleoside diphosphate kinases (NDPKs). NDPKs can directly activate G-proteins through a G-protein-coupled receptor (GPCR)-independent pathway involving GTP formation from intermediate phosphorylation of His266 on the Gβ subunit, which results from high-energy phosphate transfer from His118 of the NDPKs (#1). In addition, NDPKs can act as a scaffold, increasing the G-protein membrane content (#2), which is also expected to enhance G-protein signaling.
Besides the phosphorylation of Gβ at His266, NDPK-B contributes to the membrane targeting of G proteins (Figure 2, #2). For example, mouse embryonic fibroblasts (MEFs) of NDPK-A/B double-knockout mice have reduced membrane content of G proteins compared with wild-type MEFs. Rescuing the expression of NDPK-B in NDPK A/B knockout MEFs by overexpressing the catalytically inactive H118N-NDPK-B, where the histidine has been mutated to an asparagine, is able to rescue the G-protein content at the plasma membrane, indicating that the membrane-targeting does not depend on NDPK-B histidine kinase activity.37 This scaffolding function might additionally involve the formation of caveolae,38 which will be covered in the next section of this review. However, it should be noted that earlier studies were unable to reconstitute a complex of NDPK-B and G proteins using purified recombinant proteins. This already indicated that another protein is required to mediate this interaction.9 As NDPKs form hexamers and NDPK-C is the only isoform with an N-terminal hydrophobic domain, we recently hypothesized that NDPK-C may be a potential candidate for scaffolding the complex between NDPK-B and G proteins at the plasma membrane. In agreement with this hypothesis, far western blot experiments have shown that NDPK-C interacts with NDPK-B, but only NDPK-C, and not NDPK-B, directly interacts with the purified heterotrimeric G protein transducin (Gtαβγ) from bovine retina as well as purified stimulatory Gαs and inhibitory Gαi proteins.39 Furthermore, an increase in the overexpression of NDPK-C produces a corresponding increase in the interaction between NDPK-B and Gtαβγ.39 NDPK-C alone can also activate G-proteins directly through phosphorylation of His266. In addition, experiments with radiolabeled [γ-32P] ATP incubated with GDP and Gtαβγ have been employed to quantify NDPK activity as GTP-forming transphosphorylase histidine kinase and activator of G proteins. Addition of NDPK-C alone but not NDPK-B alone enhanced the GTP hydrolysis by Gtαβγ. The addition of both NDPK-B and NDPK-C had a synergistic effect. These data indicate that NDPK-C is required and indispensable for the interaction of NDPKs with G proteins and that both isoforms, NDPK-B and NDPK-C, work in concert to regulate G-protein activity.39
Relevance of NDPK-Mediated Regulation of G-Protein Signaling in the Heart
The regulation of G-protein signaling by NDPKs has been described for many cell types. In recent years the importance of the NDPK-mediated activation of G proteins in the heart has received considerable attention, particularly with respect to regulation of cyclic adenosine monophosphate (cAMP), a ubiquitous second messenger with an integral role in the regulation of cardiac electrophysiology and contraction. Activation of stimulatory Gαs proteins promotes cAMP production via adenylyl cyclases, whereas activation of inhibitory Gαi reduces cAMP production. Initial experiments performed in H10 cells have shown that overexpression of NDPK-B but not NDPK-A or catalytically inactive H118N-NDPK-B results in increased cAMP levels.40 Furthermore, combined overexpression of Gαs and Gβ1γ2 further increases cAMP levels in the NDPK-B-overexpressing H10 cells, which is significantly blunted in experiments overexpressing Gαs with Gβ1H266Lγ2, where the histidine at position 266 is mutated to a leucine. This mutant does not allow for intermediate histidine phosphorylation of Gβ and subsequent GTP formation. Thus, the activation of G proteins via the intermediate phosphorylation of His266 on Gβ has a significant role in the NDPK-mediated regulation of G-protein activity and promotes cAMP production. In agreement, cAMP levels are reduced in NDPK-B knockdown zebrafish. Knockdown of NDPK-C also reduces cAMP production, which is consistent with the essential role of NDPK-C mediating the interaction between G proteins and NDPK-B.39
Knockdown of NDPK-B in zebrafish also reduces G-protein expression, with a similar phenotype as the Gβ1/Gβ1-like knockdown zebrafish,41 suggesting that, in addition to acute regulation of G-protein activity, long-term modulation of NDPKs may influence the protein levels of G proteins, possibly because of the membrane-targeting effects of NDPK complexes. Consistently, studies in neonatal rat cardiomyocytes have shown that NDPK-B and NDPK-C critically regulate the membrane localization of G proteins.39, 41 Moreover, acute stimulation of rat cardiomyocytes overexpressing NDPK-C with the β-adrenoceptor agonist isoprenaline results in enhanced membrane localization of G proteins and NDPK-C,39 suggesting that recruitment of NDPK/G protein complexes to the plasma membrane may counteract a fading response to long-term isoprenaline stimulation due to desensitization of β-adrenoceptors.
