
Abstract
The complex pathophysiological interactions between heart and kidney diseases are collectively known as cardiorenal syndrome. The renin–angiotensin system (RAS) may have a pivotal role in the development of cardiorenal syndrome. The aim of this study was to elucidate the RAS activity responsible for adverse post-infarction remodeling and prognosis in mice with renal failure. To establish the type IV cardiorenal syndrome model, 5/6 nephrectomy (NTX) was performed in a surgical procedure, followed by the induction of myocardial ischemia (MI) by a coronary artery ligation 4 weeks later. NTX and MI resulted in deteriorated left ventricular remodeling and RAS activation, which was improved by an aliskiren that appeared to be independent of renal function and blood pressure (BP). Moreover, MI induced in renin and angiotensinogen double-transgenic (Tg) mice showed comparable effects to MI plus NTX mice, including advanced ventricular remodeling and enhancement of RAS, oxidative stress, and monocytes chemoattractant protein (MCP)-1. Aliskiren suppressed these changes in the MI-induced Tg mice. In in vitro study, Nox2 expression was elevated by the stimulation of plasma from NTX mice in isolated neonatal cardiomyocytes. However, Nox2 upregulation was negated when we administered plasma from aliskiren-treated-NTX mice or isolated cardiomyocytes from AT1-deficient mice. Primary mononuclear cells also showed an upregulation in the expression of Nox2 and MCP-1 by stimulation with plasma from NTX mice. Our data suggest that renal disorder results in ventricular dysfunction and deteriorates remodeling after MI through excessive RAS activation. Moreover, renin inhibition improved the changes caused by cardiorenal syndrome.
Similar content being viewed by others
Main
It is well known that there is a complex relationship between cardiovascular and renal diseases. In fact, end-stage renal disease or decreased estimated glomerular filtration rate correlates with an increased risk of cardiovascular diseases.1 This clinical condition is known as cardiorenal syndrome. In addition, the presence of chronic kidney disease (CKD) is a relatively frequent complication in patients with advanced heart failure and left ventricular (LV) dysfunction. Its presence is associated with a worse prognosis following cardiovascular diseases.2 Although preventing this condition includes identification and amelioration of the precipitating factors and connections,3 the pathophysiological mechanisms of the syndrome remain to be elucidated. It is also known that the presence of CKD increases severity, worsens the response to treatment, and is associated with poor cardiac and renal outcomes in cardiorenal syndrome.4, 5
The renin–angiotensin system (RAS) has a critical role in the development of cardiovascular and renal disease in a clinical setting. It has been reported that the role of RAS in congestive heart failure (CHF) and CKD is complex owing to involvement of multiple peptides and receptors. Increased RAS activity results in the development of CHF by stimulation of cardiac hypertrophy, apoptosis, and LV dilatation. Although RAS mediates the development of cardiorenal syndrome, clinical studies indicate that angiotensin receptor blockers do not reduce cardiovascular events in patients with nephropathy.6
Aliskiren, a new class of the first representative non-peptide direct renin inhibitor,7 is broadly used as an antihypertensive drug, as its effects directly suppress the plasma concentration of Ang peptides.8, 9 Recent papers reveal the benefit of aliskiren following myocardial ischemia (MI) independent of pressor effects in rodent and human studies.10, 11 RAS activity during sleep time, which is independent of systemic pressure, has important role in the progression of cardiac remodeling. The half-life of aliskiren is very long (24–48 h) compared with an ACE inhibitor (about 6 h). Aliskiren treatment would inhibit the RAS activity independent of systemic pressure because of its longevity. A clinical trial (ALTITUDE) in type 2 diabetes using cardiorenal endpoints is ongoing,12, 13 but the detailed pathophysiological roles of aliskiren have not yet been elucidated in the experimental models of cardiorenal syndrome.
Thus, the aim of this study was to elucidate the RAS activity responsible for adverse myocardial remodeling with renal failure and to clarify the effect of aliskiren on this condition. In this study, we demonstrated that renal disorder results in ventricular dysfunction and deteriorates remodeling after MI through excessive RAS activation in this cardiorenal model. Aliskiren treatment suppressed cardiac dysfunction, LV remodeling, and inflammatory or oxidative factors compared with the vehicle-treated group. These results may be a new methodological approach against prognosis in patients with renal failure suffering acute-MI.
