
Abstract
Toll-like receptor-2 (TLR2) is a pivotal pathogen recognition receptor that has a key role in inflammation, diabetes, and injury. Hyperglycemia, inflammation, and oxidative stress induce TLR2-myeloid differentiation factor-88 (MyD88) expression and signaling, and are major pathophysiological mechanisms in the impaired diabetic wound-healing process. The aim of the study was to examine the contribution of TLR2-MyD88 expression and signaling to the prolonged inflammation observed in diabetic wounds. Diabetes was induced in male C57BL/6J and TLR2−/− mice using streptozotocin (STZ) with matching nondiabetic mice as control. In addition, nonobese diabetic (NOD) mice were used to represent the spontaneous type 1 diabetes condition. After 2 weeks of persistent hyperglycemia in the mice, full-thickness excision wounds were made on the backs aseptically. Total RNA and protein were subjected to real-time PCR and western blot analyses. Wound sizes were measured using digital planimetry. TLR2 mRNA and protein expression increased significantly in wounds of C57BL/6J+STZ and NOD mice (P<0.05) compared with nondiabetic C57BL/6J mice. MyD88 expression, interleukin receptor-associated kinase-1 phosphorylation, and nuclear factor-κ B (NF-κB) activation were increased in diabetic wounds compared with nondiabetic wounds. Wounds of TLR2−/−+STZ mice showed less oxidative stress, decreased MyD88 signaling, NF-κB activation, and cytokine secretion. The wound closure was significant in TLR2−/−+ STZ mice compared with C57BL/6J+STZ mice. Collectively, our findings show that increased TLR2 mRNA and protein expression, signaling, and activation contribute to the prolonged inflammation in the diabetic wounds and that absence of TLR2 may result in decreased inflammation and improved wound healing.
Similar content being viewed by others


Main
With the number of diabetic patients likely to reach 300 million by the year 2030, the burden of diabetic wounds and their complications are expected to go up simultaneously.1, 2, 3 Around 40–60% of diabetic patients develop diabetic foot wounds, which account for more than 20% of all hospitalizations4 and result in an amputation every 30 s.3 Impaired wound healing is the major problem in diabetes patients and clinicians treating these wounds. An imbalance in pro-/anti-inflammatory mediators accentuates diabetic vascular complications and impedes proper wound healing.5 Excessive tumor necrosis factor-α (TNF-α) production, absence of interleukin-1 (IL-1) receptor antagonist, increased oxidative stress, decreased antioxidant capacity, and increased nuclear factor-κB (NF-κB) activity through suppression of TGF-β signaling in diabetic wounds results in impaired healing.6, 7, 8, 9, 10
Toll-like receptors (TLRs) recognize conserved pathogen-associated molecular patterns and induce innate immune responses that are essential for host defenses.11 TLRs are activated by both endogenous and exogenous agonists of microbial and nonmicrobial origins. These findings have raised the possibility that TLRs may have a role in initial inflammatory responses to injury.12, 13 TLR activation by their agonists triggers a signaling cascade leading to cytokine production and initiation of an adaptive immune response.14 Although the role of TLRs in response to infection is well defined, the mechanisms involving TLRs that regulate inflammation in skin injury are poorly understood, whereas under diabetic conditions are understudied.
In full-thickness cutaneous injury, tissue damage leads to innate immune reaction,15 but it is not known if this event is involved in the early inflammatory response that is key to normal wound repair. It is also known that uncontrolled inflammation in diabetic wounds is associated with impaired healing.16 The innate immune systems that have a role in this inflammatory response are not well defined. Given the importance of TLRs in the innate immune system and their potential role after tissue damage, we aimed to study how TLRs may be involved in the dysregulated inflammation observed in diabetic wounds.17, 18, 19, 20 Further, the inflammatory mechanisms involved in diabetic wounds have not been fully delineated. However, TLR2 expression, activation, and signaling are significantly increased in type 1 diabetes mellitus (T1DM) patients and nonobese diabetic (NOD) mice, and have been hypothesized to be significant in human health. It has been entirely unknown how TLR2 might affect inflammation in diabetic wounds. In this study, we delineate the expression and activation of TLR2 and inflammation in diabetic wounds.
MATERIALS AND METHODS
Animals
Age-matched male TLR2-deficient mice (TLR2−/−; stock no. 004650) backcrossed onto a C57BL/6J background (stock no. 000664; nine generations; wild-type control), and NOD (stock no. 001976) mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA). All the mice were housed in pathogen-free filtered air rooms in UC Davis Medical Center vivarium and were fed sterile standard chow and sterile water ad libitum and kept in individual barrier cages with daily autoclaved bedding changes. All animal experiments were approved by the UC Davis Institutional Animal Care and Use Committee.