NDPK-mediated regulation of cAMP production is expected to modulate cardiac contractility through downstream effects of cAMP-dependent protein kinase A on cardiac calcium handling. In agreement, overexpression of NDPK-C increases cAMP content and contractility of adult cardiomyocytes, whereas zebrafish with knockdown of NDPK-B or NDPK-C suffer from cardiac contractile dysfunction.39, 41 Similarly, 5-month-old NDPK-B knockout mice have reduced fractional shortening on cardiac echocardiographic analysis compared ith wild-type mice of the same age. NDPKs also appear to have important cardioprotective effects as NDPK-B knockout mice develop worse maladaptive hypertrophy and more fibrosis than wild-type mice in response to pathological stimuli such as long-term isoprenaline application.39 The mechanisms underlying these cardioprotective effects of NDPK-B remain incompletely understood but could involve both the differential modulation of cAMP pools as reflected by the reduced cardiac contractility of NDPK-B knockout mice following long-term isoprenaline stimulation,39 and direct effects of NDPKs on gene expression, which have not yet been explored in the setting of heart failure.23 Together, these data suggest that NDPKs have a critical role in the regulation of cardiac function.
Alterations in NDPK-Mediated Regulation of G-Protein Signaling in Cardiovascular Disease
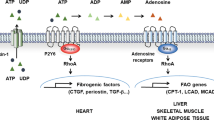
Heart failure is characterized by chronically elevated catecholamine levels, which alter β-adrenoceptor and G-protein signaling, contributing to the progressive impairment of cardiac contractility.42, 43 These alterations in G-protein signaling in end-stage heart failure patients include increased expression of the inhibitory Gαi protein by around 30% and decreased cAMP levels.44, 45, 46 Expression of NDPK-C and membrane content of NDPK-B and NDPK-C are also increased in heart failure patients.39, 47, 48 Furthermore, a similar increase in NDPK-C has also been observed in rats receiving isoprenaline via osmotic mini pumps for 4 days, mimicking the increased catecholamine levels in human heart failure, suggesting that these effects are a consequence of sympathetic stimulation.39, 49 The increased NDPK-C mRNA levels during long-term β-adrenoceptor stimulation might be due to activation of AP-2- and CREBP-binding sites in the mouse nme3 gene, which can regulate gene expression depending on cAMP levels.50, 51 Indeed, both patients and isoprenaline-stimulated rats treated with β-blockers showed a reduced NDPK content at the plasma membrane.52
The increased NDPK expression in heart failure and the stimulatory effect of NDPKs on cAMP and contractility in animal models appear at odds with the decreased cAMP levels and impaired contractility in heart failure. However, it has been suggested that NDPKs may also enhance Gi signaling in patients, contributing to a switch from NDPK-mediated regulation of Gs to Gi in heart failure, which might contribute to the decreased cAMP and impaired contractility (Figure 3).39, 47 Recent far western blot experiments indeed confirmed that NDPK-C can shift its interaction from Gαs to Gαi or vice versa, depending on the relative expression of each protein.39 Similarly, co-immunoprecipitation experiments have identified less Gαs and more Gαi2 in the NDPK-C precipitate in ventricular samples from end-stage heart failure patients compared with healthy controls, strongly suggesting a switch from predominant Gs to Gi signaling.39 Finally, experiments in neonatal rat cardiomyocytes have confirmed that overexpression of NDPK-C in the presence of elevated Gαi2 levels results in a reduction in cAMP levels, instead of the stimulatory effects of NDPK-C overexpression observed under normal conditions.39 Taken together, these experiments highlight the complexity of NDPK-mediated regulation of G-protein signaling, revealing that NDPKs are crucial for healthy heart function, but may also contribute to worsening of cardiac function, positioning them as potential novel targets for the therapy of heart failure. The dual roles of NDPKs in the heart, resulting from a switch from one interacting protein to another, may also apply to other NDPK functions. For example, NDPKs, especially NDPK-A, have been identified as tumor metastasis suppressors.19 However, in the advanced phases of tumor progression, the correlation between NDPK-A expression and cell proliferation is lost.53 Likewise, other studies have found that NDPK-A expression is positively correlated with metastatic progression in osteosarcoma.54 As it has been shown that the metastasis-suppressing role of NDPK-A depends on its interaction with various proteins,55 NDPK-A may also have a dual role in tumor metastasis depending on the type of interaction partners.

Switch from predominantly NDPK-mediated Gαs signaling in non-failing hearts, increasing cAMP production and contractility, to Gαi-based signaling in heart failure, resulting in reduced cAMP production and contributing to contractile dysfunction.
