
Abstract
The role of hepatocyte apoptosis in the physiopathology of obstructive cholestasis is still controversial. Although some data have strongly suggested that hepatocellular cholestatic injury is due to Fas-mediated hepatocyte apoptosis, some others concluded that necrosis, rather than apoptosis, represents the main type of hepatocyte death in chronic cholestasis. Moreover, it has also been suggested that the reduced liver injury observed in the absence of Fas receptor after bile duct ligation was not due to lower hepatocyte apoptosis but to the indirect role of this receptor in non-hepatocytic cells such as cholangiocytes and inflammatory cells. The aim of this work was therefore to determine whether a protection against cell death limited to hepatocytes could be sufficient to reduce liver injury and delay cholestatic fibrosis. With this purpose, we performed bile duct ligation in transgenic mice overexpressing Bcl-2 in hepatocytes and in wild-type littermates. We found that, compared with necrosis, apoptosis was negligible in this model. Our results also showed that hepatocyte Bcl-2 expression protected hepatocytes against liver injury only in the early steps of the disease. This protection was correlated with reduced mitochondrial dysfunction and lipid peroxidation. However, in contrast to Fas receptor-deficient lpr mice, fibrosis progression was not hampered and liver inflammatory response was not reduced by Bcl-2 overexpression. These results therefore comfort the hypothesis that Fas-mediated apoptotic hepatocyte pathway is not a significant contributing factor to the clinical features observed in cholestasis. Moreover, in the absence of a blunted inflammatory response in transgenic mice, Bcl-2 protection against hepatocyte mitochondrial dysfunction and lipid peroxidation was not sufficient to block fibrosis progression.
Similar content being viewed by others


Main
Cholestasis is characterized by the accumulation of toxic hydrophobic bile acids in hepatocytes and cholangiocytes causing cell death and liver damage that will progressively induce liver fibrosis. Activation of Fas receptor pathway has been associated with liver cholestasis.1, 2, 3 Fas belongs to a subfamily of death receptors that have an important role in liver homeostasis. Upon Fas activation in hepatocytes, Fas-associated death domain adaptor protein will recruit caspase-8 and induce the cleavage and translocation to the mitochondria of Bid, a proapoptotic member of the Bcl-2 family. This will activate other members such as Bax or Bak, and the release of cytochrome c. The role of Fas-mediated hepatocyte apoptosis in cholestasis-induced liver injury is rather controversial. On the one hand, toxic bile acids clearly induce hepatocyte apoptosis in vitro by direct activation of Fas.4 Moreover, the observation that Fas receptor-deficient (lpr) mice are partially protected against liver injury and fibrosis progression induced by bile duct ligation (BDL), a surgical procedure mimicking human cholestatic disorders, has suggested that Fas-induced hepatocyte apoptosis was critical for disease progression.2, 3 In the same line, a pan-caspase inhibitor attenuates hepatic injury and fibrosis after BDL.4 Similar to Fas receptor-deficient mice, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor 2-deficient mice are far less prone to fibrosis after BDL than wild-type (WT) mice.5 The authors suggested therefore that a cooperation between TRAIL receptor-2, that is upregulated during cholestasis,6 and Fas receptor potentiates hepatocytes death-inducing fibrogenesis. Importantly, in these studies, all liver cells, and not only parenchymal cells, were targeted by gene invalidation or by the pharmacological therapeutic agent. On the other hand, using the same Fas receptor-deficient lpr model, another group reached contradictory results showing that the inflammatory reaction, and not the very limited bile acid-induced hepatocyte apoptosis, accounts for liver injury after BDL.7 Fibrosis progression was unfortunately not evaluated in this work. Another work showed that necrosis rather than apoptosis represents the main type of lesion observed in cholestasis.8 Finally, a recent study demonstrated that hepatic fibrosis induced by BDL was not affected by loss of Bid,9 reinforcing the minor role of apoptosis in obstructive cholestasis. Chronic cholestasis has even been shown to induce a resistance to Fas-induced apoptosis as a consequence of stress adaptation.10, 11 To reconcile all these data, one could hypothesize that the reduced liver injury and fibrosis observed in the absence of Fas receptor in obstructive cholestasis is mainly due to a reduced inflammatory reaction and not to the lack of Fas-induced hepatocyte apoptosis.9, 12
We have developed a transgenic mouse line (Bcl-2 Tg) harboring 10 copies of the human antiapoptotic Bcl-2 cDNA and overexpressing it specifically in hepatocytes more than 10 times higher than in non-transgenic WT livers.13, 14 These mice were shown to be protected against a lethal dose of a Fas-agonist antibody, with no release of cytochrome c from mitochondria and insignificant activation of effector caspases such as caspase-3.13, 14, 15, 16 Besides its antiapoptotic functions, Bcl-2 displays antioxidant actions at the mitochondrial level.17, 18 We have indeed previously shown that Bcl-2 Tg mice presented lower lipid peroxidation after ischemia/reperfusion injury.19 Also, we have demonstrated that this expression protects hepatocytes against mitochondrial dysfunction and oxidative stress induced by CCl4 chronic administration, delaying fibrosis progression.20
These mice therefore offer a unique opportunity to determine whether a specific protection of the hepatocytes against bile acid-induced apoptotic and/or necrotic cell death could protect against liver injury and fibrogenesis after BDL.