Streptozotocin-Induced Diabetes
For inducing T1DM in mice, we injected streptozotocin (STZ; Sigma) 50 mg/kg body weight, i.p. daily for 4 consecutive days (n=10 mice per group; 8 weeks age). This method of multiple small doses of STZ injection is an established, safe, and widely accepted model to study T1DM in vivo21 with minimum STZ cytotoxicity.22 At 5 days after the injections, blood samples obtained from the tail vein were used to measure glucose concentration with the help of commercial glucometers. A glucose level >250 mg per 100 ml on 3 consecutive days and decreased insulin levels were considered as evidence of T1DM condition. Skin wounds were created 14 days after diabetes induction, a time point at which STZ is cleared from the bloodstream.23 Nondiabetic C57BL/6J and TLR2−/− mice served as additional control groups. Wounds were also created in NOD mice (male; n=10 per group; 8 weeks age) 14 days after diabetes induction. NOD mice that did not become diabetic served as controls.
Cutaneous Full-Thickness Wound Model
The advantages of using full-thickness skin wounds in mice include (1) wound with loss of tissue as often observed in clinical setting; (2) wounds can be accurately reproduced and available for multiple investigations; (3) wounds with controlled shape and size can be created; (4) wounds can be continuously observed and monitored; (5) availability of knockout animals with specific gene targeting to elucidate their role in wound healing; and (6) time required to obtain useful results, justify use of this model. We shaved the backs of all mice, thoroughly cleaned with 70% ethanol, 10% iodine pivodine, and 70% isopropanol, before wounding. Anesthesia was administered using isoflurane (Abbott Laboratories) inhalation anesthesia along with 3 mg/kg xylazine, i.p (Phoenix Scientific, St Joseph, MO, USA) as analgesic. Six 3 mm or two 8 mm diameter full-thickness wounds were placed on the dorsal shaved skin using a sterile biopsy punch (AcuDerm Inc.). Animals were monitored under heat lamps continuously until they recovered from anesthesia and then daily until the end of the experiment. To verify that the cutaneous wounding does not alter physiologic behavior patterns, we monitored the food intake for each group over the period of experiment. At 1, 3, 5, 7, and 10 days after injury, the mice were killed by pentobarbital injection and the wound tissue was collected for analysis with matched unwounded skin. The body weight and blood glucose levels were determined periodically. Wound closure was determined using a digital camera followed by planimetry of the wound gap as described earlier.24
Metabolic Assays
Serum samples were saved for measurement of glucose and insulin. Glucose was measured using a glucose HK assay from Sigma (cat. no. GAHK20) and insulin was measured using an assay from Alpco Diagnostics (cat. no. 80-INSMS-E01).25
Real-Time PCR
mRNA expression in wound tissues and unwounded skin was performed by real-time PCR (RT-PCR, using commercial sequence-specific primers and probes purchased from SA Biosciences, Gaithersburg, MD, USA). RNA was extracted from the cells using TRI reagent (Invitrogen, Carlsbad, CA, USA). The first strand of cDNA was synthesized using total RNA (1 μg per reaction). cDNA (50 ng) was amplified using primer probe sets for TLR1, TLR2, TLR4, TLR6, and 18 s (SA Biosciences) following the manufacturer's cycling parameters. Data were calculated using the 2−ΔΔCt method and are presented as ratio of transcripts for TLR gene normalized to 18 s.25, 26
Enzyme-Linked Immunosorbent Assay
The release of cytokines, interleukin-1β (IL-1β) and TNF-α were measured in wound tissue lysates and serum using an enzyme-linked immunosorbent assay (ELISA) assay (Millipore, St Charles, MO, USA). Intra- and interassay coefficient of variation (CV) were determined to be <10%.26
Nuclear/Cytoplasmic Extracts and Transcription Factor Assays
Nuclear and cytoplasmic extracts were prepared from wound tissues and normal skin as described previously.25, 26 Nuclear extracts were used to perform NF-κB (cat. no. 40096) transcription factors activation assays (Active Motif, Carlsbad, CA, USA) to verify activation of NF-κB in the diabetic wounds, indicating increased inflammation. Assays were performed in accordance to the manufacturer's protocols. Intra- and interassay CV for these assays was <7%.