NDPK-B/NME2 Influences Signal Complex Assembly at the Plasma Membrane via Caveolae Formation
Caveolae are small cholesterol-enriched invaginations at the plasma membrane, which, among other tasks, cluster and compartmentalize signaling components.21, 56, 57, 58, 59 Two classes of scaffold proteins, caveolins (Cav) and cavins are required for the formation of caveolae. Their assembly is a complex process (Figure 4). Of the three isoforms of Cav the Cav1 isoform is necessary and sufficient for caveolae formation in most tissues. In striated (skeletal and heart) muscle, however, the prominent and required isoform is Cav3.60, 61 NDPK-B has been shown to interact with Cav1 and Cav3.38, 41 After synthesis at the endoplasmatic reticulum (ER), Cav1 (or Cav3 in striated muscle), is inserted co-translationally into ER membranes and rapidly forms 8S20,w oligomers (Cav-8S). The Cav-8S oligomers translocate to the ER exit sites where a coat protein complex II (COPII)-dependent export to the Golgi apparatus occurs.62, 63, 64 If the recognition of Cavs by COPII is hampered, for example, because of mutations in the recognition sites in Cav1 (or Cav3), the export is delayed leading to accumulation in structures near the ER, eg, lipid bodies.65 In the Golgi, the Cav-8S further oligomerizes. The resulting Cav-70S complexes get enriched in cholesterol and are transported in vesicles from the Golgi.62, 66 Near the plasma membrane, Cavs in the Cav-70S complexes are palmitoylated by palmitoyl acyltransferases and, after fusion with the plasma membrane,62, 67, 68 cavin proteins aggregate on the cholesterol-rich and Cav-containing lipid rafts and thereby assist the formation of the membrane invaginations typical for caveolae.62, 69, 70

Role of nucleoside diphosphate kinases (NDPKs) in the regulation of caveolin-dependent formation of caveolae. Cav monomers are inserted into the endoplasmic reticulum (ER) membrane and they oligomerize into Cav-8S oligomers containing around 10 Cavs. These oligomers are transported to ER exit sites. NDPK-B is likely needed for COPII assembly and the transport to the Golgi apparatus. In the Golgi, cholesterol in the membranes supports the formation of Cav-70S complexes composed of about 150 Cavs. The Cav-70S-containing vesicles are transported to the plasma membrane in a phosphatidylinositol-4-phosphate (PIP4) and four phosphate-adapter protein (FAPP)-dependent manner. Near or on the plasma membrane, palmitoyl acyltransferases palmitoylate Cavs. Cavin proteins that already form trimeric complexes in the cytosol bind to the Cav-70S and induce the membrane curvature known from mature caveolae. They consist of an anionic lipid- and cholesterol-rich membrane in which the Cavs are embedded. Oligomerized cavins coat the caveolae on the inside of the cell. Additional proteins are inserted as transmembrane proteins, as lipid-modified proteins or interact directly with Cavs via their caveolin-scaffolding domain to which, for example, heterotrimeric G protein α subunits can bind.
The association of NDPK-B with Cav1 and Cav3 was first identified in the zebrafish. Knockdown of NDPK-B specifically caused not only the loss of NDPK-B and the associated heterotrimeric G proteins Gs and Gi, but also loss of the Cavs.38, 41 This close association has been confirmed in cardiomyocytes, endothelial cells, and embryonic fibroblasts.21, 38, 41, 71 In addition, the phenotypes in loss-of-expression models of NDPK-B and Cavs are overlapping. Both Cav3- and NDPK-B-depleted zebrafish larvae show an impaired cardiac contractility.38, 41 In addition, the NDPK-B-depleted zebrafish exhibited impaired vessel formation by angiogenesis, a phenotype also seen under pathological conditions in NDPK-B-deficient mice.21 Cav1-deficient mice also show impaired angiogenesis in several models.71, 72, 73 These data suggest a common, very basic function of NDPK-B in caveolae formation. Indeed, in NDPK-B-deficient fibroblasts as well as in NDPK-B-depleted endothelial cells, Cav1 does not reach the plasma membrane and gets stocked in intracellular vesicular structures that likely represent, for example, lipid bodies.21, 38, 71 In accordance, the number of caveolae at the plasma membrane was drastically reduced. This phenotype could be rescued by the re-expression of NDPK-B. Moreover, the expression of a fluorescent fusion protein of Cav1 with EGFP directly demonstrated the transport defect in the absence of NDPK-B.38 As it has been described that NDPK-B is required for the formation of the COPII complex,74 the recognition of the Cav-8S oligomers at the ER exit sites is likely hampered by NDPK-B deficiency and thus delayed. In accordance with data from recognition-deficient mutants of Cavs,64, 65 Cavs therefore accumulate in lipid bodies and similar structures. In addition, the phenotype of an acute knockdown of NDPK-B in the zebrafish as well as in cellular models can be explained by hampered caveolae formation. Heterotrimeric G proteins, GPCRs like cardiac β-adrenoceptors as well as the VEGF receptor 2 (VEGFR2) in endothelial cells, are located in caveolae.58, 59 Thus, the VEGF-induced spatial redistribution of the VEGFR2 was attenuated in NDPK-B-deficient endothelial cells and the β-adrenoceptor-induced cAMP formation was suppressed after knockdown of NDPK-B in cardiomyocytes as well as in the zebrafish.21, 38, 41