MATERIALS AND METHODS
Animals
Male C57BL/6 mice were obtained from Crea, Japan. Transgenic (Tg) mice were generated using heterozygotes carrying either the 15-kb human renin gene with the 3-kb native promoter or the 14-kb human angiotensinogen gene with the 1.3-kb promoter. Double-Tg mice were created by cross-mating between human renin-Tg and human angiotensinogen-Tg mice. Both mice were backcrossed to the C57Bl/6 background. Human renin in renin-Tg mice is found at ∼34-fold higher concentration than that obtained from normal human plasma. Plasma Ang II concentration and plasma renin activity (PRA) are ∼3.5-fold and 6–9-fold higher in double-Tg mice than in wild-type (WT) mice.14 Human renin-Tg mice and human angiotensinogen-Tg mice were provided by RIKEN BRC (Tsukuba, Japan). AT1−/− mice were also used as described previously.15
Experimental Protocols
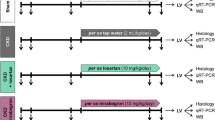
All mice were 6–8 weeks of age when the experiment started. A murine model of 5/6 nephrectomy (NTX) was generated by a two-step surgical procedure as described previously.16 Two to three arterial branches of the left kidney were ligated leaving an intact kidney segment. The right kidney was removed, and the mice received 0.9% NaCl in their drinking water. Four weeks after NTX, MI was induced by ligation of the left anterior descending (LAD) coronary artery, and this was continued for 28 days17 (Supplementary Figure 1). Double-Tg and WT-mice were induced MI without NTX. We killed the mice on day 7 (n=8–10 per each group) and day 28 (n=17–22 per each group). In total, 4–6 mice in each assay, and 7–11 mice in pathological analysis were randomly picked from each group (Supplementary Figure 1).
These experiments conform to the Guide for the Care and Use of Laboratory Animals in the Tokyo Medical and Dental University, University of Tokyo.
Treatment Protocols
Animals were assigned randomly into treatment groups and were administered a subcutaneous injection of aliskiren (25 mg/kg; Novartis Pharma), hydralazine (in drinking water, 30 mg/kg), apocynin (in drinking water, 150 mg/kg; Sigma-Aldrich), or a vehicle (PBS; Supplementary Figure 1). These drugs were administered daily for 8 weeks after NTX. The aliskiren and apocynin dosage were decided based on previously papers.8, 18 The dose of hydralazine was determined to show the equal effect of decreasing BP comparable with the aliskiren-treated group.
Physiological Examinations
Heart rate and BP (systolic pressure) were measured in conscious mice by using a tail-cuff system (BP-98A, Softron, Tokyo, Japan). Transthoracic echocardiography was performed with ultrasound equipment (Nemio, Toshiba, Tokyo, Japan) using a 14-MHz annular array transducer. Hearts were imaged in the two-dimensional mode in short-axis views at the level of papillary muscle. EF were calculated as EF=SV/EDV × 100; SV=EDV–ESV; EDV=(7 × LVDd3/1000)/{2.4+(LVDd/10)}; ESV=(7 × LVDs3/1000)/{2.4+(LVDs/10)}.
Histological Analysis
Harvested hearts were quickly dipped into 4% paraformaldehyde. The hearts were embedded in paraffin. Sample sections (1 μm) of the tissue were stained by Mallory and viewed using a light microscope with a computer-assisted analyzer (Image Pro Express software).19 To demonstrate remodeling analysis, infarct size, infarct thickness, infarct length, LV circumference, and septal thickness were measured with the computer-assisted analyzer. Collagen production was identified by the Sirius Red, and the area of collagen was determined in the LV border area. To avoid bias, the histologist was single blinded to the treatment groups of mice.
