
Abstract
As the key governors of diverse physiological processes, G protein-coupled receptors (GPCRs) have drawn attention as primary targets for several diseases, including diabetes and cardiovascular disease. Heterotrimeric G proteins converge signals from ~800 members of the GPCR family. Among the members of the G protein α family, the Gα12 family members comprising Gα12 and Gα13 have been referred to as gep oncogenes. Gα12/13 levels are altered in metabolic organs, including the liver and muscles, in metabolic diseases. The roles of Gα12/13 in metabolic diseases have been investigated. In this review, we highlight findings demonstrating Gα12/13 amplifying or dampening regulators of phenotype changes. We discuss the molecular basis of G protein biology in the context of posttranslational modifications to heterotrimeric G proteins and the cell signaling axis. We also highlight findings providing insights into the organ-specific, metabolic and pathological roles of G proteins in changes associated with specific cells, energy homeostasis, glucose metabolism, liver fibrosis and the immune and cardiovascular systems. This review summarizes the currently available knowledge on the importance of Gα12/13 in the physiology and pathogenesis of metabolic diseases, which is presented according to the basic understanding of their metabolic actions and underlying cellular and molecular bases.
Similar content being viewed by others


GPCR–G protein pathways in metabolic diseases
Metabolic syndrome has been a serious health issue in the 21st century. It is a cluster of risk factors that can lead to cardiovascular diseases, diabetes, and stroke. Insulin resistance is the major factor that induces metabolic syndrome. It has been estimated that 642 million people will have type 2 diabetes by 20401. The G protein-coupled receptor (GPCR) family is the largest membrane receptor family and serves as an attractive drug target. Currently, FDA-approved drugs, which account for approximately one-third of all drugs, target more than 100 GPCRs2. The glucagon-like peptide-1 (GLP-1) receptor, a member of the glucagon receptor family of GPCRs, is a metabolic syndrome-associated drug target. GLP-1 receptor agonists are used for glycemic control in patients with type 2 diabetes mellitus3. Because of its weight-loss effects, liraglutide (SaxendaTM) has become the first GLP-1 receptor agonist approved for the treatment of obesity4. Many GPCRs are pivotal sensors of energy metabolism. Some GPCRs are activated by energy metabolites or substrates, such as fatty acids, nucleotides, saccharides, hydroxycarboxylic acids, and citric acid cycle intermediates5. GPCRs activated by fatty acid-derived lipids have been proposed as antidiabetic drugs6. For example, the beneficial effect of omega-3 fatty acids is mediated by GPR120, also known as free fatty acid receptor 4. GPR120 is an attractive therapeutic target for the treatment of type 2 diabetes. GPR120-selective agonists improve insulin resistance and chronic inflammation and are currently under investigation for possible drug development7. Consequently, GPCR modulators are being actively investigated for their clinical applications in metabolic syndrome.
G proteins are recognized and activated by agonist-bound GPCRs. Heterotrimeric G proteins comprise the Gα, Gβ, and Gγ subunits. Gα mainly determines the functions of a G protein via the exchange of guanosine diphosphate (GDP) with guanosine triphosphate (GTP). The Gβ and Gγ subunits are synthesized separately and form a complex to function as a single functional unit. Both Gα and Gβγ subunits activate signaling molecules, thus eliciting cellular responses. Based on the characteristics of the Gα subunit, heterotrimeric G proteins are classified into four families: Gαs (s represents stimulation), Gαi (i represents inhibition), Gαq and Gα12 families. The Gαs family comprises Gαs and Gαolf. The Gαi family includes Gαi1, Gαi2, Gαi3, transducin-rod, transducin-cone, Gαo-A, Gαo-B, and Gαz. The Gαq family includes Gαq, Gα11, Gα14, Gα15 (Gα15 is the mouse ortholog of Gα16) and Gα16. The Gα12 family comprises only two members: Gα12 and Gα13. Gβ comprises five members, and Gγ comprises 12 subtypes8. Thus, many combinations of Gα, Gβ, and Gγ subunits of heterotrimeric G proteins exist.
G protein signaling regulates the complexity of diverging and converging signal transduction systems. Thus, G protein levels may have a significant effect on the suppression or amplification of physiological and biochemical activities. The roles of Gαi and Gαq in metabolite-sensing signaling pathways have been extensively demonstrated. For example, Gαi is coupled with the sweet taste receptor TAS1R2/TAS1R3 or with hydroxycarboxylic acid receptors9. Glucose, lactose, fructose, maltose, and sucrose are the ligands of TAS1R2/TAS1R310. Hydroxycarboxylic acid receptors are activated by lactate, 3-hydroxybutyrate, and 3-hydroxyoctanoate9. Gαq is the primary transducer of fatty acid receptors and citric acid cycle intermediate receptors. Some receptors, such as GPR43 and GPR91, utilize both Gαi and Gαq9. Gαs-coupled receptors play roles in glucose and lipid metabolism. For instance, Gαs-coupled GPR119 regulates GLP-1 secretion in enteroendocrine L cells and insulin secretion in pancreatic β cells11. Gαs-coupled β-adrenergic receptors stimulate lipolysis in adipocytes.
Gα12/13, also known as gep proto-oncogenes, are often overexpressed in cancers, including breast12, prostate13, and liver14 cancers. They serve as prognostic factors, and high expression of Gα12/13 is associated with poor prognosis for patients15,16. The members of the Gα12/13 family have been recently identified as direct or indirect regulators of systemic energy metabolism. In this review, we summarize the posttranslational modifications of Gα12/13, the associated signaling pathways and the currently available knowledge on the pathophysiology of the G protein family with respect to energy metabolism and metabolic diseases.
Posttranslational modifications of heterotrimeric G proteins
The Gα and Gγ subunits are posttranslationally modified by lipids. Lipid modification is essential for the functional maturation of G proteins. The functions of G proteins are also affected by phosphorylation at serine, threonine, and tyrosine residues. In addition to lipid modification and phosphorylation, G protein levels are affected by changes in stability (e.g., enhanced degradation) and increased transcription. Each topic has been described in detail in the following Section.
Lipid modifications
The functional maturation of G proteins requires posttranslational modifications. The Gα subunit is modified by palmitoylation and myristoylation17. Myristoylation occurs at Gly in the amino-terminal domain, whereas palmitoylation occurs in the Gα subunit via thioesterification of the Cys residue. Gα12/13 undergo palmitoylation but not amidical myristoylation18. Palmitoylation of Gα13 is necessary for plasma membrane localization, Rho-dependent signaling, and the redistribution of a direct effector, p115-RhoGEF, from the cytoplasm to the plasma membrane19. Gα12 resides in lipid rafts, but Gα13 has not been found in lipid rafts. Prevention of Gα12 palmitoylation by mutating Cys-11 in Gα12 partially relocalized Gα12 away from lipid rafts20. Hsp90 specifically interacts with Gα12 but not with Gα13. Hsp90 interactions and the acylation of Gα12 facilitate Gα12 movement to lipid rafts20. Receptor stimulation induces the translocation of Gαs from the plasma membrane to the cytoplasm. This translocation involves Gαs depalmitoylation, which renders Gαs less hydrophobic. However, opposite results have been reported by other research groups21. Thus, the role of depalmitoylation in membrane anchoring remains to be elucidated.
Palmitoylation is a reversible reaction; thus, receptor stimulation increases the turnover of palmitate. For instance, the turnover of palmitate attached to Gαs is faster than the turnover of Gαs itself. Isoproterenol stimulation of the β-adrenergic receptor and cholera toxin treatment increase the turnover rate without changing the total amount of palmitoylated Gαs22. Although palmitoylation is not the primary factor for the translocation of Gαs to the plasma membrane, the palmitoylation-defective mutant of Gαs is not associated with the membrane and is poorly coupled with adenylyl cyclase23. In contrast, Gαq palmitoylation is essential for the stimulation of phospholipase C23. Thus, there is no consensus showing that the palmitoyl group is directly involved in the interaction of Gα with its effector molecules. Palmitoylation and depalmitoylation enzymes have been identified; however, their regulatory mechanisms remain to be elucidated.
Phosphorylation
Gα subunits are phosphorylated by not only serine and threonine kinases but also tyrosine kinases. The Gβ subunit is phosphorylated by a kinase; however, this phosphorylation occurs on a His residue, and Gβ is an intermediate for the transfer of the phosphate group to GDP in the guanine nucleotide-binding pocket of the Gα subunit.
Phosphorylation of Gα subunits by PKA and PKC
Gα subunits are phosphorylated by kinases such as PKA and PKC, which are activated downstream of Gαs and Gαq. PKA, stimulated by a membrane-permeable cAMP analog, phosphorylates Gα13. The PKA phosphorylation site of Gα13 is tentatively assigned to Thr203. Replacement of Thr203 with Ala results in a mutant with reduced affinity for Gβγ and decreased activation of RhoA24. This phosphorylation site is conserved in Gα12, another Gα12/13 family member, but not in other G protein families such as Gαs, Gαi, and Gαq. However, most Gα subunits contain PKA phosphorylation sites other than Thr203. Whether PKA phosphorylates Gαs, Gαi and/or Gαq to modulate their functions remains to be elucidated.
During platelet activation, Gα12 and Gα13 are phosphorylated25. Treatment with thrombin and phorbol 12-myristate 13-acetate phosphorylates Gα12 and Gα13 in a PKC-dependent manner. Among PKC subtypes, PKCβ, PKCδ, and PKCε efficiently phosphorylate Gα12 and Gα1325. Gα12 is also phosphorylated by other isoforms of PKC, including PKCα and PKCζ26,27,28. Phosphorylation enhances Gα12 activation, as in the case of Gαz.
PKC also phosphorylates other G proteins (e.g., the Gαi and Gαq family). In hepatocytes, the phosphorylation of Gαi2 is enhanced by phorbol esters that activate PKC29. Insulin treatment inhibits the basal and phorbol ester-stimulated phosphorylation of Gαi2, thereby increasing the nonphosphorylated active form of Gαi2. The increased phosphorylation of Gαi2 by PKC explains adenylyl cyclase inhibition upon insulin treatment. The phosphorylation of Gαi2 at Ser44, Ser144, and Ser302 by PKC promotes morphine-induced desensitization of the mu-opioid receptor30. PKC may be critical for the phosphorylation of Gα11 at Ser154, thereby inhibiting agonist-stimulated phospholipase C activity31.