Co-Immunoprecipitation and Immunoblotting
We homogenized wound tissue (days 1 and 10 to examine persistent inflammation) and adjacent normal skin with tissue-tearor in RIPA buffer containing protease inhibitor cocktail (Roche) and then sonicated them on ice-cold water bath. We measured protein concentrations of the extracts by BCA Protein Assay kit (Pierce). Total protein (50 μg) was used for the association of TLR2, and immunoblotting with MyD88 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), phosphorylated IL-1 receptor-associated kinase (pIRAK-1; Cell Signaling, Boston, MA, USA), Toll/IL-1 receptor (TIR)-domain-containing adaptor inducing IFN-β (TRIF; Abcam, Cambridge, MA, USA), and TNF receptor-associated factor-6 (TRAF-6; Abcam). We used SDS–PAGE to separate bands, and detected TLR2, TLR2-myeloid differentiation factor-88 (MyD88), TRIF, and TRAF-6 using specific antibodies and ECL detection system.26 Appropriate antibody controls were used for each experiment and no signal was observed. Densitometric ratios were determined as reported earlier from four independent assays.26
TBAR Levels in Wounds
We measured oxidative stress through lipid peroxidations (thiobarbituric acid reactive substances, TBARS) in wound tissues on days 1 and 10, to reflect the pathogenic mechanisms in impaired wound healing in T1DM mice compared with nondiabetic mice. This is a surrogate marker of oxidative stress and malondialdehyde equivalents were determined by reading the absorbance at 532 nm using 1,1,3,3-tetramethoxypropane as an external standard.27 Results were expressed as malondialdehyde equivalents (nmol/mg protein).
Immunohistochemistry
The wound tissues from mice were collected and fixed in 4% paraformaldehyde fixative. Blocking, sectioning, and deparaffinization were carried out by routine histological staining protocol. The deparaffinization and immunostaining of selected slides were carried out by standard immunological fluorescent staining method. In brief, epitope retrieval was performed at 90°C for 30 min on citrate buffer and the primary antibody was diluted with 10% goat and donkey serum and incubated at 4°C for overnight and washed twice with sterile PBS. The primary antibody for TLR2 (1:100 dilution) and TLR6 (1:100) was raised in mouse and rabbit and obtained from InvivoGen (San Diego, CA, USA) and Abcam, respectively. Alexa 594-conjugated anti-mouse secondary antibody was used for TLR2 immunostaining and anti-rabbit, Alexa 484 secondary antibody (Invitrogen) was used as secondary antibody to visualize TLR6 expression. The slides were treated with fluorescence-conjugated secondary antibody at 10% goat and donkey serum incubated at 37°C for 1 h and washed with sterile PBS for two times. The slides were then treated with 1 μg/ml DAPI to visualize nucleus and washed and mounted on Vectashield, the slides were then visualized under appropriate filter set under fluorescent microscope (Nikon Eclipse E400) fitted with Spot RTKE camera.
Statistical Analysis
Data are presented as mean±s.d. We used two-tailed t-tests to determine significance between groups. Multiple groups were analyzed by ANOVA with appropriate post hoc analyses. P<0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism software (San Diego, CA, USA).25, 26
RESULTS
Baseline characteristics of the mice are provided in Table 1. Following STZ injection, as expected, there was significant elevation in glucose levels and decrease in insulin levels when compared with C57BL/6J mice. There were no significant differences in body weight between the four groups of mice (Table 1). We created wounds 14 days after diabetes induction in STZ22, 23 and NOD mice, keeping duration of diabetes consistent in both models. In addition, persistent hyperglycemia and decreased insulin levels were considered as an established diabetes condition in all the mice.
TLR2 Expression, Activation, and Signaling are Increased in Diabetic Wounds
In the first part of the study, we examined TLR2 expression and signaling in wound tissue of C57BL/6J+STZ (diabetic) and C57BL/6J mice (nondiabetic) to ascertain the hypothesis that increased TLR2 expression and activation accentuate inflammation in diabetic wounds. Therefore, using RT-PCR, we measured TLR2 and TLR6 mRNA expression in day 10 wound tissues, reflecting consistently increased activation in diabetic wounds compared with the nondiabetic mice wounds (Figure 1a). We also assessed TLR2 and TLR6 protein expression in the diabetic mice wounds using western blot assays. As shown in Figure 1b, wounds showed significant increases in both TLR2 and TLR6 protein expression compared with the nondiabetic mice on day 10, corroborating the mRNA data. In addition, we observed increased TLR2 expression in day 10 wounds compared day 1 wounds in spontaneously type 1 diabetic NOD mice (Figure 1c) providing strength to our STZ-diabetic model data. We used C57BL/6J+STZ (diabetic) model in all remaining experiments because of the nonavailability of TLR2−/− on NOD genetic background, the ease of inducing diabetes in knockout mice, and no difference in TLR2 expression and activity between the two animal models. TLR1 mRNA expression in the wounds remained unchanged (day 10 C57BL/6: 1±0.1; TLR2−/−: 0.8±0.2; C57BL/6+STZ: 1.2±0.4; TLR2−/−+STZ: 1.05±0.4 mRNA/18 s ratio). TLR4 mRNA expression significantly increased in the diabetic wounds compared with nondiabetic wounds (day10 C57BL/6: 0.8±0.3; TLR2−/−: 1.2±0.1; C57BL/6+STZ: 4.7±0.9*; TLR2−/−+STZ: 3.8±0.4** mRNA/18s ratio; *P<0.05 vs WT/TLR2−/− and **P<0.001 vs C57BL/6/TLR2−/−). Moreover, we found no significant differences in TLR2 mRNA and protein expression between 8 and 3 mm wounds in diabetic mice at day 10. Further analysis of adjacent normal skin (TLR2: 2.2±0.4; TLR6: 2.1±0.4 mRNA/18s ratio) and diabetic wounds (TLR2: 2.7±0.4; TLR6: 2.6±0.3 mRNA/18s ratio) in mice revealed no significant differences in mRNA expression implying that alterations in TLR2/6 expression may be because of hyperglycemia principal to diabetes, as shown earlier.26, 28 Because we did not observe significant difference in biochemical parameters, wound closure (Figure 2a), and TLR2/6 mRNA expression in wounds of nondiabetic C57BL/6J and TLR2−/− mice, we focused our molecular analyses on wounds of diabetic mice.