Hydroxyproline Assay
Collagen concentrations of whole hearts were measured using the hydroxyproline assay 1 week after MI operation. The homogenated samples in acetic buffer were hydrolyzed. Chloramine T buffer were added to sample and incubated for 20 min. Erhlich’s solution was then added to each sample, and incubated for 20 min. Absorbance 550 nm was read on a microplate reader.20
Real-Time (RT) PCR
mRNA of whole hearts were collected using the TRIzol method 1 week after MI or 4 weeks after NTX. RT-PCR was performed to determine the mRNA expression of ANP (assay ID: Mm01255747_g1), monocytes chemoattractant protein (MCP)-1 (Mm00441242_m1), collagen-type I (Mm0130243_g1), AT1a (Mm00558224_s1), NADPH oxidase (Nox)2 (Mm1287742_m1), and Nox4 (Mm00479246_m1). The mRNA expression of the target gene was normalized to 18 s ribosomal RNA (4308329). Quantitative data were calculated to normalize the control group as baseline using the comparative CT (ΔΔCT) method.21
Immunohistochemistry
Immunohistochemistry (IHC) was performed using a primary antibody against renin (no. 593), followed by the Nichirei Simple stain kit (Nichirei).19 Immunological changes were scored on a 0 to 5 basis (no detected to large quantity detected) by single-blinded histologists.
Plasma Biochemistry
Blood was collected in heparinized microtubes and subjected to centrifugation. Plasma creatinine levels were measured using the enzymatic method by SRL.
PRA Analysis
Plasma was collected from mice and diluted in an Ang I generation buffer containing 0.1 M phosphate buffer, 5 mM EDTA, and 1 mM PMSF. The mixtures were incubated for 30 min. Ang I expression was measured using the Ang I EIA kit (Peninsula Laboratories). Quantitative data were obtained to determine endogenous Ang I in each group by the following equation: quantitative data=Ang I value–endogenous Ang I value.
Cell Preparation and Culture
The hearts were isolated from neonatal mice (1–2-day-old), and minced with PBS. The hearts were then denatured by collagenase (Roche diagnosis). Isolated cells were plated for 40 min. Fibroblasts adhered to the plate, while cardiomyocytes did not. Cardiomyocytes were cultured in MEM medium containing 5% FBS on 3.5-mm plane plastic dishes. Fibroblasts were cultured in DMEM (containing 10% FBS) until confluent. Mononuclear cells were collected from the spleens using cell strainers and were plated in RPMI1640 medium.19 Isolated cardiomyocytes or mononuclear cells were serum staved, and stimulated by murine plasma (5% in the medium) isolated from NTX, aliskiren-treated NTX on week 4, or non-NTX mice as a control. Aliskiren or vehicle (PBS) was administrated to the medium for 30 min before plasma stimulation.
Statistical Analysis
All data are expressed as mean±s.e.m. A survival analysis was performed using the Kaplan–Meier method with the log-rank test. Data were compared and differences between the two groups were analyzed by the t-test. Differences in data between multiple groups were subjected to one-way or two-way ANOVA and Bonferroni post-test. A statistical test was performed using the Prism. Differences with values of P<0.05 were considered significant.
RESULTS
Aliskiren Suppressed NTX-Induced Hypertension
NTX resulted in hypertension compared with the non-NTX mice 2 weeks after the operation. Although aliskiren reduced systolic BP, hydralazine administration resulted in a decline in BP comparable to aliskiren administration (Figure 1a).

Aliskiren suppressed 5/6 nephrectomy (NTX)-induced hypertension. (a) Representative data of blood pressure in NTX mice. Mice were administrated vehicle (dilute blue line; n=10), aliskiren (red line; n=8), or hydralazine (blue line; n=10). *P<0.05 vs aliskiren, **P<0.05 vs hydralazine. (b) Representative data of serum creatinine levels in NTX. Plasma creatinine levels are shown 4 weeks after NTX in NTX and non-NTX groups (P<0.05). *P<0.05 vs each group. (c) Representative data of survival rate in NTX. Vehicle: n=26; aliskiren: n=24; hydralazine: n=13, *P<0.05.
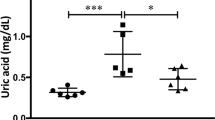
Plasma creatinine levels were elevated 4 weeks after NTX compared with the non-NTX group. Neither aliskiren nor hydralazine administration changed the plasma creatinine levels (Figure 1b).