5-HT2A receptor stimulation induces the phosphorylation of Gαq/11 at Ser154. The phosphorylation of the 5-HT2A receptor is mediated by PKC, decreasing this receptor coupling with Gαq/1131. 5-HT2A receptors couple with Gαq/11, which implies a possible desensitization mechanism for the 5-HT2A receptor.
PKC phosphorylates the Gαz and Gαq families. Phosphorylation of Gαz has been demonstrated in vitro and in permeabilized platelets32. Phosphorylation at Ser16 inhibits the binding of Gβγ. As Gβγ binding is inhibited, Gαz function is prolonged compared to that of the nonphosphorylated form.
PKC also phosphorylates Gα15, an ortholog of human Gα16, at Ser336. This phosphorylation site is located where the receptor and G protein interact. Thus, the phosphorylation-defective mutant of Gα15 is not activated by the receptor.
PAK, a p21-activated protein kinase, phosphorylates Gαz at Ser16. PAK-promoted phosphorylation of Gαz inhibits Gβγ binding, indicating that phosphorylation at the amino-terminal region regulates Gβγ binding33. This failure to bind leads to prolonged activation of Gα functions. The pathogenic bacterium Yersinia secretes Yersinia protein kinase A (YpkA), a Ser/Thr kinase, which phosphorylates Gαq and inhibits GTP binding and Gαq-mediated signaling, implicating Gαq phosphorylation by Yersinia as the cause of disease34.
Phosphorylation of Gα subunits by tyrosine kinases
The Gα subunit is phosphorylated by tyrosine kinases. Tyrosine phosphorylation of the Gαq family occurs via receptor stimulation. M1 muscarinic acetylcholine receptor stimulation increases the tyrosine phosphorylation of Gα11 at Tyr35635; this phosphorylation site is located in the receptor recognition region, and its phosphorylation increases basal phospholipase C activity in vitro. This outcome suggests that Gα11 phosphorylation plays a role not only in receptor coupling but also in effector activation. Another study showed that coexpression of the M1 muscarinic acetylcholine receptor with the tyrosine kinase Fyn enhances Gαq/11 signaling.
Stimulation of metabotropic glutamate receptor 1α (mGluR1α) by glutamine increases the Tyr phosphorylation of cellular proteins, which is inhibited by the protein tyrosine kinase inhibitors genistein and tyrphostin AG21335. Glutamate stimulation-induced inositol-triphosphate production is inhibited by genistein and AG213, supporting the idea that tyrosine phosphorylation occurs prior to the action of phospholipase C.
Gαs is phosphorylated by the proto-oncogene pp60c-src at Tyr37 and Tyr37736. The phosphorylation of Gαs stimulates GTPγS binding and receptor-stimulated GTPase activity. Because Y377 is located in receptor-coupling regions, such as Tyr356 in Gα11, pp60c-src-stimulated phosphorylation may affect receptor coupling. Hence, Src-mediated Gαs phosphorylation may be critical for cancer-associated cell growth.
The EGF receptor stimulates tyrosine phosphorylation of Gαs, increasing Gαs-mediated adenylyl cyclase activity37. The insulin receptor directly or indirectly phosphorylates Gαo and Gαi, although the functional consequences have not yet been demonstrated. It remains unknown whether Gαi, Gαq or Gα12 family G proteins are phosphorylated by tyrosine kinases.
Degradation
The expression of Gα12/13 is often deregulated in metabolic diseases. For example, hepatic Gα12 expression is decreased in humans with nonalcoholic fatty liver disease38. Hepatic Gα13 is downregulated39, whereas Gα13 is overexpressed in the skeletal muscle of patients with diabetes40. These findings suggest that the balance between Gα12/13 synthesis and degradation is disrupted under pathological conditions. However, the molecular mechanism underlying Gα12/13 degradation has not yet been fully elucidated. Here, we review the degradation process of other Gα proteins.
Ubiquitination of Gα subunits
Cholera toxin catalyzes the ADP-ribosylation of Arg201, which is critical for the GTP-hydrolyzing activity of Gαs. Because ADP-ribosylation inhibits GTPase activity, Gαs is activated even without receptor stimulation. Treatment with the cholera toxin increases the Gαs level in the cytoplasm and enhances its degradation rate. This finding suggests that Gαs is degraded after being dissociated into Gα and Gβγ. Compared to GDP-bound inactive Gαs, in the cytoplasm, GTP-bound active Gαs is subjected to an efficient degradation system41. However, it remains unknown whether Gαs degradation is mediated completely by the ubiquitin-proteasome system. The precise degradation mechanism remains to be determined.
Regulation by Ric-8
There is no definitive evidence supporting the idea that the G protein undergoes ubiquitin-mediated regulation. However, accessary G proteins were found to be controlled in a ubiquitin-dependent manner. Ric-8 was originally found to positively regulate neurotransmitter release through Gαq in Caenorhabditis elegans. Contrary to the C. elegans and Drosophila genomes, the mammalian genome contains two members of Ric-8 (Ric-8A and Ric-8B)42.
Ric-8A functions as the guanine nucleotide exchange factor (GEF) associated with Gαq, and knocking down Ric-8A by siRNA inhibits ERK activation and intracellular Ca2+ increase42. In Ric-8A-knockout fibroblasts, the protein levels of Gα13 and other G proteins such as Gαi1-2, Gαo, and Gαq were decreased to 10% those in wild-type cells. In contrast to the Ric-8A-knockout cells, Ric-8B knockout cells exhibited only reduced levels of Gαs42. Ric-8B interacts with Gαolf, Gαs, and Gαq. The coexpression of Ric-8B and Gαolf, a Gαs family member, with dopamine D1 and β2-adrenergic receptors enhanced receptor-stimulated cAMP production in HEK293 cells. In NIH-3T3 cells, knocking down Ric-8B inhibited isoproterenol-stimulated cAMP production and decreased the expression level of Gαs42. However, it did not affect the expression levels of other Gα proteins, such as Gαi and Gαq. The suppression of Ric-8B expression did not affect Gαs mRNA expression. Thus, the function of Ric-8B is to stabilize the Gαs protein.
The Ric-8 protein interacts with Gα, thus stabilizing the Gα subunit. Gα is stabilized via the inhibition of ubiquitination43. Although the E3 ubiquitin ligase for Gα proteins has not yet been identified, Ric-8 has been reported to inhibit ubiquitination and stabilize Gα proteins. Because Ric-8 controls the level of the Gα subunit, ubiquitin-mediated Ric-8 regulation is critical to G protein levels and G protein-mediated responses. Although the mechanism by which Ric-8 expression is regulated has not been reported in detail, receptor stimulation both increases and decreases the expression level of Ric-842. Thus, Ric-84 may be part of a newly discovered type of regulatory mechanism that modulates receptor-mediated signaling through G proteins. Ubiquitination regulates G protein levels in rod photoreceptors, with light-dependent translocation of transducin between the outer and inner photoreceptor segments important for light/dark adaptation and the prevention of cell damage by light. Inhibition of transducin α subunit (Tα) translocation from the inner part to the outer segment reduces light responses. In contrast, Tα is translocated to the outer segment to enhance light reception sensitivity. Cullin 3 (Cul3)-Kelch-like 18 (Klhl18) ubiquitin E3 ligase modulates the translocation of Tα44. Cul3-Klhl18 ubiquitinates UNC119, a Tα-interacting protein, and promotes the degradation of UNC119. UNC119 is a lipid-binding protein that interacts with the acylated amino-terminus of Tα. Casein kinase 2 phosphorylates UNC119, and this phosphorylation inhibits its degradation by Cul3-Klhl18. Thus, ubiquitination indirectly modulates the location and expression of Tα in rod photoreceptors.
Other regulatory mechanisms mediated by Gα subunits
Promoter analysis and the transcriptional regulation of G proteins via transcription factors have not been completely characterized to date. However, the posttranscriptional regulation of Gα12/13 by microRNA has been reported. Kim et al.45 found the reciprocal expression of miR-16 and Gα12 in liver fibrosis. miR-16 is abundantly expressed in quiescent hepatic stellate cells (HSCs), and its expression is diminished in activated HSCs. miR-16 targets Gα12. Dysregulation of miR-16 contributes to liver fibrosis by facilitating Gα12-mediated autophagy in HSCs. miR-31 and miR-30d directly target Gα13 and inhibit the invasion of breast cancer cells and colorectal cancer cells, respectively46,47. miR-182 and miR-141/200a regulate Gα13 posttranscriptionally48.
The biology of GPCRs coupled to Gα12/13
Gα12/13 transduce signals from more than 30 GPCRs. Lysophosphatidic acid receptors (LPA), sphingosine-1-phosphate receptors (S1P1–S1P5), angiotensin II type 1 receptors (AT1) and thrombin receptors (PAR1) couple with Gα12/1349,50 (Table 1). In response to ligands, the conformation of GDP-bound inactive Gα12/13 is transformed to the GTP-bound active form. The guanine nucleotide-dependent conformational change of G proteins results in the release of GTP-bound Gα12/13 from the Gβγ subunits. The Gα12 family members regulate RH-RhoGEF, which contains an RGS homology domain and activates GTPase Rho51,52. The RH-RhoGEF family contains p115RhoGEF, leukemia-associated RhoGEF (LARG) and PDZ-RhoGEF. P115RhoGEF and LARG exhibit GAP activity specifically toward Gα12/1353, whereas PDZ-RhoGEF does not affect Gα12/13. Gα13 directly stimulates p115RhosGEF and LARG54,55, and Gα12/13 are deactivated by the hydrolysis of GTP to GDP by intrinsic GTPase activity and GTPase-activating proteins (GAPs)54,55,56. Thus, Gα12/13 signaling is fine-tuned by RH-RhoGEF family.