(a) TLR2 and TLR6 mRNA expression levels in nondiabetic and diabetic mice wound tissues (3 mm) were determined using real-time PCR and expressed as ratio using 18S RNA as control. *P<0.001 compared with C57BL/6J; **P<0.05 vs TLR2−/−; n=8 mice per group. (b) Representative western blot corroborating the TLR2, TLR6 protein expression in C57BL/6J and C57BL/6J+STZ mice wound tissues (3 mm). At 10 days after injury, wound tissues were collected, lysed, and 20 μg protein was blotted for the indicated proteins and β-actin as described in Materials and methods. Densitometric ratios are indicated as described in Materials and Methods. Values are expressed as mean ratio ±s.d. *P<0.05 vs C57BL/6J. (c) Representative western blot depicting the TLR2 protein expression in nondiabetic and diabetic NOD mice wound tissues (3 mm). At 10 days after injury, wound tissues were collected, lysed, and 20 μg protein was blotted for the indicated proteins and β-actin as described in Materials and methods. Densitometric ratios are indicated as described in Materials and methods. Values are expressed as mean ratio±s.d. *P<0.05 vs day 1 wound. (d) Representative western blot demonstrating the TLR2-MyD88 signaling pathway proteins in C57BL/6J and C57BL/6J+STZ mice wound tissues (3 mm). At 10 days after injury, wound tissues were lysed, and 20 μg protein was blotted for the indicated proteins and β-actin as described in Materials and methods. Densitometric ratios are indicated as described in Materials and methods. Values are expressed as mean ratio ±s.d. *P<0.001 vs C57BL/6J.

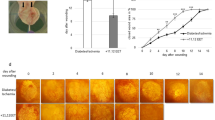
(a) TLR2−/−+STZ mice wounds have significantly smaller wounds compared with C57BL/6J+STZ mice. Wound sizes were recorded using a digital camera at days 0, 3, 5, 7, and 10 after wounding as described in Materials and Methods. *P<0.001 vs C57BL/6J+STZ, n=5 mice per group. (b) Colocalization of TLR2 (red) and TLR6 (green) in wound tissues of C57BL/6J+STZ and TLR2−/−+STZ mice on days 1 and 10 (3 mm). Red fluorescence represents TLR2, green fluorescence TLR6, whereas yellow represents the merge of both. White scale bar represents 1 μM; n=8 mice per group.
Recently, we have shown that TLR2 and TLR6 heterodimerization results in the recruitment of MyD88 and activation of the downstream signaling pathway through phosphorylation of IRAK-1, activation of TRIF, NF-κB, and upregulated expression of proinflammatory cytokines under hyperglycemia.26 Therefore, activation of MyD88-dependent pathway was used as readout of increased TLR2/6 expression in C57BL/6J+STZ (diabetic) wounds. Diabetic wounds on day 10 had significantly increased MyD88 protein, IRAK-1 phosphorylation, and TRIF protein expression compared with nondiabetic wounds, with no change in β-actin levels (Figure 1d), suggesting activation of TLR2-MyD88-mediated signaling cascade in diabetic wounds indicative of prolonged inflammation. Taken together, these results establish that TLR2-MyD88-mediated inflammation is part of the diabetic wound-healing process and suggest that inhibiting this pathway may decrease inflammation and improve healing.
Improved Wound Healing in Diabetic TLR2-Deficient Mice
In the second part of the study, having established the involvement of TLR2 in the prolonged inflammatory response of diabetic wounds, we next sought to further delineate TLR2-MyD88 pathway's physiological relevance using TLR2−/− diabetic mice (TLR2−/−+STZ). We hypothesized that attenuating TLR2 expression and activity in diabetic wounds will improve wound healing in diabetic mice. We induced diabetes in TLR2−/− and C57BL/6J mice using STZ and created wounds as described in Materials and methods. First, we determined the wound closure in all four groups of mice. Figure 2a depicts significantly improved wound closure in TLR2−/−+STZ mice compared with C57BL/6J+STZ mice, suggesting that absence of TLR2-mediated inflammation alleviates wound closure in diabetic mice. Appreciable differences in wound closure were not observed in nondiabetic C57BL/6J or TLR2−/− mice (Figure 2a).