Although many control mice died within the first 4 weeks after NTX (73.91%, n=24), aliskiren administration significantly improved the survival rates of mice with NTX (percent survival=92.31%, n=26; P<0.05 vs vehicle). However, mice treated with hydralazine showed survival rates comparable to those of vehicle-treated mice (76.92%, n=13; Figure 1c).
NTX Augmented Renin Expression and Activity
To investigate whether NTX activated renin expression and activity, we performed IHC and plasma biochemistry analysis. Four weeks post NTX, the remaining kidneys showed increased renin expression (4.5±0.3, P<0.05) at the juxtaglomerular area compared with the non-NTX kidneys (1.0±0.4; Figures 2a and b). Although NTX elevated PRA compared with the control mice, aliskiren treatment negated this effect (Figure 2c). Hydralazine treatment did not alter the PRA value. Although cardiac AT1 transcription was elevated by nephrectomy compared with the control hearts (Figure 2d), aliskiren treatment did not change the level.

NTX (5/6 nephrectomy) augmented renin expression and activity. (a) Representative immunohistochemistry (IHC) of kidneys from NTX mice. Dot-line circle indicates glomerular area. Bar shows 50 μm. (b) Quantitative data of the IHC. *P<0.05. (c) Representative plasma renin activity in NTX mice. *P<0.05 (d) Representative Real-TimePCR of hearts from NTX mice. *P<0.05.
Aliskiren Suppressed MI plus NTX-induced Myocardial Remodeling
To clarify the relationship between renal disorder and cardiac dysfunction, we induced MI by LAD ligation in mice that had undergone NTX. Cardiac function was analyzed by echocardiogram and performed 1 and 4 weeks post LAD ligation (Table 1). MI-induced hearts showed myocardial infarction in large anterior region of LV. NTX had no effect on the LV ejection fraction (LVEF) of non-infarcted hearts (86.0%±2.7%) compared with those that had not undergone NTX (86.0%±1.0%). MI hearts with NTX showed increased impairment of LVEF (at 1 week: 51.6%±4.7%, 4 weeks: 36.4%±2.2%; n=8) compared with MI hearts without NTX (at 1 week: 70.7%±5.9%, 4 weeks: 53.8%±2.6%; n=10). However, aliskiren significantly improved LVEF of MI hearts with NTX (1 week: 67.5%±3.0%, 4 weeks: 65.4%±2.4%; n=11) compared with the vehicle. Hydralazine did not improve LVEF of MI hearts with NTX (1 week: 52.1%±3.0%, 4 weeks: 36.4%±2.2%; n=7; Figures 3a and b). Aliskiren treatment also significantly suppressed ratio of lung/body weight compared with the vehicle-treated group (Table 2).

Aliskiren suppressed myocardial ischemia (MI) plus 5/6 nephrectomy (NTX)-induced myocardial remodeling. (a) Representative chronological observation of left ventricular ejection fraction (LVEF) in MI hearts with NTX (vehicle: n=8; aliskiren: n=11; and hydralazine: n=7). *P<0.05 vs vehicle and hydralazine. (b) Representative LVEF data 28 days after MI. (c) Representative light micrograph of MI heart with NTX. Black bar shows 2 mm and white bar shows 50 μm. (d) Representative collagen-type I analysis in hearts in MI with NTX mice using real-time PCR (NTX: n=4; MI: n=4; NTX-MI: n=6; NTX-MI-aliskiren: n=6; and NTX-MI-hydralazine: n=4). (e) Representative collagen concentration analysis in MI hearts with NTX (NTX: n=4; MI: n=4; NTX-MI: n=6; NTX-MI-aliskiren: n=6; NTX-MI-hydralazine: n=6). *P<0.05.