Rho kinase (ROCK) is a downstream effector of the Gα12/13-RhoA signaling pathway, which in turn phosphorylates various substrates, such as MLC phosphatase, ERM, LIMK, diaphanous, rhotekin, rhophilin, citron kinase, and CRPM-257. Both the RhoA inhibitor C3 toxin and ROCK inhibitor Y-27632 prevent Gα12/13-mediated RhoA- and ROCK-dependent MLC20 phosphorylation58. Gα12/13 also activate Jun kinase (JNK), which in turn phosphorylates Jun and ATF259,60,61. Gα12/13 play roles in inflammatory responses. Sphingosine-1-phosphate, a sphingolipid produced by platelets, stimulates cyclooxygenase-2 (COX-2), which is mediated by the S1P1/S1P3/S1P5-Gα12 pathway62. Gα12 regulates sphingosine-1-phosphate-mediated NF-κB-mediated COX-2 induction, which depends on JNK-dependent ubiquitination and the degradation of IκBα62. Thrombin induces monocyte migration via PAR1-Gα12-p115RhoGEF activation through a Gα12-mediated pathway63. Gab1 interacts with p115RhoGEF and induces monocyte F-actin cytoskeletal remodeling and migration via Rac1- and RhoA-dependent Pak2 activation63. Therefore, Gα12/13-coupled receptor signaling plays a role in cell mobility, growth, differentiation, inflammation, and transcription64.
Gα12/13 in the immune response
Inflammation is an extremely dynamic response mediated by interactions among various immune cells. Immune cell activation, migration to the site of inflammation and inflammatory effector function are largely facilitated by the coordination of various chemokine receptors that are structurally similar to GPCRs65. Examples of receptors known to interact with Gα12/13 proteins include chemokines, S1P, LPA, and thrombin receptors, which are important for recruiting immune cells to the site of inflammation and promoting entry into lymphoid organs, thereby amplifying the inflammatory response66. In addition to the abovementioned lipids and chemokines, protons can also act as Gα12/13 activators, and are highly abundant in lymphocytes. Dysregulation of Gα12/13 leads to lymphoid hyperactivation and/or malfunction. Mouse studies have shown that Gα12/13 disruption leads to lymphoid hypertrophy and adenopathy, demonstrating the importance of Gα proteins in fine-tuning immune responses67,68. Furthermore, dysregulation of Gα12/13 in immune cells may lead to pathophysiological consequences, such as cancer and autoimmunity68,69,70,71.
Gα12/13 regulation in B cells
Gα12 may be involved in B cell maturation based on its binding and activation of BTK, a kinase required for normal B cell development and activation. The reduction in MZB precursors in B cell-specific Gα12/13‐deficient mice also suggests a role for Gα12/13 in peripheral MZB maturation72. The migration of B cells in response to serum and S1P treatment is highly increased in mutant MZB cells but not in follicular B cells, indicating that Gα12/13 family members contribute to the formation of mature MZB cells by controlling precursor migration72. Gα12/13-coupled receptors, such as S1PR2 and P2RY8, have been shown to regulate B cell confinement in the germinal centers within SLOs69,73, and loss-of-function mutations have been directly associated with oncogenic proliferation and the impaired apoptosis of germinal center B cells68,69. Despite the high expression of Gα12/13-mediated receptors, precise chemokine gradients that enable accurate B cell positioning and movement throughout various developmental stages have only recently been elucidated74.
Gα12/13 regulation in T cells
Gα13‐mediated signaling, but not Gα12-mediated signaling, has been shown to be necessary for early thymocyte proliferation and survival75. The expression of a mutant form of p115RhoGEF, a Gα13 effector in progenitor T cells, leads to reduced proliferation and increased apoptosis rates at the double‐negative stage of T cell development. Accurate Gα12/13-mediated signaling has also been shown to be important to CD4 T cells in various stages after complete thymocyte development. Gα12/13-coupled receptors negatively regulate cell polarization and adhesion, thereby controlling effector T cell entry in and exit from secondary lymphoid organs67. T cells are activated via TCR signaling, and downstream cascade molecules have been shown to interact with Gα13 to mediate serum response factor transcriptional activity76. Following naive T cell activation, CD4+ T cells differentiate into effector subsets: Th1, Th2, Th17 and Tfh cells, and each of these subsets has a specified helper function that facilitates protection of the host against various types of foreign pathogens77. In particular, Tfh cells act as critical helper T cells that facilitate B cell maturation and antibody response78. Gα12/13-mediated receptor signaling is important in Tfh differentiation and functions by coordinating cell localization cues79. STAT3 activation has been suggested to be the key target of regulation by which Gα12/13-mediated signaling modulates inflammatory T cells such as Th17 cells80,81. ROCK2 has been specifically shown to directly interact with phosphorylated STAT3 and co-occupy Th17/Tfh gene promoter regions of key transcription factors such as Irf4 and Bcl6 in human Th17 cells81. ROCK2-specific inhibitor treatment led to lower levels of STAT3-mediated IL-17 and IL-21 cytokine secretions in T cells from both healthy and rheumatoid arthritis patients. In addition, ROCK inhibitor treatments in a mouse model of autoimmunity promoted STAT5-mediated Treg activity, thereby restoring disrupted immune homeostasis80. These findings indicate that the regulation of GPCR-mediated signaling in immune cells has important consequences in the context of allergy and autoimmunity in addition to its effects on metabolic diseases70,71.
Roles of Gα12/13 in physiology and pathology
Role of Gα12/13 in energy homeostasis
The liver is the key metabolic organ maintaining whole-body energy balance. The liver regulates carbohydrate, fat and protein metabolism. When excess glucose enters the blood circulation after a meal, insulin stimulates the liver to synthesize glycogen for storage. Upon a decrease in blood glucose content, glycogen is broken down into glucose through glycogenolysis. Excess carbohydrates can be converted into free fatty acids and triglycerides via hepatic de novo lipogenesis. Insulin autonomously regulates lipid synthesis in the liver82. Under insulin-resistant conditions, insulin continues to promote lipogenesis but fails to suppress hepatic glucose production82. In the catabolic pathway, the liver produces ATP via fatty acid oxidation, which is the mitochondrial aerobic process of fatty acid breakdown that generates acetyl-CoA. The liver also synthesizes nonessential amino acids and plasma proteins. When the fine-tuned regulation of energy balance is disrupted in the liver, nonalcoholic fatty liver disease can develop. A high-fat diet supplemented with fructose impairs hepatic mitochondrial function and decreases fatty acid oxidation, thereby contributing to nonalcoholic fatty liver disease83.
Hepatic expression of Gα12 is upregulated in the fasted state38. Gna12-KO mice had increased lipid accumulation in the liver after fasting than did the corresponding wild-type mice. The role of Gα12 in the regulation of mitochondrial respiration, as mediated by the SIRT1/PPARα network, was identified via microarray analyses and in vivo experiments38. SIRT1, an NAD+-dependent protein deacetylase, is an important regulator of the PPARα-mediated transcriptional network involved in fatty acid oxidation84. The Gα12 pathway governs mitochondrial respiration, lipid catabolism, acyl-CoA metabolism, ketogenesis and peroxisomal oxidation through SIRT1 stabilization38.
Fasted mice show increased serum adenosine concentrations. Adenosine is produced and released from most tissues. In addition, extracellular adenine nucleotides can be broken down into adenosine, which serves as a ligand for four distinct GPCRs (A1, A2a, A2b, and A3). Adenosine receptor agonists and antagonists have been tested for their effects against liver ischemia, liver cancer, cardiovascular disease, and Parkinson’s disease85,86,87. The adenosine receptor agonists stimulated SIRT1 expression through Gα1238 (Fig. 1). Gα12 stabilized SIRT1 protein through the HIF-1α-mediated transcriptional induction of ubiquitin-specific peptidase 22 (USP22)38. Therefore, high levels of Gα12 expression promote mitochondrial respiration. Compared with that in patients without steatosis, the Gα12 level in patients with NAFLD was diminished in the liver. Consequently, Gα12 deficiency results in high-fat diet-induced obesity and hepatic steatosis. Adenosine receptor-Gα12 coupling plays a role in lipid metabolism via the SIRT1/PPARα pathway. Gα12 is also found in mitochondria, and mitochondrial Gα12 is associated with decreased mitochondrial motility88. Gα12 binds to the inner surface of the cell membrane, and Gα12 specifically targeted to the mitochondria may control mitochondrial respiration, morphology, and dynamics in a distinct manner. During adipocyte hypertrophy and hyperplasia, white adipose tissue undergoes dynamic expansion and remodeling. The activated Gα12 mutant increased B-Raf and MEK1 expression and MAPK activity. Thus, the active form of Gα12 enhanced the proliferation of preadipocytes but prevented their differentiation89. Rho/ROCK, downstream from Gα12/13, inhibited 3T3-L1 adipogenesis in response to G-protein-deamidation of dermonecrotic toxins90. LPA4 receptors are exclusively expressed in epididymal white adipose tissue. Octadecenyl phosphate is an agonist of LPA4. The ODP–LPA4 axis activates Gα12/13 in adipocytes, and LPRA4 in adipocytes limits the continuous remodeling and healthy expansion of white adipose tissue via Gα12/13. LPA4 activation by octadecenyl phosphate treatment decreases PPARα-associated gene expression. Consistently, the loss of LPA4 promotes adipose tissue expansion and protects against high-fat diet-induced hepatic steatosis and insulin resistance91.

Intracellular ATP or UDP is released through Pannexin 1 channels. Extracellular ATP or UDP binding to the purinergic P2Y6 receptor in cardiomyocytes mediates mechanical stretch-induced Gα12/13 and Rho activation. P2Y6-Gα12/13 signaling is critical for mechanical stretch-induced fibrotic factors such as connective tissue growth factor (CTGF), periostin and transforming growth factor (TGF)-β. Extracellular ATP is actively broken down to ADP, AMP and, ultimately, adenosine by ectonucleotidases and may serve as GPCR ligands. Gα12 is involved in the fatty acid oxidation process downstream of adenosine receptors.