MATERIALS AND METHODS
Mice and Experimental Design
Bcl-2 transgenic males (Bcl-2 Tg) were generated as previously described13 and back-crossed into the C57Bl/6 background. Male WT C57Bl/6 mice (WT) were used as controls. Six to seven mice were used in each group. Blood was collected at killing and the activity of serum alanine aminotransferase18 and total bilirubin levels were measured on a Hitachi 747 analyzer. BDL and sham laparotomy were performed as previously described21 under isoflurane anesthesia and sterile conditions. Mice were killed 3 or 16 days after BDL or 16 days after laparotomy (sham animals). The six experimental groups in this study were as follows: WT sham, Bcl-2 sham, WT 3 days after BDL, Bcl-2 Tg 3 days after BDL, WT 16 days after BDL and Bcl-2 Tg 16 days after BDL. For caspase activity, WT mice submitted to Fas-induced liver injury were used as positive controls. These mice were injected intravenously with a non-lethal dose of 0.15 mg/kg of Jo2 antibody (BD, Pharmingen, San Diego, CA, USA) and livers were collected 24 h after injection. All experiments were conducted in accordance with Institutional Guidelines for the care and use of laboratory animals.
Histological Analysis
Liver samples were fixed in 10% formalin and embedded in paraffin. Five-μm thick paraffin-embedded liver sections were stained with hematoxylin and eosin (H&E) or with Sirius Red. For α-smooth muscle actin (α-SMA) immunohistochemistry, 5 μm paraffin sections were incubated with α-SMA antibody (M 0851, Dako, Glostrup, Denmark). Five-μm frozen sections were used for immunohistochemistry analyses with CD68 antibody (MCA 1957; AbD Serotec, oxford, UK) at 1/50 dilution, CD4 (clone H129.19), CD8 (clone CT-CD8a BD) and CD45 (clone 30-F11) antibodies (BD Pharmingen, San Diego, CA, USA) at 1/50 dilution. For morphometric analysis, 10 images per animal, from three different lobes, were taken at × 100 magnification. The areas of staining or inflammatory reaction were quantified using the software ImageJ 1.37v (NIH, Bethesda, MD, USA).
TUNEL Assay
Terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL) assay was used to label apoptotic nuclei in paraffin-embedded liver sections as already described.19 For quantification of individual hepatocytes, 25 images per animal, from three different lobes, were taken at × 400 magnification and the number of TUNEL-positive hepatocytes counted in each corresponding high power field (HPF). The areas of oncotic necrosis were estimated in 15 photos per animal taken at × 100 magnification by morphometric analysis using the Image J software.
Western Blot Analysis
Liver tissues of mice were homogenized in protein lysis buffer as previously described.22 Fifty μg of proteins were run on a 10% acrylamide gel and transferred to a nitrocellulose membrane. Immunoblotting was performed with the anti-SMA antibody (DAKO M0851, Glostrup, Denmark). Enhanced chemiluminescence was revealed using a horseradish peroxidase-conjugated secondary anti-mouse antibody (DAKO). The γ-tubulin-GTU-88 antibody was used as an internal control (Sigma, St Louis, MO, USA).
Real-Time RT-PCR
RNA was purified from livers using the Trizol method (Invitrogen). cDNA synthesis was carried out using the Transcriptor First Strand cDNA synthesis kit (Roche, Mannheim, Germany). Quantitative RT-PCR was performed in duplicates with the QuantiTect SYBR Green PCR kit (Qiagen, Mainz, Germany) using the LightCycler apparatus. Standard QuantiTect Primer Assays (Qiagen) were used to amplify all analyzed genes using the QuantiTect protocol. Relative expression was calculated and normalized to control gene HPRT. Sham-operated animals were used as reference.