LAD ligation resulted in significant pathological remodeling of the LV anterior wall 4 weeks after ischemia induction (Table 3). Infarction length and septal wall thickness were not significantly different between the vehicle- and aliskiren-treated group. Although vehicle-treated LAD ligation resulted in LV thinning, aliskiren administration significantly reduced this pathological change. Enlarged LV circumference was also reduced by aliskiren. Collagen expression was significantly enhanced along the border by MI with NTX (19.3%±2.4%; n=8), but this was reduced by aliskiren administration (6.8%±2.0%; n=11; P<0.05; Figure 3c and Table 3). mRNA and collagen were collected from the cardiac ventricle on week 1 post MI. Although MI hearts elevated collagen-type I mRNA levels and collagen concentration compared with the hearts of control mice, MI with NTX hearts showed enhanced collagen levels compared with MI hearts without NTX (Figures 3d and e). Aliskiren suppressed the levels of collagen in MI hearts with NTX.
NTX Enhanced ANP, Oxidative Stress, and MCP-1 in MI Mice
RAS is known to have a key role in myocardial hypertrophy and fibrosis via increased oxidative stress and the expression of inflammatory chemokines. Thus, we examined the levels of ANP, Nox2, Nox4, and MCP-1 mRNA in MI or non-MI hearts treated by NTX. mRNA was collected from the cardiac ventricle 1 week after MI. ANP levels were elevated in the MI hearts compared with the non-MI hearts. Moreover, hearts with MI treated by NTX showed enhanced ANP mRNA levels compared with the MI hearts without NTX. The mRNA levels of Nox2 and Nox4 were elevated in MI hearts compared with non-MI hearts. Nox2 and Nox4 are isoforms of Nox, a major enzyme that produces reactive oxygen species (ROS). In addition, MI-induced hearts treated with NTX showed further elevation of Nox2, Nox4, and MCP-1 mRNA levels as compared with the MI hearts that were not treated with NTX (Figures 4a–d). Aliskiren treatment significantly suppressed these levels compared with the NTX plus MI group.

NTX (5/6 nephrectomy) enhanced ANP, oxidative stress, and monocytes chemoattractant protein (MCP)-1 in myocardial ischemia (MI). Real-time (RT) PCR. We performed RT-PCR to analyze the expression of ANP (a), oxidative stresses (b and c) and MCP-1 (d) in hearts from MI with NTX mice. *P<0.05. (e) Representative left ventricular ejection fraction analysis of MI hearts with NTX. Mice were administrated apocynin (red line; n=6) or vehicle (blue line; n=8). (f) Representative light micrograph of apocynin-treated hearts from MI with NTX mice. *P<0.05 vs vehicle.
It is known that Nox enzymes can be activated by RAS. To reveal the role of Nox enzymes in MI-induced myocardial remodeling in renal dysfunction, we administrated apocynin as a Nox inhibitor to the NTX plus MI mice. Although apocynin did not alter BP, attenuation of Nox improved LVEF and myocardial fibrosis in the 28 days after LAD ligation mice as compared with the vehicle-treated group (Figures 4e and f).
Double-Tg Mice Deteriorated After MI
Although we demonstrated that NTX deteriorated LV dysfunction through RAS activation, we did not clarify the relationship between upregulated circulating RAS and the progression of LV remodeling. To clarify the pathophysiological mechanism, we used human renin and angiotensinogen Tg mice. Systolic BP was consistently elevated in the double-Tg mice compared with the WT mice (Supplementary Figure 2A). The high BP resulted in mild cardiac hypertrophy, however, cardiac function was comparable between the Tg and WT mice. (Table 2 and Supplementary Table 1). The creatinine level was not affected in the double-Tg mice compared with the age-matched WT mice (creatinine: WT 0.13±0.01 mg/dl and double-Tg 0.18±0.04 mg/dl). We induced MI in double-Tg and WT mice to analyze the effect of increasing RAS activation including renin in this model. We showed basal data of double-Tg mice in Supplementary Table 1. LAD ligation expanded remodeling at the LV anterior wall in the double-Tg mice compared with the WT mice. No Tg mice died after MI operation. MI hearts in the double-Tg mice showed impaired LVEF compared with the WT mice (Figure 5b). Collagen concentration from the cardiac ventricle was analyzed using hydroxyproline assay. MI hearts in double-Tg mice elevated collagen concentration compared with the WT+MI mice. Aliskiren treatment suppressed the concentration of collagen in double-Tg mice (Supplementary Figures 2C and D). The double-Tg mice showed increased deterioration of pathological myocardial remodeling after MI compared with WT+MI mice. We then examined levels of ANP, collagen-type I, Nox2, Nox4, and MCP-1. MI hearts in the double-Tg mice showed additionally elevated levels of ANP, collagen-type I, Nox2, Nox4, and MCP-1 mRNA compared with the WT+MI mice. Aliskiren treatment suppressed BP increasing, cardiac dysfunction, LV remodeling, and inflammatory or oxidative factors in double-Tg mice compared with the non-treated double-Tg mice (Supplementary Figures 2E–I).