Skeletal muscle is one of the major organs in systemic energy homeostasis because it requires a high amount of nutrients. Mammalian skeletal muscles have two types of myofibers: oxidative and nonoxidative; these myofibers show distinct metabolic characteristics. Endurance exercise training reprograms myofiber types into oxidative fibers, which have a higher capacity for mitochondrial respiration and fatty acid oxidation92. Obese individuals or patients with type 2 diabetes possess fewer oxidative myofibers. GPR56 is an adhesion GPCR, and it transduces signals through activated Gα12/13-Rho. GPR56 is involved in mechanical overload-induced muscle hypertrophy93. PGC-1α4, an alternatively spliced form of PGC1α, transcriptionally regulates GPR56 induced by resistance exercise. Consistently, Gα12 and Gα13 mRNA levels increase in human subjects performing resistance training than in sedentary individuals.
Studies investigating Gα13 have been limited because the gene-knockout animal model showed a significant defect in vasculogenesis during development, which led to embryo lethality94. Using the Cre–loxP system, muscle-specific Gα13-knockout mice were generated40. In this study, knocking out skeletal muscle-specific Gna13 promoted the reprogramming of oxidative-type myofibers, with resultant increases in mitochondrial biogenesis. Gα13 and its effector RhoA suppressed NFATc1 by increasing Rock2 and was critical for the phosphorylation at Ser243 in NFATc1; this suppression and phosphorylation were reduced after exercise but were higher in HFD-fed obese animals. Consequently, the muscle-specific ablation of Gα13 increased whole-body energy metabolism, protecting animals from obesity and liver steatosis. In the absence of Gα13, Gα12 plays a role in mitochondrial regulation in skeletal muscle38. Thus, the Gα12 signaling pathway controls mitochondrial energy expenditure via SIRT1-mediated and HIF-1α-dependent USP22 induction38 (Fig. 2).

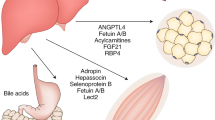
Gα12 levels are lower in the liver of high-fat diet (HFD)-fed mice and in patients with steatosis and/or nonalcoholic steatohepatitis. Gα12 transduces signals of deubiquitination and stabilization of SIRT1 through HIF-1α-mediated transcriptional control of ubiquitin-specific peptidase 22 (USP22). SIRT1 governs the PPARα transcriptional network in metabolic processes, particularly fatty acid oxidation. The Gα12 pathway facilitates whole-body energy expenditure through USP22/SIRT1-regulated mitochondrial respiration. Gα13 levels in skeletal muscle are decreased in the exercise-induced state (a condition of energy deficiency) but are increased in mice fed an HFD or in patients with type 2 diabetes. Gα13-RhoA-ROCK2 phosphorylates nuclear factor of activated T cells 1 (NFATc1) at Ser243 to inhibit NFATc1 activation. NFATc1 contributes to the transformation of fibers into oxidative-type fibers. Deficiency of Gα13 in skeletal muscle promotes energy expenditure, thereby protecting mice from metabolic challenge induced by NFATc1-dependent myofiber-type reprogramming.
Even though both Gα12 and Gα13 commonly activate RhoA, the downstream effectors and functions are diverse among tissues. In the postprandial state, dietary triglycerides are transported to the liver from the intestines, and then, the liver utilizes fatty acids and glycerol to synthesize triglycerides. Excessive amounts of nutrients are mainly stored in fat and liver tissues. Obesity increases HIF-1α levels and decreases the sinusoidal blood flow rate and velocity in the liver95. Hypoxia enhances nonalcoholic fatty liver disease96. The inhibition of the RhoA/Rock pathway attenuated the effect of Gα12 overexpression on HIF-1α. Gα13 activates Rho/Rock2. Rock2 phosphorylates NFATc1 in skeletal muscle. NFATc1 is not expressed in the liver (https://www.proteinatlas.org/ENSG00000131196-NFATC1/tissue/liver). Therefore, we presume that Gα12 activates HIF-1α in the liver and that Gα13 inactivates NFAT1c in skeletal muscle.
Role of Gα12/13 in glucose metabolism
Because insulin resistance causes diabetes and contributes to metabolic syndrome in multiple organs, approaches targeting single organs have limitations. The liver senses extracellular nutritional availability and regulates overall glucose metabolism. Sustained excessive intake of calories leads to fat accumulation and liver steatosis. Steatosis, frequently accompanied by hyperglycemia, usually leads to metabolic dysfunction in other organs97,98,99, suggesting a causal role of liver pathophysiology in the dysregulation of systemic energy homeostasis. The increased hepatocellular lipid content causes hepatic insulin resistance. Excessive free fatty acids and imbalanced adipocytokines cause not only insulin resistance but also promote the progression of hepatic steatosis to nonalcoholic steatohepatitis and cirrhosis. Hepatic fat content is a key determinant of metabolic flux in insulin resistance and type 2 diabetes mellitus100. Nevertheless, the evidence that the liver may be the origin and driver of the systemic disruption of energy metabolism primarily involving insulin resistance in the setting of metabolic disease progression has drawn little attention.
According to a phase III placebo-controlled study, fasiglifam (TAK-875), a partial GPR40 agonist, effectively lowers HbA1c in people with type 2 diabetes101. Due to off-target liver toxicity, the clinical development of fasiglifam was terminated102. Subsequently, GPR40 full agonists have been under development in preclinical settings. A GPR40 allosteric full agonist enhanced the glucose-stimulated insulin secretion in pancreatic β cells via the GPR40-mediated activation of Gα12103. However, the overexpression of Gα12 decreased insulin secretion through JNK38. Gna12-KO mice fed a HFD displayed lower fasting glucose levels with hyperinsulinemia. Nevertheless, whole-body glucose flux was not significantly altered by Gα12 deficiency.
Hyperglycemia decreases Gα13 in the liver, eventually contributing to glucose intolerance and insulin resistance in other metabolic organs by overproducing liver-secretory O-GlcNAc protein (Fig. 3)39. With HFD feeding, hepatocyte-specific Gα13-knockout mice exhibited exacerbated glucose tolerance and insulin resistance, although a normal diet had no effect on the metabolic phenotypes, such as body weight gain and fasting blood glucose content39. Therefore, the decrease in Gα13 in hepatocytes was clearly manifested by metabolic challenges and was distinctively associated with glucose utilization.

Hepatic Gα13 levels are downregulated in high-fat diet (HFD)-fed or genetically obese mice and patients with diabetes. In response to a decrease in Gα13, the inter-α-trypsin inhibitor heavy chain 1 (ITIH1) is O-GlcNAcylated by O-GlucNAc transferase (OGT) induced in hepatocytes and then excessively secreted into the bloodstream. ITIH1 is a binding partner of HA, one of the major extracellular matrix components. In mice deficient in Gα13 in hepatocytes, increased ITIH1 is deposited onto the hyaluronan surrounding skeletal muscle and white adipose tissue. Overproduction of ITIH1 from the liver after the loss of Gα13 causes systemic insulin resistance. Treatment with ST045849 (an inhibitor of O-GlcNAC transferase) or antibody neutralization of ITIH1 ameliorates systemic insulin resistance.
The extracellular matrix (ECM) is a highly dynamic compartment consisting of different extracellular proteins. The ECM modulates not only biological processes, including cell growth and migration, but also physiological communication. Thus, ECM remodeling in peripheral tissues affects glucose metabolism and insulin signaling under diabetic conditions. A number of pathological conditions affect aberrant ECM remodeling and deposition. Notably, the stiffness or rigidity of the ECM also significantly affects cellular function and is highly dependent on various interacting proteins, stabilizing and potentiating their binding properties with other ECM proteins. Hyaluronan (HA) is one of the major components of the ECM, and its level is increased in insulin-resistant tissues. Excessive accumulation of HA in metabolic tissues has been observed in obese diabetic mice104. Moreover, serum HA levels are increased in patients with diabetes and liver fibrosis105,106. Depletion of HA via the intravenous administration of hyaluronidase in mice led to improvements in systemic glucose tolerance and insulin sensitivity, indicating the crucial role of HA in the pathogenesis of insulin resistance104. Furthermore, an HA synthesis inhibitor attenuated NASH-mediated liver fibrosis106 (Fig. 4).

miR-16 directly targets Gα12. In activated hepatic stellate cells, miR-16 is dysregulated. Overexpression of Gα12 due to a decrease in miR-16 promotes autophagy through JNK-mediated ATG12-5 conjugation. The Gα12 signaling pathway contributes to hepatic stellate activation. In response to transforming growth factor-β (TGF-β), a central mediator of fibrogenesis, hyaluronan synthase 2 (HAS2) is transcriptionally upregulated. HAS2 synthesizes high-molecular-weight HA (HMW-HA). Reactive oxygen species or hyaluronan-degrading enzymes facilitate the conversion of HMW-HA into low molecular weight (LMW)-HA. LMW-HA treatment activates Notch1, which is critical for liver fibrosis.
The differential abundance of the proteins regulated by Gα13 in the liver was determined using proteomics-based approaches39. Among the liver-enriched secretory proteins, ITIH1 was revealed as a key molecule associated with metabolic defects. ITIH1, an HA-binding protein (i.e., SHAP–HA complex), is predominantly expressed in hepatocytes under diabetic conditions. Several studies have demonstrated changes in ITIH1 levels under pathological situations107. ITIH1 levels were decreased in patients with hepatic fibrosis108. Circulating ITIH1 mRNA levels were elevated in rats with D-galactosamine-induced liver injury. Bleomycin affects ITIH1 expression in a lung fibrosis model109. Hepatic Gα13 levels were diminished under diabetic conditions in which ITIH1 was the major driver of organ cross talk controlled by the liver. ITIH1 is overexpressed in the absence of hepatic Gα13, secreted through the circulation and directly binds to HA in the adipose tissue and skeletal muscle to stabilize the integrity of each, thereby aggravating peripheral insulin resistance. Hepatic Gα13 increased ITIH1 overexpression through O-linked β-N-acetylglucosamine transferase-catalyzed O-GlcNAcylation. The Gα13-mediated signaling cascade evident in systemic glucose intolerance provides a new conceptual framework implicating the liver as the primary metabolic organ critical for whole-body glucose metabolism under diabetic conditions.