Caspase Activity
Livers were minced and homogenized in 1 mM EDTA, 5 mM dithiothreitol, 50 mM Hepes, 0.1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propane-sulfonate, 4 mg/ml leupeptin and 4 mg/ml pepstatin, pH 7.4. After centrifugation at 14 000 g for 10 min at 4°C, the supernatant was recovered. Caspases-2, 8 and 3 activities were measured with the fluorescent ApoAlert Caspase Assay plate (Clontech, Mountain View, CA, USA) following the manufacturer's instructions.
GSH levels, Lipid Peroxidation and Activity of Mitochondrial Respiratory Chain Complex I
Reduced glutathione (GSH) levels were determined as described previously.20 To evaluate hepatic lipid peroxidation, we measured thiobarbituric acid reactants (TBARs). Briefly, the reaction mixture consisted of 0.2 ml of an 8.1% sodium lauryl sulfate solution, 1.5 ml of a 20% acetic acid solution, pH 3.5, and 1.5 ml of a 0.8% aqueous solution of thiobarbituric acid. To this mixture was added 0.2 ml of liver homogenate prepared in 0.1 mM phosphate buffer containing 3 mM EDTA. The mixture was brought up to 4.0 ml with 0.5 ml of distilled water and 0.1 ml of 10 mM butylated hydroxytoluene, and was heated at 95°C for 60 min. After cooling, the absorbance was measured at 532 nm. TBAR contents were expressed as nmoles of malondialdehyde (MDA) equivalents per mg of protein.
The mitochondrial respiratory chain complex I (NADH-ubiquinone oxidoreductase) activity was measured by spectrophotometry at 30°C, as previously described.20 Briefly, liver homogenates were resuspended in 0.1 mM phosphate buffer and the decrease in absorbance due to oxidation of NADH to NAD+ was followed at 340 nm for 4 min in the presence of decylubiquinone and antimycin A. The reaction mixture contained 40 mM potassium phosphate (pH 7.5), 2 mM KCN, 5 mM MgCl2, 100 μM decylubiquinone in DMSO, 100 μM NADH, 2 μg antimycin A with or without 5 μg/ml rotenone.
Statistical Analysis
Statistical analyses were performed using the software Statview 4.5. Data sets were compared by Student's t-test. Data are represented as mean±s.e.m. P-values <0.05 were considered significant.
RESULTS
Liver Injury in WT and Bcl-2 Transgenic Mice after BDL
Cholestasis was induced by surgical BDL in Bcl-2 transgenic mice and WT littermates. It has been previously demonstrated that acute hepatocellular injury peaks at days 2–3 after BDL, whereas fibrosis reaches a plateau from 14 days onward.23 Thus, in order to analyze early events leading to fibrosis and also the established fibrotic scars, we killed the animals at two time points, namely, 3 and 16 days after BDL. The overall survival of mice after BDL until 16 days was equivalent in both experimental groups. We first determined the level of liver injury and cholestasis by measuring serum ALT and total bilirubin levels, respectively. Sixteen days after BDL, animals showed jaundice characterized by a yellow staining of the skin easily detectable on paws and confirmed by hyperbilirubinemia that was found equivalent in transgenic and non-transgenic animals (Figure 1a). In WT mice, plasma ALT levels increased about 15-fold 3 days after BDL, whereas only 7-fold in Bcl-2 mice, compared with respective sham-operated animals (Figure 1a). However, this partial protection against hepatocyte cytolysis by Bcl-2 liver expression was not maintained 16 days after ligation.

Liver injury (apoptosis vs necrosis) after bile duct ligation (BDL) in Bcl-2 Tg and WT mice. (a) Serum levels of total bilirubin and ALT were measured as markers of cholestasis and hepatic damage, respectively. Wild-type (WT) and Bcl-2 transgenic mice (Bcl-2) underwent BDL and were sacrificed after 3 or 16 days. Sham-operated animals were used as controls. Mean values+s.e.m. are presented for all time points (*P<0.05). (n=6 mice in each group). Quantitative assessment of cell death is presented at 3 and 16 days after BDL. Mean values+s.e.m. are presented for all time points (*P<0.05). (b) Activity of caspases 2, 3 and 8 was measured in the liver of WT and Bcl-2 Tg mice at 3 and 16 days after BDL (n=6 per group). As positive control (Fas control), animals were submitted to Fas-induced liver injury (0.15 mg/kg i.v. of Jo2 antibody) and livers were harvested 24 h after injection.