Renin–angiotensin system enhanced oxidative stress and monocytes chemoattractant protein (MCP)-1 in in vitro. Cardiomyocytes were isolated from wild type (WT) (a) or AT1 KO (b) and stimulated by murine plasma. (a) Representative real-time PCR data detecting the expression of Nox2. (b) Nox2 levels in AT1-deficient cardiomyocytes with stimulation of 5/6 nephrectomy (NTX) plasma. Mononuclear cells were isolated from WT (c and e) or AT1 KO (d and f) and stimulated by the specified plasma. *P<0.05; NS, not significant.
RAS Enhanced Oxidative Stress and MCP-1 in In Vitro
Isolated cardiomyocytes were stimulated by murine plasma, and mRNA was collected 24 h after stimulation. Significantly, enhanced expression of Nox2 mRNA levels was observed by NTX murine plasma stimulation compared with plasma from non-NTX mice. Plasma stimulation from aliskiren-treated NTX mice decreased expression of Nox2 levels, whereas aliskiren-pretreated cardiomyocytes did not show altered mRNA levels. The AT1-deficient cardiomyocytes with stimulation of NTX mice plasma did not show increased Nox2 levels (Figures 5a and b).
Plasma from NTX mice elevated expression of Nox2 and MCP-1 in primary mononuclear cells. Although plasma from aliskiren-treated mice did not alter these levels, aliskiren pre-treated mononuclear cells showed significantly suppressed Nox2 and MCP-1 levels (Figures 5c and e). AT1-deficient mononuclear cells did not have suppressed Nox2 and MCP-1 expression, but aliskiren-treated mononuclear cells showed markedly suppressed expression of these factors compared with the NTX plasma-treated group (Figures 5d and f).
DISCUSSION
In this study, we investigated the pathophysiology of ventricular remodeling after MI associated with renal failure. RAS activation by renal failure augmented myocardial remodeling, expression of inflammatory factors, and oxidative stress. Double-Tg mice showed myocardial remodeling and Nox2 and Nox4 elevation comparable to those of WT MI mice with NTX. In addition, a Nox inhibitor attenuated LV remodeling in MI plus NTX mice. We revealed that upregulated Nox induced by RAS stimulation has a pivotal role in the development of myocardial remodeling associated with renal dysfunction in this type IV cardiorenal syndrome (or chronic renocardiac symdrome) model.
It is known that oxidative stress, which results in cell damage, is increased in various cardiovascular diseases. The Nox protein family consists of well-known factors that characterize ROS-generating systems. Nox2 and Nox4 can also be activated by a number of stimuli, such as RAS, inflammatory cytokines, sheer stress, or hypoxia. Recent studies indicate that similar Nox systems are present in a wide variety of cells. Nox2 and Nox4 are known to be expressed in cardiomyocytes and fibroblasts and may potentially contribute to adverse remodeling. Previous studies showed that Nox2–/– reduced LV remodeling after aortic constriction or MI, suggesting that Nox2 exerted specific effects on the extracellular matrix.22 Nox4 is known to be a key contributor in converting fibroblasts to myofibroblasts, which are a main contributor to cardiac fibrosis.23 Nox4 overexpression in cardiomyocytes induces apoptosis resulting in expanded myocardial remodeling.24 Therefore, elevated myocardial levels of Nox in NTX mice deteriorated MI-induced cardiac collagen synthesis and ventricular remodeling. We demonstrated that the MI plus NTX hearts showed upregulation of Nox2 and Nox4 compared with the MI plus non-NTX hearts. Moreover, in vitro studies revealed that stimulation of plasma from NTX mice enhanced Nox2 expression from WT cardiomyocytes, but not AT1-deficient cells. These results suggest that Nox expression was increased through the AT1 pathway and resulted in deteriorated myocardial remodeling through collagen synthesis.