Generally, Cav1-deficient as well as Cav3-deficient mice display severe alterations and a shortened lifespan.75, 76, 77, 78 In contrast, NDPK-B-deficient mice are rather healthy and have a normal life expectancy. In contrast to the acute NDPK-B knockdown in zebrafish larvae, the cardiac function of NDPK-B−/− mice is normal up to 5 months of age and thereafter cardiac contractility is only mildly attenuated.29, 39 Although in the knockdown fish angiogenic vessel formation was severely hampered, the physiological angiogenesis in the retina of newborn NDPK-B−/− mice was not altered, whereas retinal vessel formation in Cav1−/− mice was delayed.21, 71 These data argue for a compensation of the lack of NDPK-B, which might rescue caveolae formation in newborn NDPK-B−/− mice. Indeed, as detected in brain endothelial cells of NDPK-B−/− mice, the amount of cellular Cav1 was increased and the number of caveolae was normalized.71 Interestingly, the amount of intracellular vesicles was also substantially increased, which indicates that the slower transport of Cav-containing vesicles was compensated by a higher number of such vesicles.71 This mechanism, whereas it might be able to counteract the NDPK-B deficiency and uphold the required amount of caveolae under physiological conditions, is apparently not able to cope with pathological stress conditions. Thus, NDPK-B-deficient mice show aggravated deterioration in cardiac as well as vascular stress models when compared with wild-type littermates. Further research is clearly needed to identify the specific function of NDPK-B in the ER exit and Cav transport and whether its enzymatic activity is required, for example, for the activation of so far not identified cofactors, eg, monomeric GTPases. It is also unclear whether NDPK hetero-oligomers (for example, NDPK-B/NDPK-C complexes) are involved and required for the interaction with and transport of heterotrimeric G proteins.
FUTURE PERSPECTIVES
Chamber-Specific Regulation of G-Protein Signaling
Although most studies so far have addressed G-protein signaling in ventricular cardiomyocytes, for example, in the setting of heart failure, it is likely that regulation of G-protein signaling by NDPKs and caveolins is also involved in other cardiovascular diseases. For example, cAMP-dependent regulation of calcium handling in atrial cardiomyocytes has been implicated in the initiation of atrial arrhythmias including atrial fibrillation (AF).79 Furthermore, AF produces pronounced structural, electrical, calcium-handling, and autonomic remodeling that contribute to AF maintenance and progression.79 In patients with chronic AF, cAMP levels are increased, suggesting potential alterations in the regulation of G-protein signaling.80 Preliminary data have shown that NDPK-B and NDPK-C protein levels are increased in patients with chronic AF,81 suggesting a potential role in AF-related changes in cAMP signaling that warrant further studies. Importantly, the subcellular structure of atrial cardiomyocytes is significantly different from ventricular cardiomyocytes.82 For example, atrial cardiomyocytes have a less-developed t-tubular system. As such, it is likely that local signaling domains regulating G-protein signaling are also distinct between atrial and ventricular cardiomyocytes,83 although this has not yet been extensively investigated.
Elucidation of the NDPK Interactome and its Subcellular Localization
Recent work has shown that regulation of G-protein signaling is highly localized and orchestrated by macromolecular complexes of numerous proteins, including NDPK-B, NDPK-C, and caveolins. In addition, for NDPK-A, a variety of interacting proteins have been identified.55 Nevertheless, the exact composition of the NDPK interactome under specific experimental and/or clinical conditions is still unknown. Future biochemical, proteomics, and mass spectroscopy studies are expected to provide a more comprehensive understanding of the NDPK interactome and its roles in biochemical pathways. Furthermore, advances in electron microscopy and super-resolution live-cell imaging methodologies will help to elucidate the subcellular localization of these signaling complexes and their organization in caveolae and other structures. For example, the switch from predominantly Gs to Gi protein signaling in heart failure39 showed that the composition and localization of these macromolecular complexes can change over time in response to diverse stimuli. Thus, future biochemical and live-cell imaging studies will need to take into account these changes and provide insights into the temporal dynamics of the NDPK-interactome and -oligomer composition.