Hepatocyte Apoptosis and Necrosis in WT and Bcl-2 Transgenic Mice after BDL
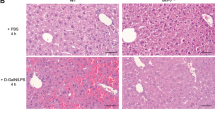
Hematoxylin and eosin staining showed a periportal inflammatory reaction in both experimental groups and small areas of degenerated hepatocytes corresponding to bile infarcts 3 days after BDL (Figure 2). We performed TUNEL analysis and found isolated positive hepatocytes disseminated throughout the different lobules at this time point. We quantified these isolated positive cells and found them significantly higher in WT mice compared with Bcl-2 Tg mice (Figure 2). However, 3 days after BDL, these cells represent about 0.6 cell per HPF ( × 400) and 0.12 cell per HPF in Bcl-2 mice, and they were even less numerous after 16 days. In contrast, large and numerous typical bile infarcts were found 16 days after BDL (Figure 2). These foci were made of swollen necrotic hepatocytes with disruption of plasma membranes characteristic of necrosis, and their area represent 7.5% of the liver parenchyma of WT mice. This proportion was not significantly different in Bcl-2 Tg mice. As previously reported,9 a minority of these necrotic foci also stained positive in the TUNEL assay (Figure 2). As TUNEL assays cannot distinguish between apoptosis and necrosis,24 we looked for caspase activation in order to determine the extent of apoptotic cell death. Activation of caspases 2, 3 and 8 was not statistically different at 3 days after BDL compared with sham-operated animals. Sixteen days after BDL, activation of caspases 2 and 3 was noticed compared with sham controls, but no significant difference was observed between Bcl-2 Tg and WT mice (Figure 1b). Moreover, compared with positive control livers isolated from mice injected with the Fas agonist antibody Jo2, these inductions were negligible. All these data indicate that necrosis represents the main type of hepatocyte death in cholestasis-induced liver injury and that Bcl-2 transiently protects against this hepatocyte cytolysis.

Hematoxylin and eosin (H&E) and TUNEL staining of representative serial liver sections. H&E and TUNEL staining 3 days and 16 days after BDL, revealed liver damage with periportal inflammatory reaction, bile ducts proliferation and isolated TUNEL-positive cells in both experimental groups (original magnification × 100 for the upper panel and × 400 for the lower panel). Sixteen days after BDL, staining revealed large bile infarcts (white asterisks) that were quantified and found equivalent in both experimental groups. Occasionally, bile infarcts stained positive for TUNEL assay in both groups, as illustrated here in a Bcl-2 Tg or WT liver (black arrowheads).
Fibrosis in WT and Bcl-2 Transgenic Mice after BDL
To investigate the consequence of Bcl-2 overexpression on fibrosis progression, liver sections were stained with picrosirius, a marker of collagen deposition (Figure 3a) and for hepatic stellate cells activation (Figure 3b). There was no significant increase of collagen staining nor α-SMA staining 3 days after BDL compared with sham-operated mice. Sixteen days after BDL, Bcl-2 Tg mice presented slightly less fibrosis than WT mice, but this difference did not reach statistical significance (Figure 3a). This observation was confirmed both by α-SMA immunostaining (Figure 3b) and western blot (Figure 3c), as well as by real-time RT-PCR for α-SMA, collagen α-I and TGF-β1 (Figure 3d). All these markers were reduced in Bcl-2 Tg mice 16 days after BDL, but this difference reached statistical significance only for TGF-β1. Therefore, even if Bcl-2 overexpression potentially had a slight impact in fibrogenesis, it was not enough to block the progression of fibrosis induced by BDL, despite its protection against liver injury in the early steps of cholestasis.

Induction of cholestatic fibrosis after BDL in Bcl-2 Tg and WT mice. (a) Sirius Red staining of representative liver sections and its quantification in WT and Bcl-2 Tg mice (Original magnification, × 100) after BDL. (b) α-SMA staining of representative liver sections and its quantification in WT and Bcl-2 Tg mice (Original magnification, × 100) after BDL. (c) Liver samples of WT and Bcl-2 Tg mice were analyzed for α-SMA expression by immunoblot. (d) Assessment of mRNA expression of fibrosis markers TGF-β1, collagen α-I and α-SMA by real-time RT-PCR (*P<0.05).