Second, immunoflourescent staining of TLR2 (red) and TLR6 (green) showed intense signal on days 1 and 10 in wound bed and wound edges of diabetic mice (Figure 2b). On the contrary, TLR6 staining in the TLR2−/−+STZ mouse wound tissue expression was limited on day 1 and significantly reduced on day 10, thus, indicating the colocalization of TLR2/TLR6 in the wound tissues of C57BL/6J+STZ mice and its amelioration in wounds of TLR2−/−+STZ mice. Further, we did not observe appreciable signal in wounds of nondiabetic C57BL/6J or TLR2−/− mice (data not shown).
Then, using co-IP assays with TLR2 antibody, we confirmed the recruitment of MyD88-dependent signaling by TLR2 in vivo. Here, we presented data for two points (days 1 and 10) emphasizing the length of TLR2 pathway activation and its inhibition on wound-healing process. As shown in Figure 3a, MyD88, pIRAK-1, and TRIF protein expression levels were significantly increased in days 1 and day 10 wound tissues of C57BL/6J+STZ mice and decreased in TLR2−/−+STZ mice, further supporting the hypothesis. Interestingly, expression of TRAF-6, a key signaling protein mediating MyD88 signaling, did not change significantly in TLR2−/−+ STZ mice, in part due to alternate mechanisms being active, which requires further investigation.

(a) Representative western blot showing expression of MyD88, pIRAK-1, TRIF, and TRAF-6 in day 1 and day 10 wound tissue lysates (8 mm) immunoprecipitated with TLR2 antibody as detailed in Materials and methods. β-Actin was used as a loading control, n=6 mice per group. (b) The DNA-binding activity of nuclear NF-κB p65 in day 1 and day 10 wound tissues (8 mm) was assessed by ELISA as detailed in Materials and methods. Values are normalized to mg nuclear protein and expressed as mean±s.d. *P<0.01 vs C57BL/6J; n=8 mice per group. (c) Lipid peroxidation in wound tissue (8 mm) lysates was measured using thiobarbituric acid reactive substances (TBARS) assay as described in Materials and methods. Values are normalized to mg cell protein and expressed as mean±s.d. *P<0.01 vs C57BL/6J; **P<0.05 vs C57BL/6J+STZ; n=8 mice per group.
To determine the functional significance of TLR2-MyD88-NF-κB signaling on inflammatory mediator expression in diabetic wounds, we investigated the local and systemic IL-1β and TNF-α release, known to be activated by TLR2. Table 2 shows significantly increased concentrations of IL-1β and TNF-α in diabetic mice (wound tissue and serum) compared with nondiabetic mice at days 1 and 10. Furthermore, there was significant reduction in IL-1β and TNF-α concentrations in TLR2−/−+ STZ mice compared with C57BL/6J+STZ mice (P<0.001). Levels of nuclear NF-κB were examined in day 1 and day 10 wounds. C57BL/6J+STZ wounds had significantly increased nuclear NF-κB activity compared with nondiabetic wounds on days 1 and 10 (Figure 3b). Furthermore, compared with C57BL/6J+STZ mice, there was a significant decrease in NF-κB activity in the TLR2−/−+STZ mice (P<0.001).
Because oxidative stress and inflammation are linked by TLRs,29 as a surrogate index of oxidative stress, we measured TBARS formation during an acid-heating reaction in wound tissues as described earlier.27 Figure 3c depicts TBAR levels in day 1 and day 10 wounds. Wounds in C57BL/6J+STZ mice had significantly increased levels compared with nondiabetic mice and this was significantly attenuated in TLR2−/−+STZ mice (P<0.005).
DISCUSSION
Diabetes impairs several aspects of wound healing, including hemostasis and inflammation, matrix deposition, and angiogenesis. These impairments are present in different tissues including myocardium, skeletal muscle, nerve, and skin. Skin wounds in diabetics have been shown to have altered blood flow, impaired neutrophil antimicrobial activity, and a dysfunctional inflammatory state associated with abnormal chemokine expression.16 Diabetic wounds show exacerbated inflammation and extensive necrosis often leading to amputation of the affected body parts.17, 18, 19, 20 Here, we hypothesized that TLR2 can specifically modulate inflammatory responses in cutaneous diabetic wounds and in part contributes to the impaired healing process. Our study results confirmed this hypothesis in two animal models of diabetic wounds, while also revealing that TLR2 is a key element in the induction of inflammation in the diabetic wounds. Inhibition of this inflammatory response using specific gene-deficient mice further implicated that inflammation in diabetic wounds is mediated in part by TLR2 and contributes to impaired healing.