The roles of Gα13 in energy metabolism differ substantially in the skeletal muscle (prodiabetic) and liver (antidiabetic)39,40. GNA13 (encoding Gα13) is predominantly expressed in muscles compared with its expression in other metabolically active organs40. Exercise diminished Gα13 expression but increased Gα12 expression39. Deficiency of Gα13 in muscles causes skeletal muscle to acquire the oxidative phenotype, and the accompanying reciprocal increase in Gα12 promotes fatty acid oxidation. Koo et al. found that muscle-specific deficiency of Gα13 protected mice from diet-induced adiposity with increased fatty acid metabolism. Because intramyocellular lipids are primary contributors to insulin resistance, muscle-specific deficiency of Gα13 enhances muscle glucose metabolism and insulin sensitivity40. In contrast, deficiency of Gα13 in the liver does not affect glucose metabolism in the liver but causes overproduction of ITIH1 in hepatocytes. Circulating ITIH1 consequently binds to HA on the surface of adipose tissue and skeletal muscle, culminating in systemic insulin resistance39.
Interestingly, Gα12 is also present in the endoplasmic reticulum and serves as an activator of the endoplasmic reticulum export machinery110. The COPII subunit Sec24 senses cargo folding and acts as a GEF to activate Gα12110. Activation of Gα12 in endoplasmic reticulum exit sites responding to a folded cargo protein load facilitates cargo export and suppresses protein synthesis; this process is also known to autoregulate endoplasmic reticulum export (AREX)110. AREX signaling regulates a fraction of the secretome110.
Role of Gα12 in liver fibrosis
Under the condition of liver fibrosis, HSCs are activated and transdifferentiated into myofibroblasts, which produce aberrant ECM in response to liver injury111. HSCs are activated by mediators such as platelet-derived growth factor (PDGF) and transforming growth factor-beta (TGF-β). Certain GPCR pathways promote liver fibrogenesis. Levels of thrombin, lysophosphatidic acid, endothelin-1, sphingosine-1-phosphate, angiotensin II and acetylcholine are all elevated during liver fibrosis, and most GPCRs activated by these ligands are coupled to Gα12. Moreover, among the G protein members, Gα12 is particularly overexpressed in activated HSCs45. Thus, signals from activated GPCRs in the HSCs are augmented. Gα13 is not significantly affected in these cells.
The transdifferentiation of HSCs and the resultant changes in ECM proteins are also controlled by miR-29b, miR-150, and miR-194, suggesting the pleiotropic action of multiple microRNAs on HSC activation. As a liver-enriched miRNA, miR-16 has been shown to affect Bcl-2 and cyclin D1 to control HSC proliferation and apoptosis resistance112,113,114. In addition, miR-16 dysregulation contributes to the activation of HSCs through Gα12 overexpression (Fig. 4).
Autophagy is the key process for organelle turnover and nutrient recycling. Under nutrient deprivation, autophagy is initiated. As a result, metabolites and macromolecules such as amino acids, glucose, fatty acids and nucleic acids are made available as energy sources. Dysregulation of autophagy is often associated with metabolic diseases, including obesity, diabetes and cardiac diseases. Impaired autophagy results in triglyceride accumulation and insulin resistance in the liver115. Autophagy regulates beige adipocyte maintenance and adipocyte differentiation116 and controls glucose tolerance and muscle mass117. Therefore, the dysregulation of autophagy inhibits adipocyte differentiation and muscle atrophy. Autophagy supplies the energy necessary to support HSC transdifferentiation by mobilizing lipids and inducing mitochondrial oxygen consumption, which allows HSCs to cope with energy demands and maintain phenotype and cell homeostasis. Hence, autophagy may be an important modulator of signaling pathways in HSCs. Autophagy is accompanied by changes in ATG5/12, consistent with reports that ligands known to activate GPCRs coupled with Gα12 (e.g., thrombin, sphingosine-1-phosphate and angiotensin II) stimulate autophagy. Because ATG5/12 are the key mediators in late stage autophagy118, it is inferred that Gα12 plays a role in the signal amplification during autophagy in HSCs and that its dysregulation contributes to liver fibrosis.
Previously, JNK was identified as a kinase regulated by the Gα12 signaling pathway during different biological events62; further, JNK activation promotes α-SMA expression in response to TGF-β, PDGF and angiotensin II, thereby activating HSCs. The Gα12-mediated JNK pathway participates in multiple autophagy steps, such as ATG12-5 conjugation. Gα12/13 increased Rho/Rac-dependent AP-1 activity. In another study, Gα12 signaling enhanced Nrf2 ubiquitination and degradation119. Nrf2 may be a promising target for the suppression of HSC activation; this possibility is supported by the finding that liver injuries caused by toxicants promote HSC activation through increased oxidative stress and/or decreased Nrf2. Moreover, Nrf2 activation may elicit an antifibrotic effect by inhibiting TGF-β/Smad signaling114. Together, HSC activation induced by Gα12 overexpression may be associated with increased Nrf2 degradation and TGF-β/Smad pathway activation.
Role of Gα12 in cardiovascular disease
Metabolic syndrome is defined as a combination of cardiovascular risk factors associated with obesity, diabetes, dyslipidemia and hypertension. Gα12 plays a role in the cardiovascular system. Baseline blood pressure is regulated by Gαq/11 but not Gα12/1358,120. However, salt-triggered hypertension is dependent on the Gα12/13 signaling pathway121. The vasoactive compound lysophosphatidic acid is a blood-derived bioactive lipid. Lysophosphatidic acid promotes transient hypertension mainly via the Gα12/13-coupling LPA4 receptor, one of five LPA receptors (LPA1–4 and LPA6). The ROCK inhibitor Y-27632 successfully suppresses LPA-induced hypertension, suggesting that LPA increases blood pressure via the Gα12/13-Rho/ROCK pathway122. Angiotensin II is an important factor for increasing blood pressure via AT1. In vascular smooth muscle cells, AT1-Gα12 activates Rho/ROCK in the rostral ventrolateral medulla in the brain. Inhibition of Gα12 via oligodeoxynucleotide infusion attenuated angiotensin II-mediated hypertension123. While the angiotensin II AT1 receptor antagonist has an anti-hypertensive effect in both young and old animals, the endothelin ETA antagonist darusentan can reduce blood pressure in aged animals. Endothelin increases the vascular tone in the smooth muscle of aged mice in a Gαq/11- and Gα12/13-dependent manner124.
Under conditions of vascular injury, the hyperplastic proliferation of vascular smooth muscle cells occurs and causes neointimal hyperplasia, a condition of exaggerated intimal thickness. The GPCR ligand sphingosine-1-phosphate stimulates abnormal vascular smooth muscle cell proliferation by inducing the secretion of ECM-associated proteins, particularly cysteine-rich protein 61 (CYR61). CYR61 is a member of the connective tissue growth factor family. Sphingosine-1-phosphate regulates Gα12/13-Rho-dependent CYR61 induction, leading to hyperplastic vascular abnormalities125. Thromboxane A2 can also induce vascular smooth muscle cell proliferation and migration. The thromboxane A2 receptor activates Yes-associated protein (YAP)/transcriptional coactivator with PDZ-binding motif (TAZ) to activate Gα12/13 and thus enhance these processes126. The Hippo pathway inhibits YAP/ TAZ, which sense mechanical cues, such as ECM stiffness127. During atherogenesis, the lysophosphatidic acid levels are increased, which then activates YAP/ TAZ through Gα12/13-coupled receptors128,129. Integrins are noncanonical Gα13-coupled receptors that mediate cell-ECM adhesion. Gα13 directly binds to integrin β3 and regulates integrin outside-in signaling125. Upon unidirectional shear stress, integrin is activated, leading to an interaction between integrin and Gα13130. This interaction inhibits RhoA and YAP/TAZ activity, thereby delaying atherogenesis.
Pressure overload induces cardiac fibrosis. Mechanical stretching enhances the release of nucleotides such as ATP and UDP from cardiac myocytes via pannexin-1. Nucleotides outside the cell control the mechanical stretch-induced activation of Gα12/13 through the P2Y6 receptor. Inhibition of Gα12/13-coupled P2Y6 receptors lowers fibrogenic factors and angiotensin-converting enzyme levels, inhibiting cardiac fibrosis131. P2Y6 receptors heterodimerize with AT1 receptors. The formation of AT1R-P2Y6 receptor heterodimers enhances vascular hypertrophy and angiotensin II-induced hypertension132.
Concluding remarks
Diverse activation pathways in many GPCRs converge through heterotrimeric G proteins. In contrast to the mechanisms of GPCR regulation, the regulatory mechanisms of G proteins have not been completely elucidated. For more than 25 years, Gα12 has been of great interest in the field of cancer biology. Gα12/13 play multifunctional and distinct roles at multiple stages in different organs and in the development of metabolic diseases. In pathological states, abnormal expression of GPCR ligands, Gα12/13-coupled receptors and G proteins is often observed. G proteins are important mediators that transduce the signals through GPCRs to intracellular secondary messengers, leading to cellular responses. The regulation of GPCRs by phosphorylation and ubiquitination and the consequent degradation of GPCRs have been extensively analyzed. The cell-targeted gene delivery system and phenotyping of cell-specific knockout mice revealed novel roles for Gα12/13 (Fig. 5). Based on their pathological role in most prominent tissues and cells, Gα12/13 are associated with obesity, glucose intolerance, hepatic steatosis and cardiovascular disease, and the GPCR–Gα12/13 axis is considered an attractive biomarker and therapeutic target for the diagnosis and treatment of metabolic diseases. We suggest that Gα12/13-coupled receptors or downstream effectors may be of use as druggable targets. For example, ITIH1 antibodies can be developed for the treatment of type 2 diabetes. In several studies, it has been shown that targeting GPCR-G protein signaling pathways may provide opportunities to overcome certain metabolic diseases. A better understanding of the ligand-receptor-G protein signaling network may provide us with new strategies and methods for the prevention and treatment of metabolic diseases.