Mitochondrial Dysfunction and Oxidative Stress after BDL in WT and Bcl-2 Transgenic Mice
Cumulative evidence indicates that oxidative stress is involved in the physiopathology of liver fibrogenesis. To understand the mechanisms behind this partial protection by Bcl-2 against hepatocyte death, we then examined mitochondrial dysfunction and liver lipid peroxidation induced by bile acids toxicity. We first measured total hepatic GSH content. However, we found no reduction of GSH content at the two time points whatever the genotypes (Figure 4a). GSH levels are known to be very rapidly modulated after liver injury25 and increased hepatic glutathione levels have even been reported after BDL in mice,26 possibly as a compensatory response to lipid peroxidation. We therefore evaluated the global oxidative stress induced in the liver by BDL through the quantification of MDA, a marker of lipid peroxidation (Figure 4b). There was a significant induction of lipid peroxidation in WT mice only 16 days after BDL. In contrast, MDA levels in the liver of Bcl-2 Tg mice remained low at this time point (Figure 4b). Finally, we confirmed, by measuring liver citrate synthase activity, that the mitochondrial mass was unchanged either before or after ligation in both groups of animals (data not shown) and we measured the activity of complex I of the mitochondrial respiratory chain. We found it significantly decreased in WT mice 3 and 16 days after BDL (Figure 4c). In contrast, Bcl-2 Tg livers showed no significant mitochondrial dysfunction (Figure 4c). These data show that Bcl-2 was able to protect hepatocytes against BDL-induced mitochondrial respiratory chain dysfunction and lipid peroxidation.

Oxidative stress after BDL in Bcl-2 Tg and WT mice. (a) Reduced glutathione (GSH) levels measured in the liver of WT and Bcl-2 transgenic mice. (b) Lipid peroxidation in WT and Bcl-2 livers assessed by MDA levels and compared to sham-operated mice (*P<0.05). (c) Activity of mitochondrial respiratory chain complex I, at 3 and 16 days after BDL in WT and Bcl-2 transgenic animals compared with sham-operated mice (**P<0.001).
Inflammatory Reaction after BDL in WT and Bcl-2 Transgenic Mice
As inflammation is also known to have an important role in this model, we analyzed the hepatic expression of several cytokines by real-time RT-PCR at 3 and 16 days after BDL. We found a massive induction of the expression of TNF-α, MCP-1, MIP-2 and to a lesser extent, IL-6 and IP-10 already at 3 days after BDL (Table 1). Bcl-2 Tg mice presented a weaker induction of IL-6 and TNF-α compared with WT mice at 3 days after BDL, but this difference was not significant. Moreover, the overexpression of Bcl-2 had no impact on this inflammatory reaction 16 days after BDL, as attested by the similar expression of inflammation markers in WT and Bcl-2 Tg mice. This was corroborated by the histological analysis of the global inflammatory area upon H&E staining (Figure 5a). Moreover, we checked the activation of Kupffer cells. Although in sham animals, CD68-positive macrophages remained isolated, LVB caused recruitment of Kupffer cells into clusters in the inflammatory area (Figure 5c). Quantification of these cells showed no significant difference between transgenic and WT mice (Figure 5b). Finally, the nature of inflammatory cells (CD45/CD4/CD8/B220/NK1.1) was similar in both experimental groups (Figure 5c and data not shown). Altogether, these results suggest that Bcl-2 was able to protect against BDL-induced lipid peroxidation and hepatocyte mitochondrial dysfunction. Despite the fact that this protection was responsible for a lower liver injury in the early stages of cholestasis, it was not sufficient to block fibrosis progression and to counteract the inflammatory reaction in this model.

Analysis of inflammatory reaction. (a) Quantification of the global inflammatory area mainly found around portal tracts 16 days after BDL. (b) Quantification of CD68 staining before and after surgery. (c) Immunostaining of representative sections for CD68 (macrophages), CD45 (common leukocyte antigen), CD4 and CD8 (T lymphocytes) before and 16 days after BDL in WT and Bcl-2 transgenic mice (original magnification, × 200). Some positive cells are indicated by an arrow.
DISCUSSION
Cholestasis is a common syndrome in biliary atresia, primary biliary cirrhosis, primary sclerosing cholangitis and various other forms of biliary tract diseases. It has been previously reported that liver injury induced by cholestasis was mediated by death receptors1, 2, 27, 28 and particularly by Fas-induced hepatocyte apoptosis. This conclusion was recently challenged by data suggesting that, rather than hepatocyte apoptosis, it was hepatocyte necrosis,8, 12 induced by an intense inflammatory reaction7, 9, 12, 29, 30 that was responsible for fibrosis progression. The objective of our study was therefore to analyze whether Bcl-2 overexpression, by protecting hepatocytes against mitochondrial dysfunction and potential Fas-induced apoptosis, could affect liver injury and hamper the progression of liver fibrosis.