The inflammatory mechanisms involved in wound healing of T1DM are not clear. Although TLR2 has been shown to be upregulated in NOD mice30 and T1DM patients,28 its role in contributing to the exacerbated inflammatory state of diabetic wounds is unclear. Furthermore, increasing experimental evidence indicates that engagement of the TLR2 by endogenous ligands may be a major trigger of inflammation in response to skin injury, ischemia, hemorrhagic shock, and ischemia/reperfusion injuries. Examples of TLR2 deficiency in injury models improving outcomes include decreased NF-κB activation, IL-6, and TNF-α levels in partial hepatectomized mice; decreased renal dysfunction and tubular damage in mice following ischemia/reperfusion injury; maintained coronary endothelial function or left ventricular function during ischemia/reperfusion injury in mice.31, 32, 33 Thus, we show that TLR2 contributes to hyperinflammation and its inhibition improves healing in diabetic wounds in line with above studies.
There are several models of cutaneous injury in diabetic animals.34, 35 The models include all-sized and types of injuries in different animals. In the context of our hypothesis and the foremost question on the status of TLR2 expression and activation in wounds (both diabetic and nondiabetic), we have chosen two sizes (3 and 8 mm) for full-thickness injuries (avoiding the skin thickness problem), to address the issue. On the basis of the results obtained, we believe that this is as an optimal model, to understand and answer our specific questions. Although the use of STZ for inducing diabetes has been frequently questioned because of its cytotoxicity problems, we developed a low-dose multiple injection STZ regimen in which all the mice developed diabetes with persistent hyperglycemia yet with negligible weight loss and toxicity. In parallel, we also showed similar results using spontaneously diabetic NOD mice. Another variable that may obscure the interpretation of increased healing in diabetic TLR2−/−mice is contraction due to smaller wound size. However, our findings showed no differences in TLR2-mediated hyperinflammation in large or small wounds suggesting TLR2 role in wound healing is independent of size. In addition, fibroblasts, keratinocytes, and macrophages are critical in wound-healing process and all these cells express TLR2. Histological analysis of wound sections show increased number of macrophages (CD68+ cells) in C57BL/6+STZ compared with C57BL/6 or TLR2−/− mice and their number reduced markedly in TLR2−/−+STZ mice (data not shown). Our current work is aimed at elucidating the contribution of macrophages to the novel findings we report on in the elevated TLR2 expression and signaling in diabetic wounds in vivo.
We observed a significant abrogation of lipid peroxidation (TBARS) in wound tissues of diabetic TLR2−/− mice compared with diabetic control mice consistent with other studies. Arbibe et al36 have shown that TLR2-mediated NF-κB activation in monocytic cells requires Rac1, a key mediator of oxidative stress in monocytes. Thus, it appears that in diabetic wounds, TLR2 may have a key role in oxidative stress through Rac1 activation, leading to activation of NF-κB and pro-inflammatory cytokines. We also show that wounds of diabetic TLR2−/−mice show significantly decreased inflammation as evidenced by decreased NF-κB, IL-1β, and TNF-α levels compared with diabetic control mice. Similar results were observed in atherosclerotic ApoE/TLR2−/− mice37 in agreement with our findings and the mechanisms for this reduction need further investigation.
Dimerization is a major event in the functional activation of TLRs and results in cytokine production and TLR2 requires heterodimerization with TLR1 or TLR6 for activity.38 We have previously shown that under hyperglycemia, TLR2/TLR6 association results in cytokine secretion in human monocytes26 consistent with the present data. Also, we did not observe changes in TLR1 mRNA expression and it is not known if TLR6 by itself is inflammatory. TLR2 primarily activates MyD88-dependent signaling pathway.39, 40, 41, 42, 43 MyD88 is an immediate and common downstream adaptor molecule recruited by activated TLR2 through their TIR domain. MyD88, in turn, recruits IRAK-1 and induces its phosphorylation. IRAK-1 associates with TRIF, leading to the activation of IKK complex and resulting in the activation of NF-κB transcription factor. The activation of MyD88-dependent signaling pathway leads to the induction of inflammatory cytokines. There are studies showing delayed dermal wound healing in nondiabetic MyD88-deficient mice,44 suggesting that alternate TLR pathways may be active in diabetic milieu (for example, TLR3 and TLR4). Here, we provide the first evidence, using the STZ-induced diabetes model, that TLR2 deficiency results in significant decrease in diabetes-induced inflammation in wounds and improves healing. The decreased inflammation in diabetic