Clinical Applications
Modulation of kinases and phosphatases is increasingly explored as therapeutic strategy.84 There are rather unspecific pharmacological modulators of NDPK activity like ellagic acid available,85 but no NDPK-subtype-specific inhibitors have been reported. Hypothetically, overexpression of the histidine phosphatase PHP could be employed to counteract detrimental effects of overactive NDPKs (eg, in the setting of heart failure39 or arteriosclerosis86). However, the recently identified dual roles of NDPKs in G-protein signaling, involving the switch from Gs to Gi,39 suggest that effective NDPK-based therapy requires modulation of the affinity of NDPKs for their individual targets and interacting proteins. Moreover, given the ubiquitous expression of NDPKs, this modulation would have to be targeted to specific cell types and subcellular regions to prevent unwanted side effects. Future studies about the molecular interactions between NDPKs and target proteins will likely provide the mechanistic understanding necessary to develop effective NDPK-based therapeutic options.
References
Rodbell M, Birnbaumer L, Pohl SL et al. The glucagon-sensitive adenyl cyclase system in plasma membranes of rat liver. V. An obligatory role of guanylnucleotides in glucagon action. J Biol Chem 1971;246:1877–1882.
Hamm HE . The many faces of G protein signaling. J Biol Chem 1998;273:669–672.
Neves SR, Ram PT, Iyengar R . G protein pathways. Science 2002;296:1636–1639.
Syrovatkina V, Alegre KO, Dey R et al. Regulation, signaling, and physiological functions of G-proteins. J Mol Biol 2016;428:3850–3868.
Gilman AG . G proteins: transducers of receptor-generated signals. Annu Rev Biochem 1987;56:615–649.
Wieland T, Michel MC . Can a GDP-liganded G-protein be active? Mol Pharmacol 2005;68:559–562.
Oldham WM, Hamm HE . Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol 2008;9:60–71.
Hollinger S, Hepler JR . Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol Rev 2002;54:527–559.
Cuello F, Schulze RA, Heemeyer F et al. Activation of heterotrimeric G proteins by a high energy phosphate transfer via nucleoside diphosphate kinase (NDPK) B and Gβ subunits. Complex formation of NDPK B with Gβγ dimers and phosphorylation of His-266 in Gβ. J Biol Chem 2003;278:7220–7226.
Hippe HJ, Lutz S, Cuello F et al. Activation of heterotrimeric G proteins by a high energy phosphate transfer via nucleoside diphosphate kinase (NDPK) B and Gβ subunits. Specific activation of Gsα by an NDPK B Gβγ complex in H10 cells. J Biol Chem 2003;278:7227–7233.
Herbert E, Potter VR, Takagi Y . Nucleotide metabolism: IV. The phosphorylation of 5'-uridine nucleotides by cell fractions from rat liver. J Biol Chem 1955;213:923–940.
Pedersen PL . Coupling of adenosine triphosphate formation in mitochondria to the formation of nucleoside triphosphates. Involvement of nucleoside diphosphokinase. J Biol Chem 1973;248:3956–3962.
Tepper AD, Dammann H, Bominaar AA et al. Investigation of the active site and the conformational stability of nucleoside diphosphate kinase by site-directed mutagenesis. J Biol Chem 1994;269:32175–32180.
Morera S, Chiadmi M, LeBras G et al. Mechanism of phosphate transfer by nucleoside diphosphate kinase: X-ray structures of the phosphohistidine intermediate of the enzymes from Drosophila and Dictyostelium. Biochemistry 1995;34:11062–11070.
Boissan M, Dabernat S, Peuchant E et al. The mammalian Nm23/NDPK family: from metastasis control to cilia movement. Mol Cell Biochem 2009;329:51–62.
Barraud P, Amrein L, Dobremez E et al. Differential expression of nm23 genes in adult mouse dorsal root ganglia. J Comp Neurol 2002;444:306–323.
Mitchell KA, Szabo G, de S Otero A . Direct binding of cytosolic NDP kinases to membrane lipids is regulated by nucleotides. Biochim Biophys Acta 2009;1793:469–476.
Milon L, Meyer P, Chiadmi M et al. The human nm23-H4 gene product is a mitochondrial nucleoside diphosphate kinase. J Biol Chem 2000;275:14264–14272.
Steeg PS, Bevilacqua G, Kopper L et al. Evidence for a novel gene associated with low tumor metastatic potential. J Natl Cancer Inst 1988;80:200–204.
Fan Z, Beresford PJ, Oh DY et al. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell 2003;112:659–672.
Feng Y, Gross S, Wolf NM et al. Nucleoside diphosphate kinase B regulates angiogenesis through modulation of vascular endothelial growth factor receptor type 2 and endothelial adherens junction proteins. Arterioscler Thromb Vasc Biol 2014;34:2292–2300.
Qiu Y, Zhao D, Butenschon VM et al. Nucleoside diphosphate kinase B deficiency causes a diabetes-like vascular pathology via up-regulation of endothelial angiopoietin-2 in the retina. Acta Diabetol 2016;53:81–89.
Postel EH . Multiple biochemical activities of NM23/NDP kinase in gene regulation. J Bioenerg Biomembr 2003;35:31–40.