The roles of Gα12/13 and Gα12/13-coupled receptors, such as G-protein-coupled receptor 40 (GPR40), lysophosphatidic acid receptor (LPA4/6), sphingosine-1-phosphate receptor (S1P3), and the purinergic P2Y6 receptor in metabolic organs, the cardiovascular system, and the immune system, are summarized. Gna12-knockout (KO) mice showed enhanced fasting-induced fat accumulation and diet-induced steatosis in the liver and insulin secretion after high-fat diet (HFD) feeding in the pancreas and promoted an increase in fat mass but decreased fatty acid oxidation in skeletal muscle. Liver-specific Gna13-KO mice developed systemic insulin resistance, whereas skeletal muscle-specific Gna13-KO mice showed myofiber reprogramming and thereby increased whole-body metabolism. T cell-specific Gna12/13-double-knockout (DKO) mice had lymphadenopathy, whereas B cell-specific Gna12/13-DKO mice showed reduced marginal zone B cell (MZB cell) maturation and GC architecture.
References
Chatterjee, S., Khunti, K. & Davies, M. J. Type 2 diabetes. Lancet 389, 2239–2251 (2017).
Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schioth, H. B. & Gloriam, D. E. Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842 (2017).
Diamant, M. et al. Once weekly exenatide compared with insulin glargine titrated to target in patients with type 2 diabetes (DURATION-3): an open-label randomised trial. Lancet 375, 2234–2243 (2010).
Astrup, A. et al. Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet 374, 1606–1616 (2009).
Blad, C. C., Tang, C. & Offermanns, S. G protein-coupled receptors for energy metabolites as new therapeutic targets. Nat. Rev. Drug Discov. 11, 603–619 (2012).
Gendaszewska-Darmach, E., Drzazga, A. & Koziolkiewicz, M. Targeting GPCRs activated by fatty acid-derived lipids in type 2 diabetes. Trends Mol. Med. 25, 915–929 (2019).
Oh, D. Y. et al. A Gpr120-selective agonist improves insulin resistance and chronic inflammation in obese mice. Nat. Med. 20, 942–947 (2014).
Syrovatkina, V., Alegre, K. O., Dey, R. & Huang, X. Y. Regulation, signaling, and physiological functions of G-proteins. J. Mol. Biol. 428, 3850–3868 (2016).
Husted, A. S., Trauelsen, M., Rudenko, O., Hjorth, S. A. & Schwartz, T. W. GPCR-mediated signaling of metabolites. Cell Metab. 25, 777–796 (2017).
Li, X. et al. Human receptors for sweet and umami taste. Proc. Natl Acad. Sci. USA 99, 4692–4696 (2002).
Lauffer, L. M., Iakoubov, R. & Brubaker, P. L. GPR119 is essential for oleoylethanolamide-induced glucagon-like peptide-1 secretion from the intestinal enteroendocrine L-cell. Diabetes 58, 1058–1066 (2009).
Kelly, P. et al. The G12 family of heterotrimeric G proteins promotes breast cancer invasion and metastasis. Proc. Natl Acad. Sci. USA 103, 8173–8178 (2006).
Kelly, P. et al. A role for the G12 family of heterotrimeric G proteins in prostate cancer invasion. J. Biol. Chem. 281, 26483–26490 (2006).
Yang, Y. M. et al. Galpha12 gep oncogene deregulation of p53-responsive microRNAs promotes epithelial-mesenchymal transition of hepatocellular carcinoma. Oncogene 34, 2910–2921 (2015).
Yang, Y. M. et al. Galpha12 overexpressed in hepatocellular carcinoma reduces microRNA-122 expression via HNF4alpha inactivation, which causes c-Met induction. Oncotarget 6, 19055–19069 (2015).
Xu, Y. et al. High expression of GNA13 is associated with poor prognosis in hepatocellular carcinoma. Sci. Rep. 6, 35948 (2016).
Wang, M. & Casey, P. J. Protein prenylation: unique fats make their mark on biology. Nat. Rev. Mol. Cell Biol. 17, 110–122 (2016).
Veit, M. et al. The alpha-subunits of G-proteins G12 and G13 are palmitoylated, but not amidically myristoylated. FEBS Lett. 339, 160–164 (1994).
Bhattacharyya, R. & Wedegaertner, P. B. Galpha 13 requires palmitoylation for plasma membrane localization, Rho-dependent signaling, and promotion of p115-RhoGEF membrane binding. J. Biol. Chem. 275, 14992–14999 (2000).
Waheed, A. A. & Jones, T. L. Hsp90 interactions and acylation target the G protein Galpha 12 but not Galpha 13 to lipid rafts. J. Biol. Chem. 277, 32409–32412 (2002).
Wedegaertner, P. B., Wilson, P. T. & Bourne, H. R. Lipid modifications of trimeric G proteins. J. Biol. Chem. 270, 503–506 (1995).
Degtyarev, M. Y., Spiegel, A. M. & Jones, T. L. Increased palmitoylation of the Gs protein alpha subunit after activation by the beta-adrenergic receptor or cholera toxin. J. Biol. Chem. 268, 23769–23772 (1993).
Wedegaertner, P. B., Chu, D. H., Wilson, P. T., Levis, M. J. & Bourne, H. R. Palmitoylation is required for signaling functions and membrane attachment of Gq alpha and Gs alpha. J. Biol. Chem. 268, 25001–25008 (1993).
Manganello, J. M., Huang, J. S., Kozasa, T., Voyno-Yasenetskaya, T. A. & Le Breton, G. C. Protein kinase A-mediated phosphorylation of the Galpha13 switch I region alters the Galphabetagamma13-G protein-coupled receptor complex and inhibits Rho activation. J. Biol. Chem. 278, 124–130 (2003).
Offermanns, S., Hu, Y. H. & Simon, M. I. Galpha12 and galpha13 are phosphorylated during platelet activation. J. Biol. Chem. 271, 26044–26048 (1996).
Suzuki, N., Hajicek, N. & Kozasa, T. Regulation and physiological functions of G12/13-mediated signaling pathways. Neurosignals 17, 55–70 (2009).
Fields, T. A. & Casey, P. J. Phosphorylation of Gz alpha by protein kinase C blocks interaction with the beta gamma complex. J. Biol. Chem. 270, 23119–23125 (1995).
Kozasa, T. & Gilman, A. G. Protein kinase C phosphorylates G12 alpha and inhibits its interaction with G beta gamma. J. Biol. Chem. 271, 12562–12567 (1996).
Morris, N. J., Bushfield, M., Lavan, B. E. & Houslay, M. D. Multi-site phosphorylation of the inhibitory guanine nucleotide regulatory protein Gi-2 occurs in intact rat hepatocytes. Biochem J. 301(Pt 3), 693–702 (1994).
Chu, J., Zheng, H., Zhang, Y., Loh, H. H. & Law, P. Y. Agonist-dependent mu-opioid receptor signaling can lead to heterologous desensitization. Cell Signal 22, 684–696 (2010).
Shi, J. et al. Agonist induced-phosphorylation of Galpha11 protein reduces coupling to 5-HT2A receptors. J. Pharm. Exp. Ther. 323, 248–256 (2007).
Lounsbury, K. M., Casey, P. J., Brass, L. F. & Manning, D. R. Phosphorylation of Gz in human platelets. Selectivity and site of modification. J. Biol. Chem. 266, 22051–22056 (1991).
Wang, J., Frost, J. A., Cobb, M. H. & Ross, E. M. Reciprocal signaling between heterotrimeric G proteins and the p21-stimulated protein kinase. J. Biol. Chem. 274, 31641–31647 (1999).
Navarro, L. et al. Identification of a molecular target for the Yersinia protein kinase A. Mol. Cell 26, 465–477 (2007).
Umemori, H. et al. Activation of the G protein Gq/11 through tyrosine phosphorylation of the alpha subunit. Science 276, 1878–1881 (1997).
Moyers, J. S., Linder, M. E., Shannon, J. D. & Parsons, S. J. Identification of the in vitro phosphorylation sites on Gs alpha mediated by pp60c-src. Biochem J. 305(Pt 2), 411–417 (1995).
Poppleton, H., Sun, H., Fulgham, D., Bertics, P. & Patel, T. B. Activation of Gsalpha by the epidermal growth factor receptor involves phosphorylation. J. Biol. Chem. 271, 6947–6951 (1996).
Kim, T. H. et al. Galpha12 ablation exacerbates liver steatosis and obesity by suppressing USP22/SIRT1-regulated mitochondrial respiration. J. Clin. Invest. 128, 5587–5602 (2018).
Kim, T. H. et al. Overproduction of inter-alpha-trypsin inhibitor heavy chain 1 after loss of Galpha13 in liver exacerbates systemic insulin resistance in mice. Sci. Transl. Med. 11, eaan4735 (2019).
Koo, J. H. et al. Galpha13 ablation reprograms myofibers to oxidative phenotype and enhances whole-body metabolism. J. Clin. Invest 127, 3845–3860 (2017).
Levis, M. J. & Bourne, H. R. Activation of the alpha subunit of Gs in intact cells alters its abundance, rate of degradation, and membrane avidity. J. Cell Biol. 119, 1297–1307 (1992).
Papasergi, M. M., Patel, B. R. & Tall, G. G. The G protein alpha chaperone Ric-8 as a potential therapeutic target. Mol. Pharm. 87, 52–63 (2015).
Jenie, R. I. et al. Increased ubiquitination and the crosstalk of G protein signaling in cardiac myocytes: involvement of Ric-8B in Gs suppression by Gq signal. Genes Cells 18, 1095–1106 (2013).
Chaya, T. et al. Cul3-Klhl18 ubiquitin ligase modulates rod transducin translocation during light-dark adaptation. EMBO J. 38, e101409 (2019).