First of all, we confirmed here that apoptosis is negligible in the BDL model. Previous data have already failed to detect any caspase activation after BDL.7, 9 We also found no caspase 8 activation at any time point in both experimental groups, whereas only a marginal activation of caspases 2 and 3, equivalent in both groups, was observed 16 days after BDL. This activation was about 100 times lower than the one induced by a sublethal dose of Jo2, a Fas-agonist antibody. We quantified isolated TUNEL-positive cells in BDL-operated livers, and found them significantly less numerous in Bcl-2 Tg than in WT mice 3 days after BDL. However, we also observed some TUNEL-stained macrophages scattered throughout the liver sections both in Bcl-2 and WT mice that could explain why caspase activity, albeit low, does not differ between transgenic and WT mice (data not shown). We also observed TUNEL-positive staining of typical necrotic bile infarcts, confirming that TUNEL assays can reflect not only apoptosis but also late necrosis. Altogether, our data suggest that the transient protection conferred by Bcl-2 overexpression that translated into lower plasma ALT most probably reflects a protection against necrosis.
To determine whether this effect had an impact in fibrogenesis, we looked at various fibrosis markers. Consistently with the literature,23 there was no significant collagen deposition or α-SMA-positive area 3 days after BDL in WT mice. Sixteen days after surgery, all markers were found lower in Bcl-2 Tg mice compared with WT mice, but these differences did not reach significance except for TGFβ1 mRNA expression. Interestingly, in accordance with our data, Bid-deficient mice, although protected against Fas-induced apoptosis also showed no modification of hepatic fibrosis induced by BDL.9
We then set out to investigate how Bcl-2 exerted its protection against BDL-induced liver injury at early stages of the disease. It has been shown that hydrophobic bile acids produced during cholestasis stimulate the generation of reactive oxygen species (ROS) in hepatocytes interfering with normal mitochondrial electron transport in inhibiting the activity of complexes I and III.31 Bcl-2 is a guardian of the mitochondrial membrane and displays antioxidant functions. Using our Bcl-2 transgenic mouse model, we had previously demonstrated that Bcl-2 overexpression protects hepatocytes against mitochondrial damages and oxidative stress induced by chronic administration of CCl4.20 In this latter model, hepatocyte protection against ROS was correlated with reduced fibrotic scarring after 3 weeks of chronic injury. In contrast, even though the mitochondria of Bcl-2 Tg mice were still protected after 6 weeks of chronic administration of CCl4, fibrogenesis was no longer reduced in Bcl-2 mice. These data indicated that the sole protection of hepatocytes against oxidative damage was not sufficient to block the vicious circle of ROS production, particularly by non-parenchymal cells, and in consequence the fibrotic process.20 Interestingly, the expression of inflammation markers, such as MIP-2, MCP-1, TNFα and IP-10, was clearly upregulated after BDL, but was equivalent between Bcl-2 Tg and WT mice. When we compared this activation of inflammatory markers with the one induced by chronic CCl4 toxic injury, we found it to be significantly higher in the BDL model, particularly for MCP-1 and MIP2 that were found 10- to 20-fold more induced 3 or 16 days after BDL compared with CCl4.20 The upregulation of hepatocyte MCP-1 induced by bile acids has recently been shown to be a key early event in liver fibrogenesis in recruiting hepatic stellate cells.29 This could explain why the protection against the fibrotic scar is less important after BDL than after chronic CCl4 injections. We therefore propose a model (Figure 6) in which Bcl-2, by protecting hepatocyte mitochondria, reduces ROS generation and lipid peroxidation induced by bile salts accumulation or CCl4 chronic injections. In the CCl4 model, inflammation is low in the early stages and Bcl-2, by protecting against mitochondrial damage, delays the progression of fibrosis. In contrast, in the BDL model, inflammation is already prominent at 3 days post-ligature, limiting the protective effect of Bcl-2 in the early stages of fibrosis, although hepatocyte mitochondrial dysfunction and oxidative stress are reduced. The relative role of inflammation and mitochondrial dysfunction in the establishment of fibrosis is therefore most probably dependent on the disease etiology. However, in a non-mutually exclusive hypothesis, bile acids may also have had a direct role in the activation of hepatic stellate cells, as it has been recently reported in vitro.32

Schematic model depicting the role of Bcl-2 expression in fibrosis physiopathology in two models: bile duct ligation and toxic liver injury by CCl4 administration. BDL induces a high level of inflammation and cytokines (large black arrows) in WT and Bcl-2 Tg mice. Although Bcl-2 protects against hepatocyte mitochondrial dysfunction and reduces lipid peroxidation, it is not sufficient to delay fibrosis progression. In contrast, in chronic CCl4 liver injury, the level of inflammation is lower (thin black arrows) allowing Bcl-2 protection against mitochondrial dysfunction to temporarily delay fibrosis progression.