Kimura N, Shimada N . Evidence for complex formation between GTP binding protein (Gs) and membrane-associated nucleoside diphosphate kinase. Biochem Biophys Res Commun 1990;168:99–106.
Wieland T, Jakobs KH . Receptor-regulated formation of GTP[γS] with subsequent persistent Gs-protein activation in membranes of human platelets. FEBS Lett 1989;245:189–193.
Wieland T, Jakobs KH . Evidence for nucleoside diphosphokinase-dependent channeling of guanosine 5'-(gamma-thio)triphosphate to guanine nucleotide-binding proteins. Mol Pharmacol 1992;42:731–735.
Wieland T, Attwood PV . Alterations in reversible protein histidine phosphorylation as intracellular signals in cardiovascular disease. Front Pharmacol 2015;6:173.
Panda S, Srivastava S, Li Z et al. Identification of PGAM5 as a mammalian protein histidine phosphatase that plays a central role to negatively regulate CD4+ T cells. Mol Cell 2016;63:457–469.
Di L, Srivastava S, Zhdanova O et al. Nucleoside diphosphate kinase B knock-out mice have impaired activation of the K+ channel KCa3.1, resulting in defective T cell activation. J Biol Chem 2010;285:38765–38771.
Cai X, Srivastava S, Surindran S et al. Regulation of the epithelial Ca2+ channel TRPV5 by reversible histidine phosphorylation mediated by NDPK-B and PHPT1. Mol Biol Cell 2014;25:1244–1250.
Attwood PV, Wieland T . Nucleoside diphosphate kinase as protein histidine kinase. Naunyn Schmiedebergs Arch Pharmacol 2015;388:153–160.
Fuhs SR, Meisenhelder J, Aslanian A et al. Monoclonal 1- and 3-phosphohistidine antibodies: new tools to study histidine phosphorylation. Cell 2015;162:198–210.
Hippe HJ, Wieland T . High energy phosphate transfer by NDPK B/Gβγ complexes—an alternative signaling pathway involved in the regulation of basal cAMP production. J Bioenerg Biomembr 2006;38:197–203.
Wieland T . Interaction of nucleoside diphosphate kinase B with heterotrimeric G protein βγ dimers: consequences on G protein activation and stability. Naunyn Schmiedebergs Arch Pharmacol 2007;374:373–383.
Hsu T, Steeg PS, Zollo M et al. Steering Committee on Nme-related research and the organizers of the International Congresses of the NKNaGF. Progress on Nme (NDP kinase/Nm23/Awd) gene family-related functions derived from animal model systems: studies on development, cardiovascular disease, and cancer metastasis exemplified. Naunyn Schmiedebergs Arch Pharmacol 2015;388:109–117.
Fuhs SR, Hunter T . pHisphorylation: the emergence of histidine phosphorylation as a reversible regulatory modification. Curr Opin Cell Biol 2017;45:8–16.
Hippe HJ, Abu-Taha I, Wolf NM et al. Through scaffolding and catalytic actions nucleoside diphosphate kinase B differentially regulates basal and β-adrenoceptor-stimulated cAMP synthesis. Cell Signal 2011;23:579–585.
Hippe HJ, Wolf NM, Abu-Taha HI et al. Nucleoside diphosphate kinase B is required for the formation of heterotrimeric G protein containing caveolae. Naunyn Schmiedebergs Arch Pharmacol 2011;384:461–472.
Abu-Taha IH, Heijman J, Hippe HJ et al. Nucleoside diphosphate kinase-C suppresses cAMP formation in human heart failure. Circulation 2017;135:881–897.
Hippe HJ, Luedde M, Lutz S et al. Regulation of cardiac cAMP synthesis and contractility by nucleoside diphosphate kinase B/G protein βγ dimer complexes. Circ Res 2007;100:1191–1199.
Hippe HJ, Wolf NM, Abu-Taha I et al. The interaction of nucleoside diphosphate kinase B with Gβγ dimers controls heterotrimeric G protein function. Proc Natl Acad Sci USA 2009;106:16269–16274.
Movsesian MA . Cyclic AMP-mediated signal transduction in heart failure: molecular pathophysiology and therapeutic implications. J Investig Med 1997;45:432–440.
El-Armouche A, Eschenhagen T . β-adrenergic stimulation and myocardial function in the failing heart. Heart Fail Rev 2009;14:225–241.
Feldman AM, Cates AE, Veazey WB et al. Increase of the 40,000- mol wt pertussis toxin substrate (G protein) in the failing human heart. J Clin Invest 1988;82:189–197.
Neumann J, Schmitz W, Scholz H et al. Increase in myocardial Gi-proteins in heart failure. Lancet 1988;2:936–937.