Kim, K. M. et al. Galpha12 overexpression induced by miR-16 dysregulation contributes to liver fibrosis by promoting autophagy in hepatic stellate cells. J. Hepatol. 68, 493–504 (2018).
Rasheed, S. A. et al. MicroRNA-31 controls G protein alpha-13 (GNA13) expression and cell invasion in breast cancer cells. Mol. Cancer 14, 67 (2015).
Muhammad, S. et al. miRNA-30d serves a critical function in colorectal cancer initiation, progression and invasion via directly targeting the GNA13 gene. Exp. Ther. Med. 17, 260–272 (2019).
Rasheed, S. A., Teo, C. R., Beillard, E. J., Voorhoeve, P. M. & Casey, P. J. MicroRNA-182 and microRNA-200a control G-protein subunit alpha-13 (GNA13) expression and cell invasion synergistically in prostate cancer cells. J. Biol. Chem. 288, 7986–7995 (2013).
Kihara, Y., Mizuno, H. & Chun, J. Lysophospholipid receptors in drug discovery. Exp. Cell Res. 333, 171–177 (2015).
Macrez-Lepretre, N., Kalkbrenner, F., Morel, J. L., Schultz, G. & Mironneau, J. G protein heterotrimer Galpha13beta1gamma3 couples the angiotensin AT1A receptor to increases in cytoplasmic Ca2+ in rat portal vein myocytes. J. Biol. Chem. 272, 10095–10102 (1997).
Chen, Z., Singer, W. D., Sternweis, P. C. & Sprang, S. R. Structure of the p115RhoGEF rgRGS domain-Galpha13/i1 chimera complex suggests convergent evolution of a GTPase activator. Nat. Struct. Mol. Biol. 12, 191–197 (2005).
Fukuhara, S., Murga, C., Zohar, M., Igishi, T. & Gutkind, J. S. A novel PDZ domain containing guanine nucleotide exchange factor links heterotrimeric G proteins to Rho. J. Biol. Chem. 274, 5868–5879 (1999).
Kozasa, T. et al. p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13. Science 280, 2109–2111 (1998).
Hart, M. J. et al. Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Galpha13. Science 280, 2112–2114 (1998).
Suzuki, N., Nakamura, S., Mano, H. & Kozasa, T. Galpha 12 activates Rho GTPase through tyrosine-phosphorylated leukemia-associated RhoGEF. Proc. Natl Acad. Sci. USA 100, 733–738 (2003).
Ross, E. M. & Wilkie, T. M. GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev. Biochem 69, 795–827 (2000).
Siehler, S. G12/13-dependent signaling of G-protein-coupled receptors: disease context and impact on drug discovery. Expert Opin. Drug Discov. 2, 1591–1604 (2007).
Gohla, A., Schultz, G. & Offermanns, S. Role for G(12)/G(13) in agonist-induced vascular smooth muscle cell contraction. Circ. Res. 87, 221–227 (2000).
Marinissen, M. J. et al. The small GTP-binding protein RhoA regulates c-jun by a ROCK-JNK signaling axis. Mol. Cell 14, 29–41 (2004).
Dermott, J. M., Ha, J. H., Lee, C. H. & Dhanasekaran, N. Differential regulation of Jun N-terminal kinase and p38MAP kinase by Galpha12. Oncogene 23, 226–232 (2004).
Chaveroux, C. et al. Identification of a novel amino acid response pathway triggering ATF2 phosphorylation in mammals. Mol. Cell Biol. 29, 6515–6526 (2009).
Ki, S. H., Choi, M. J., Lee, C. H. & Kim, S. G. Galpha12 specifically regulates COX-2 induction by sphingosine 1-phosphate. Role for JNK-dependent ubiquitination and degradation of IkappaBalpha. J. Biol. Chem. 282, 1938–1947 (2007).
Gadepalli, R. et al. Novel role for p21-activated kinase 2 in thrombin-induced monocyte migration. J. Biol. Chem. 288, 30815–30831 (2013).
Liu, S. C. et al. G(alpha)12-mediated pathway promotes invasiveness of nasopharyngeal carcinoma by modulating actin cytoskeleton reorganization. Cancer Res. 69, 6122–6130 (2009).
Lammermann, T. & Kastenmuller, W. Concepts of GPCR-controlled navigation in the immune system. Immunol. Rev. 289, 205–231 (2019).
Cho, H. & Kehrl, J. H. Regulation of immune function by G protein-coupled receptors, trimeric G proteins, and RGS proteins. Prog. Mol. Biol. Transl. Sci. 86, 249–298 (2009).
Herroeder, S. et al. Guanine nucleotide-binding proteins of the G12 family shape immune functions by controlling CD4+ T cell adhesiveness and motility. Immunity 30, 708–720 (2009).
Healy, J. A. et al. GNA13 loss in germinal center B cells leads to impaired apoptosis and promotes lymphoma in vivo. Blood 127, 2723–2731 (2016).
Muppidi, J. R. et al. Loss of signalling via Galpha13 in germinal centre B-cell-derived lymphoma. Nature 516, 254–258 (2014).
Druey, K. M. Regulation of G-protein-coupled signaling pathways in allergic inflammation. Immunol. Res. 43, 62–76 (2009).
Zhang, L. & Shi, G. Gq-Coupled Receptors in Autoimmunity. J. Immunol. Res. 2016, 3969023 (2016).
Rieken, S. et al. G12/G13 family G proteins regulate marginal zone B cell maturation, migration, and polarization. J. Immunol. 177, 2985–2993 (2006).
Muppidi, J. R., Lu, E. & Cyster, J. G. The G protein-coupled receptor P2RY8 and follicular dendritic cells promote germinal center confinement of B cells, whereas S1PR3 can contribute to their dissemination. J. Exp. Med. 212, 2213–2222 (2015).
Lu, E. & Cyster, J. G. G-protein coupled receptors and ligands that organize humoral immune responses. Immunol. Rev. 289, 158–172 (2019).
Coffield, V. M., Helms, W. S., Jiang, Q. & Su, L. Galpha13 mediates a signal that is essential for proliferation and survival of thymocyte progenitors. J. Exp. Med. 200, 1315–1324 (2004).
Huang, W. et al. The zinc-binding region of IL-2 inducible T cell kinase (Itk) is required for interaction with Galpha13 and activation of serum response factor. Int. J. Biochem. Cell Biol. 45, 1074–1082 (2013).
Hirahara, K. & Nakayama, T. CD4+ T-cell subsets in inflammatory diseases: beyond the Th1/Th2 paradigm. Int. Immunol. 28, 163–171 (2016).
Crotty, S. T follicular helper cell differentiation, function, and roles in disease. Immunity 41, 529–542 (2014).
Moriyama, S. et al. Sphingosine-1-phosphate receptor 2 is critical for follicular helper T cell retention in germinal centers. J. Exp. Med. 211, 1297–1305 (2014).
Zanin-Zhorov, A. et al. Selective oral ROCK2 inhibitor down-regulates IL-21 and IL-17 secretion in human T cells via STAT3-dependent mechanism. Proc. Natl Acad. Sci. USA 111, 16814–16819 (2014).
Chen, W. et al. ROCK2, but not ROCK1 interacts with phosphorylated STAT3 and co-occupies TH17/TFH gene promoters in TH17-activated human T cells. Sci. Rep. 8, 16636 (2018).
Titchenell, P. M. et al. Direct hepatocyte insulin signaling is required for lipogenesis but is dispensable for the suppression of glucose production. Cell Metab. 23, 1154–1166 (2016).
Softic, S. et al. Dietary sugars alter hepatic fatty acid oxidation via transcriptional and post-translational modifications of mitochondrial proteins. Cell Metab. 30, 735–753 e734 (2019).
Purushotham, A. et al. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 9, 327–338 (2009).
Peleli, M., Fredholm, B. B., Sobrevia, L. & Carlstrom, M. Pharmacological targeting of adenosine receptor signaling. Mol. Asp. Med. 55, 4–8 (2017).
Fernandez, H. H. et al. Istradefylline as monotherapy for Parkinson disease: results of the 6002-US-051 trial. Parkinsonism Relat. Disord. 16, 16–20 (2010).
Massie, B. M. et al. Rolofylline, an adenosine A1-receptor antagonist, in acute heart failure. N. Engl. J. Med. 363, 1419–1428 (2010).
Andreeva, A. V., Kutuzov, M. A. & Voyno-Yasenetskaya, T. A. G alpha12 is targeted to the mitochondria and affects mitochondrial morphology and motility. FASEB J. 22, 2821–2831 (2008).
Denis-Henriot, D., de Mazancourt, P., Morot, M. & Giudicelli, Y. Mutant alpha-subunit of the G protein G12 activates proliferation and inhibits differentiation of 3T3-F442A preadipocytes. Endocrinology 139, 2892–2899 (1998).
Bannai, Y., Aminova, L. R., Faulkner, M. J., Ho, M. & Wilson, B. A. Rho/ROCK-dependent inhibition of 3T3-L1 adipogenesis by G-protein-deamidating dermonecrotic toxins: differential regulation of Notch1, Pref1/Dlk1, and beta-catenin signaling. Front Cell Infect. Microbiol. 2, 80 (2012).
Yanagida, K. et al. The Galpha12/13-coupled receptor LPA4 limits proper adipose tissue expansion and remodeling in diet-induced obesity. JCI Insight 3, e97293 (2018).
Egan, B. & Zierath, J. R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 17, 162–184 (2013).
White, J. P. et al. G protein-coupled receptor 56 regulates mechanical overload-induced muscle hypertrophy. Proc. Natl Acad. Sci. USA 111, 15756–15761 (2014).
Offermanns, S., Mancino, V., Revel, J. P. & Simon, M. I. Vascular system defects and impaired cell chemokinesis as a result of Galpha13 deficiency. Science 275, 533–536 (1997).
Lee, Y. S., Riopel, M., Cabrales, P. & Bandyopadhyay, G. K. Hepatocyte-specific HIF-1alpha ablation improves obesity-induced glucose intolerance by reducing first-pass GLP-1 degradation. Sci. Adv. 5, eaaw4176 (2019).