MCP-1 is well known to be contributor to the development of LV remodeling following MI. The reduction against MCP-1 and the receptor suppress LV remodeling in MI models.25, 26 Our data, therefore, suggest that MCP-1 may have contributed to the development of LV remodeling in this model. Nox2 and MCP-1 were upregulated in mononuclear cells stimulated with NTX plasma or with NTX plus aliskiren plasma. However, cells stimulated with NTX that were pretreated with aliskiren did not show upregulation of these factors. Pre-treatment of aliskiren inhibits renin activation and Ang II expression, but does not have an inhibitory effect on already-produced Ang II. These results indicate that MCP-1 expression was not increased through the AT1 pathway. A previous study showed that renin deficiency of macrophage or aliskiren-reduced macrophage adhesion to endothelial cells resulted in suppression of atherosclerosis in mice.8 Our data may indicate that aliskiren has a direct role on the anti-inflammatory effect through inactivation of macrophages. However, the underlying mechanism is unknown, and further investigation is needed to determine the effect of aliskiren on LV remodeling in AT1−/− mice and the mechanism of aliskiren action on monocytes.
This study demonstrated that NTX deteriorated MI-induced LV remodeling through RAS activation. Oxidative stresses such as increased Nox2, Nox4, and MCP-1 induced the activation of RAS. These proteins are known as key factors contributing to the development of LV remodeling through enhanced interstitial fibrosis and inflammation. These results suggested that the increase of inflammatory response affected development following MI. Aliskiren suppressed these factors and improved LV function and remodeling. Therefore, further investigation is needed to determine the clinical usefulness of RAS inhibition on cardiorenal syndrome. Interestingly, upregulation of cardiac AT1a receptor was evident after NTX in mice, and in vitro study showed a lack of AT1a receptor benefit on oxidative stress. This data suggest that AT1 blockers may also benefit the cardiac remodeling caused by MI plus CKD.
Our data showed that aliskiren treatment improved cardiac remodeling and dysfunction in NTX-MI mice without alteration of renal function. However, creatinine is imperfect in mice as a measure of renal function. We need further investigation using more solid methods to analyze the renal function.
In conclusion, RAS activation has a pivotal role in adverse MI-induced myocardial remodeling associated with renal failure, and aliskiren treatment improved the myocardial remodeling and suppressed the expression of oxidative stress and inflammatory factors in this cardiorenal models. Aliskiren treatment may be a new methodological approach about patients with cardiorenal syndrome.
References
Go AS, Chertow GM, Fan D et al. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004;351:1296–1305.
Virzì GM, Corradi V, Panagiotou A et al. ADPKD: prototype of cardiorenal syndrome type 4. Int J Nephrol 2010;2011:490795.
McCullough P . Prevention of cardiorenal syndromes. Contrib Nephrol 2010;165:101–111.
McCullough PA . Cardiorenal syndromes: pathophysiology to prevention. Int J Nephrol 2011;2011:762590.
McCullough PA, Ahmad A . Cardiorenal syndromes. World J Cardiol 2011;3:1–9.
Strauss M, Hall A . Angiotensin receptor blockers may increase risk of myocardial infarction: unraveling the ARB-MI paradox. Circulation 2006;114:838–854.
Wood JM, Maibaum J, Rahuel J et al. Structure-based design of aliskiren, a novel orally effective renin inhibitor. Biochem Biophys Res Commun 2003;308:698–705.
Lu H, Rateri DL, Feldman DL et al. Renin inhibition reduces hypercholesterolemia-induced atherosclerosis in mice. J Clin Invest 2008;118:984–993.
Fisher ND, Jan Danser AH, Nussberger J et al. Renal and hormonal responses to direct renin inhibition with aliskiren in healthy humans. Circulation 2008;117:3199–3205.
Solomon SD, Shin SH, Shah A et al. Effect of the direct renin inhibitor aliskiren on left ventricular remodelling following myocardial infarction with systolic dysfunction. Eur Heart J 2011;32:1227–1234.