Bohm M, Reiger B, Schwinger RH et al. cAMP concentrations, cAMP dependent protein kinase activity, and phospholamban in non-failing and failing myocardium. Cardiovasc Res. 1994;28:1713–1719.
Lutz S, Mura R, Baltus D et al. Increased activity of membrane-associated nucleoside diphosphate kinase and inhibition of cAMP synthesis in failing human myocardium. Cardiovasc Res 2001;49:48–55.
Lutz S, Hippe HJ, Niroomand F et al. Nucleoside diphosphate kinase-mediated activation of heterotrimeric G proteins. Methods Enzymol 2004;390:403–418.
Mende U, Eschenhagen T, Geertz B et al. Isoprenaline-induced increase in the 40/41 kDa pertussis toxin substrates and functional consequences on contractile response in rat heart. Naunyn Schmiedebergs Arch Pharmacol 1992;345:44–50.
Müller FU, Loser K, Kleideiter U et al. Transcription factor AP-2α triggers apoptosis in cardiac myocytes. Cell Death Differ 2004;11:485–493.
Müller FU, Lewin G, Baba HA et al. Heart-directed expression of a human cardiac isoform of cAMP-response element modulator in transgenic mice. J Biol Chem 2005;280:6906–6914.
Lutz S, Mura RA, Hippe HJ et al. Plasma membrane-associated nucleoside diphosphate kinase (nm23) in the heart is regulated by β-adrenergic signaling. Br J Pharmacol 2003;140:1019–1026.
Caligo MA, Cipollini G, Berti A et al. NM23 gene expression in human breast carcinomas: loss of correlation with cell proliferation in the advanced phase of tumor progression. Int J Cancer 1997;74:102–111.
Oda Y, Naka T, Takeshita M et al. Comparison of histological changes and changes in nm23 and c-MET expression between primary and metastatic sites in osteosarcoma: a clinicopathologic and immunohistochemical study. Hum Pathol 2000;31:709–716.
Marino N, Marshall JC, Steeg PS . Protein-protein interactions: a mechanism regulating the anti-metastatic properties of Nm23-H1. Naunyn Schmiedebergs Arch Pharmacol 2011;384:351–362.
Couet J, Sargiacomo M, Lisanti MP . Interaction of a receptor tyrosine kinase, EGF-R, with caveolins. Caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. J Biol Chem 1997;272:30429–30438.
Fujita T, Toya Y, Iwatsubo K et al. Accumulation of molecules involved in α1-adrenergic signal within caveolae: caveolin expression and the development of cardiac hypertrophy. Cardiovasc Res 2001;51:709–716.
Insel PA, Head BP, Patel HH et al. Compartmentation of G-protein-coupled receptors and their signalling components in lipid rafts and caveolae. Biochem Soc Trans 2005;33:1131–1134.
Liao WX, Feng L, Zhang H et al. Compartmentalizing VEGF-induced ERK2/1 signaling in placental artery endothelial cell caveolae: a paradoxical role of caveolin-1 in placental angiogenesis in vitro. Mol Endocrinol 2009;23:1428–1444.
Li S, Galbiati F, Volonte D et al. Mutational analysis of caveolin-induced vesicle formation. Expression of caveolin-1 recruits caveolin-2 to caveolae membranes. FEBS Lett 1998;434:127–134.
Song KS, Scherer PE, Tang Z et al. Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J Biol Chem 1996;271:15160–15165.
Hayer A, Stoeber M, Bissig C et al. Biogenesis of caveolae: stepwise assembly of large caveolin and cavin complexes. Traffic 2010;11:361–382.
Monier S, Parton RG, Vogel F et al. VIP21-caveolin, a membrane protein constituent of the caveolar coat, oligomerizes in vivo and in vitro. Mol Biol Cell 1995;6:911–927.
Mora R, Bonilha VL, Marmorstein A et al. Caveolin-2 localizes to the golgi complex but redistributes to plasma membrane, caveolae, and rafts when co-expressed with caveolin-1. J Biol Chem 1999;274:25708–25717.
Ostermeyer AG, Paci JM, Zeng Y et al. Accumulation of caveolin in the endoplasmic reticulum redirects the protein to lipid storage droplets. J Cell Biol 2001;152:1071–1078.
Godi A, Di Campli A, Konstantakopoulos A et al. FAPPs control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns(4)P. Nat Cell Biol 2004;6:393–404.
Das AK, Dasgupta B, Bhattacharya R et al. Purification and biochemical characterization of a protein-palmitoyl acyltransferase from human erythrocytes. J Biol Chem 1997;272:11021–11025.
Parat MO, Fox PL . Palmitoylation of caveolin-1 in endothelial cells is post-translational but irreversible. J Biol Chem 2001;276:15776–15782.
Kovtun O, Tillu VA, Jung W et al. Structural insights into the organization of the cavin membrane coat complex. Dev Cell 2014;31:405–419.