Chen, J. et al. Hypoxia exacerbates nonalcoholic fatty liver disease via the HIF-2alpha/PPARalpha pathway. Am. J. Physiol. Endocrinol. Metab. 317, E710–E722 (2019).
Day, C. P. & James, O. F. Steatohepatitis: a tale of two “hits”? Gastroenterology 114, 842–845 (1998).
Petersen, K. F. et al. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 54, 603–608 (2005).
Turner, N. et al. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 56, 1638–1648 (2013).
Roden, M. Mechanisms of disease: hepatic steatosis in type 2 diabetes-pathogenesis and clinical relevance. Nat. Clin. Pr. Endocrinol. Metab. 2, 335–348 (2006).
Marcinak, J., Cao, C., Lee, D. & Ye, Z. Fasiglifam for glycaemic control in people with type 2 diabetes: a phase III, placebo-controlled study. Diabetes Obes. Metab. 19, 1714–1721 (2017).
Marcinak, J. F., Munsaka, M. S., Watkins, P. B., Ohira, T. & Smith, N. Liver safety of Fasiglifam (TAK-875) in patients with type 2 diabetes: review of the global clinical trial experience. Drug Saf. 41, 625–640 (2018).
Rives, M. L. et al. GPR40-mediated Galpha12 Activation by allosteric full agonists highly efficacious at potentiating glucose-stimulated insulin secretion in human islets. Mol. Pharm. 93, 581–591 (2018).
Kang, L. et al. Hyaluronan accumulates with high-fat feeding and contributes to insulin resistance. Diabetes 62, 1888–1896 (2013).
Dasu, M. R., Devaraj, S., Park, S. & Jialal, I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 33, 861–868 (2010).
Yang, Y. M. et al. Hyaluronan synthase 2-mediated hyaluronan production mediates Notch1 activation and liver fibrosis. Sci. Transl. Med. 11, eaat9284 (2019).
Hamm, A. et al. Frequent expression loss of Inter-alpha-trypsin inhibitor heavy chain (ITIH) genes in multiple human solid tumors: a systematic expression analysis. BMC Cancer 8, 25 (2008).
Caillot, F. et al. Novel serum markers of fibrosis progression for the follow-up of hepatitis C virus-infected patients. Am. J. Pathol. 175, 46–53 (2009).
Garantziotis, S. et al. Serum inter-alpha-trypsin inhibitor and matrix hyaluronan promote angiogenesis in fibrotic lung injury. Am. J. Respir. Crit. Care Med. 178, 939–947 (2008).
Subramanian, A. et al. Auto-regulation of secretory flux by sensing and responding to the folded cargo protein load in the endoplasmic reticulum. Cell 176, 1461–1476 e1423 (2019).
Brenner, D. A. et al. Origin of myofibroblasts in liver fibrosis. Fibrogenes. Tissue Repair 5, S17 (2012).
Guo, C. J., Pan, Q., Li, D. G., Sun, H. & Liu, B. W. miR-15b and miR-16 are implicated in activation of the rat hepatic stellate cell: An essential role for apoptosis. J. Hepatol. 50, 766–778 (2009).
Guo, C. J., Pan, Q., Jiang, B., Chen, G. Y. & Li, D. G. Effects of upregulated expression of microRNA-16 on biological properties of culture-activated hepatic stellate cells. Apoptosis 14, 1331–1340 (2009).
Pan, Q., Guo, C., Sun, C., Fan, J. & Fang, C. Integrative analysis of the transcriptome and targetome identifies the regulatory network of miR-16: an inhibitory role against the activation of hepatic stellate cells. Biomed. Mater. Eng. 24, 3863–3871 (2014).
Schneider, J. L., Suh, Y. & Cuervo, A. M. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 20, 417–432 (2014).
Altshuler-Keylin, S. et al. Beige adipocyte maintenance is regulated by autophagy-induced mitochondrial clearance. Cell Metab. 24, 402–419 (2016).
Masiero, E. et al. Autophagy is required to maintain muscle mass. Cell Metab. 10, 507–515 (2009).
Noda, T., Fujita, N. & Yoshimori, T. The late stages of autophagy: how does the end begin? Cell Death Differ. 16, 984–990 (2009).
Cho, M. K. et al. Role of Galpha12 and Galpha13 as novel switches for the activity of Nrf2, a key antioxidative transcription factor. Mol. Cell Biol. 27, 6195–6208 (2007).
Guilluy, C. et al. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat. Med. 16, 183–190 (2010).
Wirth, A. et al. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med. 14, 64–68 (2008).
Kano, K. et al. Molecular mechanism of lysophosphatidic acid-induced hypertensive response. Sci. Rep. 9, 2662 (2019).
Gao, J., Denys, I., Shahien, A., Sutphen, J. & Kapusta, D. R. Down-regulation of brain Galpha12 attenuates angiotensin II dependent hypertension. Am. J. Hypertens. 33, 198–204 (2019).
Wirth, A. et al. Age-dependent blood pressure elevation is due to increased vascular smooth muscle tone mediated by G-protein signalling. Cardiovasc. Res. 109, 131–140 (2016).
Kim, Y. M. et al. G(alpha)12/13 induction of CYR61 in association with arteriosclerotic intimal hyperplasia: effect of sphingosine-1-phosphate. Arterioscler. Thromb. Vasc. Biol. 31, 861–869 (2011).
Feng, X. et al. Thromboxane A2 activates YAP/TAZ protein to induce vascular smooth muscle cell proliferation and migration. J. Biol. Chem. 291, 18947–18958 (2016).
Dupont, S. et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011).
Bot, M. et al. Lysophosphatidic acid triggers mast cell-driven atherosclerotic plaque destabilization by increasing vascular inflammation. J. Lipid Res. 54, 1265–1274 (2013).
Yu, F. X. et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 150, 780–791 (2012).
Wang, L. et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 540, 579–582 (2016).
Nishida, M. et al. P2Y6 receptor-Galpha12/13 signalling in cardiomyocytes triggers pressure overload-induced cardiac fibrosis. EMBO J. 27, 3104–3115 (2008).
Nishimura, A. et al. Purinergic P2Y6 receptors heterodimerize with angiotensin AT1 receptors to promote angiotensin II-induced hypertension. Sci Signal 9, ra7 (2016).
Olivera, A. et al. Sphingosine kinase type 1 induces G12/13-mediated stress fiber formation, yet promotes growth and survival independent of G protein-coupled receptors. J. Biol. Chem. 278, 46452–46460 (2003).
Dusaban, S. S., Chun, J., Rosen, H., Purcell, N. H. & Brown, J. H. Sphingosine 1-phosphate receptor 3 and RhoA signaling mediate inflammatory gene expression in astrocytes. J. Neuroinflammation 14, 111 (2017).
Sobel, K. et al. FTY720 phosphate activates sphingosine-1-phosphate receptor 2 and selectively couples to Galpha12/13/Rho/ROCK to induce myofibroblast contraction. Mol. Pharm. 87, 916–927 (2015).
Yung, B. S. et al. Selective coupling of the S1P3 receptor subtype to S1P-mediated RhoA activation and cardioprotection. J. Mol. Cell Cardiol. 103, 1–10 (2017).
Marinissen, M. J., Servitja, J. M., Offermanns, S., Simon, M. I. & Gutkind, J. S. Thrombin protease-activated receptor-1 signals through Gq- and G13-initiated MAPK cascades regulating c-Jun expression to induce cell transformation. J. Biol. Chem. 278, 46814–46825 (2003).
Gavard, J. & Gutkind, J. S. Protein kinase C-related kinase and ROCK are required for thrombin-induced endothelial cell permeability downstream from Galpha12/13 and Galpha11/q. J. Biol. Chem. 283, 29888–29896 (2008).
Kang, K. W., Choi, S. Y., Cho, M. K., Lee, C. H. & Kim, S. G. Thrombin induces nitric-oxide synthase via Galpha12/13-coupled protein kinase C-dependent I-kappaBalpha phosphorylation and JNK-mediated I-kappaBalpha degradation. J. Biol. Chem. 278, 17368–17378 (2003).
Gohla, A., Offermanns, S., Wilkie, T. M. & Schultz, G. Differential involvement of Galpha12 and Galpha13 in receptor-mediated stress fiber formation. J. Biol. Chem. 274, 17901–17907 (1999).
Yanagida, K. et al. The Galpha12/13-coupled receptor LPA4 limits proper adipose tissue expansion and remodeling in diet-induced obesity. JCI Insight 3, 97293 (2018).
Yasuda, D. et al. Lysophosphatidic acid-induced YAP/TAZ activation promotes developmental angiogenesis by repressing Notch ligand Dll4. J. Clin. Invest. 130, 4332–4349 (2019).
Fujii, T. et al. Galpha12/13-mediated production of reactive oxygen species is critical for angiotensin receptor-induced NFAT activation in cardiac fibroblasts. J. Biol. Chem. 280, 23041–23047 (2005).
Shatanawi, A. et al. Angiotensin II-induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase pathway. Am. J. Physiol. Cell Physiol. 300, C1181–C1192 (2011).
Acknowledgements
This study was supported by the National Research Foundation (NRF) funded by the Korean government (Ministry of Science, ICT and Future Planning) (#2017K1A1A2004511), and Y.M.Y. was supported by the National Research Foundation (#2020R1C1C1004185).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, Y.M., Kuen, DS., Chung, Y. et al. Gα12/13 signaling in metabolic diseases. Exp Mol Med 52, 896–910 (2020). https://doi.org/10.1038/s12276-020-0454-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-020-0454-5
This article is cited by
-
Purinergic signaling during Marek’s disease in chickens
Scientific Reports (2023)
-
Insights into divalent cation regulation and G13-coupling of orphan receptor GPR35
Cell Discovery (2022)
-
Non-alcoholic fatty liver disease and liver secretome
Archives of Pharmacal Research (2022)
-
Regulatory roles of G-protein coupled receptors in adipose tissue metabolism and their therapeutic potential
Archives of Pharmacal Research (2021)