Westermann D, Riad A, Lettau O et al. Renin inhibition improves cardiac function and remodeling after myocardial infarction independent of blood pressure. Hypertension 2008;52:1068–1075.
Burney BO, Kalaitzidis RG, Bakris GL . Novel therapies of diabetic nephropathy. Curr Opin Nephrol Hypertens 2009;18:107–111.
Parving HH, Brenner BM, McMurray JJ et al. Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints (ALTITUDE): rationale and study design. Nephrol Dial Transplant 2009;24:1663–1671.
Fukamizu A, Sugimura K, Takimoto E et al. Chimeric renin-angiotensin system demonstrates sustained increase in blood pressure of transgenic mice carrying both human renin and human angiotensinogen genes. J Biol Chem 1993;268:11617–11621.
Sugaya T, Nishimatsu S, Tanimoto K et al. Angiotensin II type 1a receptor-deficient mice with hypotension and hyperreninemia. J Biol Chem 1995;270:18719–18722.
Tapia E, Franco M, Sánchez-Lozada L et al. Mycophenolate mofetil prevents arteriolopathy and renal injury in subtotal ablation despite persistent hypertension. Kidney Int 2003;63:994–1002.
Shimazaki M, Nakamura K, Kii I et al. Periostin is essential for cardiac healing after acute myocardial infarction. J Exp Med 2008;205:295–303.
Harraz MM, Marden JJ, Zhou W et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest 2008;118:659–670.
Ogawa M, Suzuki J, Hishikari K et al. Clarithromycin attenuates acute and chronic rejection via matrix metalloproteinase suppression in murine cardiac transplantation. J Am Coll Cardiol 2008;51:1977–1985.
Reddy GK, Enwemeka CS . A simplified method for the analysis of hydroxyproline in biological tissues. Clin Biochem 1996;29:225–229.
Ogawa M, Suzuki J, Kosuge H et al. The mechanism of anti-inflammatory effects of prostaglandin E2 receptor 4 activation in murine cardiac transplantation. Transplantation 2009;87:1645–1653.
Looi YH, Grieve DJ, Siva A et al. Involvement of Nox2 NADPH oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension 2008;51:319–325.
Cucoranu I, Clempus R, Dikalova A et al. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res 2005;97:900–907.
Ago T, Kuroda J, Pain J et al. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res 2010;106:1253–1264.
Hayashidani S, Tsutsui H, Shiomi T et al. Anti-monocyte chemoattractant protein-1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation 2003;108:2134–2140.
Kaikita K, Hayasaki T, Okuma T et al. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am J Pathol 2004;165:439–447.
Acknowledgements
We especially thank Ms Noriko Tamura and Ms Yasuko Matsuda for their excellent technical assistance. This study was supported by the Japan Heart Foundation Young Investigator’s Research Grant, a grant of the Research Fellow of the Japan Society for the Promotion of Science; Yokohama Research Grant from Research Foundation for Pharmaceutical Sciences; Takeda Science Foundation; and Suzuken Memorial Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
The renin-angiotensin system (RAS) has a critical role in the development of cardiorenal syndrome, the complex pathophysiological interactions between heart and kidney diseases. A novel animal model for cardiorenal syndrome suggests that nephrectomy exacerbates myocardial ischemia-induced left ventricular dysfunction, and that renin activity is responsible for adverse myocardial remodeling.
Rights and permissions
About this article
Cite this article
Ogawa, M., Suzuki, Ji., Takayama, K. et al. Impaired post-infarction cardiac remodeling in chronic kidney disease is due to excessive renin release. Lab Invest 92, 1766–1776 (2012). https://doi.org/10.1038/labinvest.2012.136
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2012.136
Keywords
This article is cited by
-
A peptide vaccine targeting angiotensin II attenuates the cardiac dysfunction induced by myocardial infarction
Scientific Reports (2017)
-
Angiotensin II receptor blocker irbesartan attenuates cardiac dysfunction induced by myocardial infarction in the presence of renal failure
Hypertension Research (2016)
-
Crosstalk between the heart and peripheral organs in heart failure
Experimental & Molecular Medicine (2016)

{kind=link}
{kind=link}
{kind=link}