Ludwig A, Nichols BJ, Sandin S . Architecture of the caveolar coat complex. J Cell Sci 2016;129:3077–3083.
Gross S, Devraj K, Feng Y et al. Nucleoside diphosphate kinase B regulates angiogenic responses in the endothelium via caveolae formation and c-Src-mediated caveolin-1 phosphorylation. J Cereb Blood Flow Metab 2016;37:2471–2484.
Santibanez JF, Blanco FJ, Garrido-Martin EM et al. Caveolin-1 interacts and cooperates with the transforming growth factor-β type I receptor ALK1 in endothelial caveolae. Cardiovasc Res 2008;77:791–799.
Sonveaux P, Martinive P, DeWever J et al. Caveolin-1 expression is critical for vascular endothelial growth factor-induced ischemic hindlimb collateralization and nitric oxide-mediated angiogenesis. Circ Res 2004;95:154–161.
Kapetanovich L, Baughman C, Lee TH . Nm23H2 facilitates coat protein complex II assembly and endoplasmic reticulum export in mammalian cells. Mol Biol Cell 2005;16:835–848.
Hagiwara Y, Sasaoka T, Araishi K et al. Caveolin-3 deficiency causes muscle degeneration in mice. Hum Mol Genet 2000;9:3047–3054.
Park DS, Woodman SE, Schubert W et al. Caveolin-1/3 double-knockout mice are viable, but lack both muscle and non-muscle caveolae, and develop a severe cardiomyopathic phenotype. Am J Pathol 2002;160:2207–2217.
Razani B, Combs TP, Wang XB et al. Caveolin-1-deficient mice are lean, resistant to diet-induced obesity, and show hypertriglyceridemia with adipocyte abnormalities. J Biol Chem 2002;277:8635–8647.
Schubert W, Frank PG, Woodman SE et al. Microvascular hyperpermeability in caveolin-1 (-/-) knock-out mice. Treatment with a specific nitric-oxide synthase inhibitor, L-NAME, restores normal microvascular permeability in Cav-1 null mice. J Biol Chem 2002;277:40091–40098.
Heijman J, Voigt N, Nattel S et al. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res 2014;114:1483–1499.
Voigt N, Li N, Wang Q et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 2012;125:2059–2070.
Schäfer M, Abu-Taha HI, Naud P et al. Abstract 13235: enhanced receptor-independent nucleoside diphosphate kinase mediated Gs protein signaling contributes to arrhythmogenic calcium mishandling in atrial fibrillation. Circulation 2016;134:A13235.
Dobrev D, Teos LY, Lederer WJ . Unique atrial myocyte Ca2+ signaling. J Mol Cell Cardiol 2009;46:448–451.
Brandenburg S, Kohl T, Williams GS et al. Axial tubule junctions control rapid calcium signaling in atria. J Clin Invest 2016;126:3999–4015.
Heijman J, Dewenter M, El-Armouche A et al. Function and regulation of serine/threonine phosphatases in the healthy and diseased heart. J Mol Cell Cardiol 2013;64:90–98.
Malmquist NA, Anzinger JJ, Hirzel D et al. Ellagic acid inhibits nucleoside diphosphate kinase-B activity. Proc West Pharmacol Soc 2001;44:57–59.
Zhou XB, Feng YX, Sun Q et al. Nucleoside diphosphate kinase B-activated intermediate conductance potassium channels are critical for neointima formation in mouse carotid arteries. Arterioscler Thromb Vasc Biol 2015;35:1852–1861.
Acknowledgements
This work is supported by grants from the Deutsche Forschungsgemeinschaft (Wi1373/9-1,2,3 and SFB/TR23 TP B6 to TW; Do 769/4-1 to DD), the European Foundation for the Study of Diabetes (EFSD to YF), the Netherlands Organization for Scientific Research (NWO/ZonMW Veni 91616057 to JH), the National Institutes of Health (HL131517 to DD), and the German Federal Ministry of Education and Research through the DZHK (German Center for Cardiovascular Research to DD and TW). Elements from Servier Medical Art were used in the preparation of the figures.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
NDPKs are translocated and/or upregulated in human heart failure. Here the authors describe recent advances in the current understanding of NDPK functions and how they impact local regulation of G-protein signaling.
Rights and permissions
About this article

Cite this article
Abu-Taha, I., Heijman, J., Feng, Y. et al. Regulation of heterotrimeric G-protein signaling by NDPK/NME proteins and caveolins: an update. Lab Invest 98, 190–197 (2018). https://doi.org/10.1038/labinvest.2017.103
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2017.103
This article is cited by
-
NDK/NME proteins: a host–pathogen interface perspective towards therapeutics
Current Genetics (2022)
-
Activation of Nm23-H1 to suppress breast cancer metastasis via redox regulation
Experimental & Molecular Medicine (2021)
-
The NDPK/NME superfamily: state of the art
Laboratory Investigation (2018)