In conclusion, using an original transgenic mouse model, we show that Bcl-2 hepatocyte overexpression is not sufficient to prevent fibrosis, even though it reduces liver injury in the initial steps of cholestasis by protecting against hepatocyte oxidative stress and mitochondrial dysfunction. Moreover, we comfort the hypothesis that necrosis represents the main cause of hepatocyte death in acute and chronic cholestasis in mice. Finally, our data reinforce the role of inflammatory cells in the progression of fibrosis. An anti-inflammatory approach, in addition to an antioxydant treatment, could therefore constitute an interesting synergistic therapeutic strategy, during cholestasis-induced liver injury.
References
Faubion WA, Guicciardi ME, Miyoshi H, et al. Toxic bile salts induce rodent hepatocyte apoptosis via direct activation of Fas. J Clin Invest 1999;103:137–145.
Canbay A, Higuchi H, Bronk SF, et al. Fas enhances fibrogenesis in the bile duct ligated mouse: a link between apoptosis and fibrosis. Gastroenterology 2002;123:1323–1330.
Miyoshi H, Rust C, Roberts PJ, et al. Hepatocyte apoptosis after bile duct ligation in the mouse involves Fas. Gastroenterology 1999;117:669–677.
Canbay A, Feldstein A, Baskin-Bey E, et al. The caspase inhibitor IDN-6556 attenuates hepatic injury and fibrosis in the bile duct ligated mouse. J Pharmacol Exp Ther 2004;308:1191–1196.
Kahraman A, Barreyro FJ, Bronk SF, et al. TRAIL mediates liver injury by the innate immune system in the bile duct-ligated mouse. Hepatology (Baltimore, MD) 2008;47:1317–1330.
Higuchi H, Bronk SF, Taniai M, et al. Cholestasis increases tumor necrosis factor-related apoptotis-inducing ligand (TRAIL)-R2/DR5 expression and sensitizes the liver to TRAIL-mediated cytotoxicity. J Pharmacol Exp Ther 2002;303:461–467.
Gujral JS, Liu J, Farhood A, et al. Reduced oncotic necrosis in Fas receptor-deficient C57BL/6J-lpr mice after bile duct ligation. Hepatology (Baltimore, MD) 2004;40:998–1007.
Fickert P, Trauner M, Fuchsbichler A, et al. Oncosis represents the main type of cell death in mouse models of cholestasis. J Hepatol 2005;42:378–385.
Nalapareddy P, Schungel S, Hong JY, et al. The BH3-only protein bid does not mediate death-receptor-induced liver injury in obstructive cholestasis. Am J Pathol 2009;175:1077–1085.
Schoemaker MH, Gommans WM, Conde de la Rosa L, et al. Resistance of rat hepatocytes against bile acid-induced apoptosis in cholestatic liver injury is due to nuclear factor-kappa B activation. J Hepatol 2003;39:153–161.
Vogel A, Aslan JE, Willenbring H, et al. Sustained phosphorylation of Bid is a marker for resistance to Fas-induced apoptosis during chronic liver diseases. Gastroenterology 2006;130:104–119.
Gujral JS, Farhood A, Bajt ML, et al. Neutrophils aggravate acute liver injury during obstructive cholestasis in bile duct-ligated mice. Hepatology (Baltimore, MD) 2003;38:355–363.
Lacronique V, Mignon A, Fabre M, et al. Bcl-2 protects from lethal hepatic apoptosis induced by an anti-Fas antibody in mice. Nat Med 1996;2:80–86.
De la Coste A, Fabre M, McDonell N, et al. Differential protective effects of Bcl-xL and Bcl-2 on apoptotic liver injury in transgenic mice. Am J Physiol 1999;277 (3 Part 1):G702–G708.
Mitchell C, Mignon A, Guidotti JE, et al. Therapeutic liver repopulation in a mouse model of hypercholesterolemia. Hum Mol Genet 2000;9:1597–1602.
Mignon A, Guidotti JE, Mitchell C, et al. Selective repopulation of normal mouse liver by Fas/CD95-resistant hepatocytes. Nat Med 1998;4:1185–1188.