wounds, ie, release of pro-inflammatory cytokines was associated with significant reduction in NF-κB activity and TLR2-MyD88 signaling pathway, including MyD88, pIRAK, TRIF. Previously, we have shown that diabetic patients have increased monocytic TLR2 and TLR4 expression, which correlated to increased NF-κB activity, increased expression of downstream adapter proteins such as MyD88 and pIRAK, resulting in increased pro-inflammatory cytokines.28 Similar findings were found when cells were treated in vitro under hyperglycemic conditions.26 Thus, we suggest that abrogating inflammation in diabetic wounds using TLR2 as a target appears to be a reasonable approach to alleviate inflammation accelerating diabetic wound-healing process. Furthermore, absence of TLR2 may result in attenuated downstream signaling through MyD88, and consequent inflammation.31, 32, 33, 45
Taken together, these findings are best valued when recognizing that diabetes-induced TLR2 activation, signaling, and inflammation is undesirable in wounds and that genetic deficiency of TLR2 significantly abrogates the hyperinflammatory state of diabetic wounds. Future studies will focus on examining the role of the TLR2 on the increased inflammation in diabetic wounds on NOD and db/db genetic backgrounds, and will address the status of TLR2-mediated inflammation at intermediate time points (days 4 and 7 after wounding) to see the effects when granulation is maximal. Prolonged and dysregulated cytokine production leads to sustained inflammatory responses and impaired healing, causing extensive tissue damage (amputations in case of diabetic wounds). Therefore, it is important to understand local inflammatory mechanisms that might be useful in developing beneficial strategies for the management of difficult wounds burdened by excessive inflammation. Our findings show for the first time the role of TLR2 in the diabetic wound repair process and emphasize the importance of understanding the various pathogenic inflammatory signaling mechanisms in a complicated wound-healing process.
References
Jeffcoate WJ, Harding KG . Diabetic foot ulcers. Lancet 2003;361:1545–1551.
Boulton JM, Vileikyte L, Ragnarson-Tennvall G, et al. The global burden of diabetic foot disease. Lancet 2005;366:1719–1724.
Hogan P, Dall T, Nikolov P, et al. Economic costs of diabetes in the US. Diabetes Care 2002;26:917–932.
Johannesson A, Larsson GU, Ramstrand N, et al. Incidence of lower-limb amputation in the diabetic and nondiabetic general population: a 10-year population-based cohort study of initial unilateral and contralateral amputations and reamputations. Diabetes Care 2009;32:275–280.
Falanga V . Wound healing and its impairment in diabetic foot. Lancet 2005;366:1736–1743.
Goova MT, Li J, Kislinger T, et al. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am J Pathol 2001;159:513–525.
Liu R, Bal HS, Desta T, et al. Tumor necrosis factor-alpha mediates diabetes-enhanced apoptosis of matrix-producing cells and impairs diabetic healing. Am J Pathol 2006;168:757–764.
Goren I, Müller E, Pfeilschifter J, et al. Severely impaired insulin signaling in chronic wounds of diabetic ob/ob mice: a potential role of tumor necrosis factor-alpha. Am J Pathol 2006;168:765–777.
Mertz PM, Sauder DL, Davis SC, et al. IL-1 as a potent inducer of wound re-epithelization. Prog Clin Biol Res 1991;365:473–480.
Ishida Y, Kondo T, Kimura A, et al. Absence of IL-1 receptor antagonist impaired wound healing along with aberrant NF-κB activation and a reciprocal suppression of TGF-beta signal pathway. J Immunol 2006;176:5598–5606.
Uematsu S, Akira S . Toll-like receptors and innate immunity. J Mol Med 2006;84:712–725.
Matzinger P . Tolerance, danger, and the extended family. Annu Rev Immunol 1994;12:991–1045.
Marshak-Rothstein A . Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol 2006;6:823–835.
Beutler B . Inferences, questions and possibilities in Toll-like receptor signaling. Nature 2004;430 (6996):257–263.
Mollen KP, Anand RJ, Tsung A, et al. Emerging paradigm: Toll-like receptor 4-sentinel for the detection of tissue damage. Shock 2006;26:430–437.
Wetzler C, Kampfer H, Stallmeyer B, et al. Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse: prolonged persistence of neutrophils and macrophages during the late phase of repair. J Invest Dermatol 2000;115:245–253.
Armstrong DG, Lipsky BA . Advances in the treatment of diabetic foot infections. Diabetes Technol Ther 2004;6:167–177.
Lipsky BA . Medical treatment of diabetic foot infections. Clin Infect Dis 2004;39 (Suppl 2):S104–S114.
Miyajima S, Shirai A, Yamamoto S, et al. Risk factors for major limb amputations in diabetic foot gangrene patients. Diabetes Res Clin Pract 2006;71:272–279.
Markowitz JS, Gutterman EM, Magee G, et al. Risk of amputation in patients with diabetic foot ulcers: a claims-based study. Wound Repair Regen 2006;14:11–17.
Wu KK, Huan Y . Diabetic atherosclerosis mouse models. Atherosclerosis 2007;191:241–249.