Hockenbery DM, Oltvai ZN, Yin XM, et al. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 1993;75:241–251.
Kane DJ, Sarafian TA, Anton R, et al. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science (New York, NY) 1993;262:1274–1277.
Peralta C, Perales JC, Bartrons R, et al. The combination of ischemic preconditioning and liver Bcl-2 overexpression is a suitable strategy to prevent liver and lung damage after hepatic ischemia-reperfusion. Am J Pathol 2002;160:2111–2122.
Mitchell C, Robin MA, Mayeuf A, et al. Protection against hepatocyte mitochondrial dysfunction delays fibrosis progression in mice. Am J Pathol 2009;175:1929–1937.
Serviddio G, Pereda J, Pallardo FV, et al. Ursodeoxycholic acid protects against secondary biliary cirrhosis in rats by preventing mitochondrial oxidative stress. Hepatology (Baltimore, MD) 2004;39:711–720.
Mitchell C, Couton D, Couty JP, et al. Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice. Am J Pathol 2009;174:1766–1775.
Georgiev P, Jochum W, Heinrich S, et al. Characterization of time-related changes after experimental bile duct ligation. Br J Surg 2008;95:646–656.
Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, et al. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology (Baltimore, MD) 1995;21:1465–1468.
Szymanska JA . Hepatotoxicity of brominated benzenes: relationship between chemical structure and hepatotoxic effects in acute intoxication of mice. Arch Toxicol 1998;72:97–103.
Song M, Song Z, Barve S, et al. Tetrathiomolybdate protects against bile duct ligation-induced cholestatic liver injury and fibrosis. J Pharmacol Exper Therap 2008;325:409–416.
Gabele E, Froh M, Arteel GE, et al. TNFalpha is required for cholestasis-induced liver fibrosis in the mouse. Biochem Biophys Res Commun 2009;378:348–353.
Kahraman A, Mott JL, Bronk SF, et al. Overexpression of mcl-1 attenuates liver injury and fibrosis in the bile duct-ligated mouse. Dig Dis Sci 2009;54:1908–1917.
Ramm GA, Shepherd RW, Hoskins AC, et al. Fibrogenesis in pediatric cholestatic liver disease: role of taurocholate and hepatocyte-derived monocyte chemotaxis protein-1 in hepatic stellate cell recruitment. Hepatology (Baltimore, MD) 2009;49:533–544.
Takeda K, Kojima Y, Ikejima K, et al. Death receptor 5 mediated-apoptosis contributes to cholestatic liver disease. Proc Natl Acad Sci USA 2008;105:10895–10900.
Sokol RJ, McKim Jr JM, Goff MC, et al. Vitamin E reduces oxidant injury to mitochondria and the hepatotoxicity of taurochenodeoxycholic acid in the rat. Gastroenterology 1998;114:164–174.
Sommerfeld A, Reinehr R, Haussinger D . Bile acid-induced epidermal growth factor receptor activation in quiescent rat hepatic stellate cells can trigger both proliferation and apoptosis. J Biol Chem 2009;284:22173–22183.
Acknowledgements
We thank Dr Patrick Jaffray for serum ALT measurements, H. Strick-Marchand and J.E. Guidotti for fruitful discussions. This study was supported by ANR (Agence Nationale de la Recherche) and INCA (National Institute of Cancer), France.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Mitchell, C., Mahrouf-Yorgov, M., Mayeuf, A. et al. Overexpression of Bcl-2 in hepatocytes protects against injury but does not attenuate fibrosis in a mouse model of chronic cholestatic liver disease. Lab Invest 91, 273–282 (2011). https://doi.org/10.1038/labinvest.2010.163
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2010.163
Keywords
This article is cited by
-
Apoptosis-inducing factor plays a role in the pathogenesis of hepatic and renal injury during cholestasis
Naunyn-Schmiedeberg's Archives of Pharmacology (2021)
-
Mechanisms and in vitro models of drug-induced cholestasis
Archives of Toxicology (2019)
-
In vitro prediction of drug-induced cholestatic liver injury: a challenge for the toxicologist
Archives of Toxicology (2018)
-
MicroRNA-29a protects against acute liver injury in a mouse model of obstructive jaundice via inhibition of the extrinsic apoptosis pathway
Apoptosis (2014)
-
Drug-Induced Inhibition of Mitochondrial Fatty Acid Oxidation and Steatosis
Current Pathobiology Reports (2013)