Like AA, Rossini AA . Streptozotocin-induced pancreatic insulitis: new model of diabetes mellitus. Science 1976;193 (4251):415–417.
Schein PS, Kahn R, Gordon P, et al. Streptozotocin for malignant insulinomas and carcinoid tumor: report of eight cases and review of the literature. Arch Intern Med 1973;132:555–561.
Wilgus TA, Ferreira AM, Oberyszyn TM, et al. Regulation of scar formation by vascular endothelial growth factor. Lab Invest 2008;88:579–590.
Dasu MR, Park S, Devaraj S, et al. Pioglitazone inhibits Toll-like receptor expression and activity in human monocytes and db/db mice. Endocrinology 2009;150:3457–3464.
Dasu MR, Devaraj S, Zhao L, et al. High glucose induces toll-like receptor expression in human monocytes: mechanism of activation. Diabetes 2008;57:3090–3098.
Jialal I, Devaraj S . Low density lipoprotein oxidation, antioxidants, and atherosclerosis: a clinical biochemistry perspective. Clin Chem 1996;42:496–506.
Devaraj S, Dasu MR, Rockwood J, et al. Increased Toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: further evidence of a proinflammatory state. J Clin Endocrinol Metab 2008;93:578–583.
Gill R, Tsung A, Billiar T . Linking oxidative stress to inflammation: Toll-like receptors. Free Radic Biol Med 2010;48:1121–1132.
Kim HS, Han MS, Chung KW, et al. Toll-like receptor 2 senses beta-cell death and contributes to the initiation of autoimmune diabetes. Immunity 2007;27:321–333.
Leemans JC, Stokman G, Claessen N, et al. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest 2005;115:2894–2903.
Favre J, Musette P, Douin-Echinard V, et al. Toll-like receptors 2-deficient mice are protected against postischemic coronary endothelial dysfunction. Arterioscler Thromb Vasc Biol 2007;27:1064–1071.
Sakata Y, Dong JW, Vallejo JG, et al. Toll-like receptor 2 modulates left ventricular function following ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 2007;292:H503–H509.
Greenhalgh DG . Models of wound healing. J Burn Care Rehabil 2005;26:293–305.
Greenhalgh DG . Wound healing and diabetes mellitus. Clin Plast Surg 2003;30:37–45.
Arbibe L, Mira JP, Teusch N, et al. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat Immunol 2000;1:533–540.
Mullick AE, Tobias PS, Curtiss LK . Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest 2005;115:3149–3156.
Drage MG, Pecora ND, Hise AG, et al. TLR2 and its co-receptors determine responses of macrophages and dendritic cells to lipoproteins of Mycobacterium tuberculosis. Cell Immunol 2009;258:29–37.
Medzhitov R, Janeway Jr CA . Decoding the patterns of self and nonself by the innate immune system. Science 2002;296:298–300.
Takeda K, Kaisho T, Akira S . Toll-like receptors. Annu Rev Immunol 2003;21:335–376.
Uematsu S, Akira S . Toll-like receptors and innate immunity. J Mol Med 2006;84:712–725.
Kawai T, Akira S . TLR signaling. Cell Death Differ 2006;13:8156–8825.
Kawai T, Adachi O, Ogawa T, et al. Unresponsiveness of MyD88 deficient mice to endotoxin. Immunity 1999;11:115–122.
Macedo L, Pinhal-Enfield G, Alshits V, et al. Wound healing is impaired in MyD88-deficient mice: a role for MyD88 in the regulation of wound healing by adenosine A2A receptors. Am J Pathol 2007;171:1774–1788.
Lai Y, Di Nardo A, Nakatsuji T, et al. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat Med 2009;15:1377–1382.
Acknowledgements
This study was supported by grant nos. ADA-JF-07-16, JDRF-2007-585, NIH K24 AT 00596.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Authors declare no conflicts of interest.
Rights and permissions
About this article
Cite this article
Dasu, M., Thangappan, R., Bourgette, A. et al. TLR2 expression and signaling-dependent inflammation impair wound healing in diabetic mice. Lab Invest 90, 1628–1636 (2010). https://doi.org/10.1038/labinvest.2010.158
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2010.158
Keywords
This article is cited by
-
Hyaluronan, a double-edged sword in kidney diseases
Pediatric Nephrology (2022)
-
Plantaricin NC8 αβ prevents Staphylococcus aureus-mediated cytotoxicity and inflammatory responses of human keratinocytes
Scientific Reports (2021)
-
TLR3 activation induces S100A7 to regulate keratinocyte differentiation after skin injury
Science China Life Sciences (2017)
-
Oestrogen promotes healing in a bacterial LPS model of delayed cutaneous wound repair
Laboratory Investigation (2016)
-
Transition from inflammation to proliferation: a critical step during wound healing
Cellular and Molecular Life Sciences (2016)
