
Abstract
Atrial fibrillation (AF) is the most common cardiac rhythm disorder and the prevalence is increasing. The disease confers an increased risk of severe complications such as heart failure, stroke and death, yet the treatment options available are insufficient. AF can develop secondary to other diseases, but there is also evidence of a heritable component. The molecular basis for the heritability of AF has been explored in depth over the past decade. Rare variants have been identified in ion channels, transcription factors and a wide range of other genes. More recently, common variant analyses have identified 14 genetic loci associated with AF. Thus, the genetics of AF is complex and heterogeneous. In this review, we describe the common and rare variants identified for AF, the potential clinical implications of these variants and the future directions in this field. Increasing our understanding of the pathophysiology of AF will aid the development of new and improved treatment strategies and risk prediction of AF, the first steps toward a more individualized treatment of the arrhythmia.
Similar content being viewed by others
Introduction
Atrial fibrillation (AF) is the most common cardiac arrhythmia, affecting 1–2% of the general population. At present there are about 30 million individuals affected worldwide.1 Some individuals experience only mild symptoms while others are left disabled because of complications such as heart failure or stroke. In addition, individuals with AF have nearly twice the risk of death compared with the general population.2 As the prevalence of AF increases, the economic burden on the society is rising. Currently, the incremental AF cost in the US alone is over $26 billion annually.3 AF can develop secondary to other diseases, but there is also a substantial heritable component. The first report of familial AF was made in 1936, when Orgain described three brothers diagnosed with AF at a young age.4 Several studies have since then supported the heritability of AF.5, 6, 7, 8, 9, 10, 11, 12 In the Framingham Heart Study, familial AF has been associated with 40% increased risk of AF.5 Similarly, in a study in Iceland, the relative risk of AF was 1.77 in individuals with a first-degree relative with AF.6 Finally, in a study of Danish twins, over 60% of the variance of AF was estimated to be explained by genetic effects.12 Unraveling the molecular basis of AF has been the aim of many investigators during the past decade, resulting in the findings of both rare, disease-causing variants and common variants that increase the susceptibility to AF. In this review, we present an overview of common and rare variants associated with AF, and we discuss the potential investigational, therapeutic and clinical implications of these findings.
Family studies and linkage analysis
In the initial era of genetic research on AF, researchers relied on the studies of families with AF, and used linkage analysis to identify genetic markers co-segregating with the phenotype of interest. This method takes advantage of the fact that genetic markers located close to each other on a chromosome are more likely to co-segregate through the generations of a family than genetic markers located far from one another, because of meiotic genetic recombination. A genetic marker that co-segregates with the AF phenotype in a family will thus be a marker for the disease gene, which typically will be located in the locus or a few million base pair region around the linked marker. Many genetic loci associated with AF have been discovered using genetic linkage analysis,13, 14, 15, 16 but the considerable size of the loci identified has made it difficult to identify the actual causal variants. However, a few studies have had success identifying the variant causing AF in families with an autosomal dominant pattern of inheritance.17, 18 For example, Chen et al.17 reported the first gene for AF in 2003, when they identified an S140G mutation in KCNQ1 in a multi-generation Chinese kindred.
Candidate genes and AF
Using previous knowledge of gene function to guide the selection of certain genes for resequencing is typically referred to as ‘candidate gene sequencing’ and has an inherent selection bias. Given the initial identification of a mutation in KCNQ1, many investigators then resequenced ion channel genes involved in the cardiac action potential, and a cascade of mutations have been identified in the aftermath. Table 1 provides a comprehensive list of the reported variants associated with AF.
Potassium channel genes
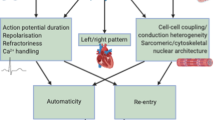
The S140G mutation in KCNQ1 led to a gain-of-function in the IKs channel complex.17, 19 The increase in this repolarizing current was hypothesized to cause more rapid repolarization of the cardiac cells, leading to a shorter effective refractory period, which in turn would leave the cells susceptible to depolarization by a subsequent electrical impulse. This could increase the risk of formation of re-entry loops which is thought to be a trigger for AF, according to the multiple wavelet theory (Figures 1a and b).20 The significance of KCNQ1 has been underlined by the many reports of gain-of-function mutations in this gene in families21, 22, 23, 24, 25, 26 and unrelated individuals with AF.27, 28, 29, 30 Similarly, mutations identified in KCNE1, KCNE2, KCNE3 and KCNE5, encoding regulatory β-subunits of Kv7.1, have been shown to exert gain-of-function effects on IKs.31, 32, 33, 34, 35, 36 The mutation E141A in KCNE4 was identified in one individual with AF and reported by Mann et al.37 to have potential effect on Ito, IKs and IKr.

(a) Rare variants in ion channel genes have been shown to produce both gain-of-function and loss-of-function, leading to both shortening (dotted line) and prolongation (dashed line) of the atrial action potential and thus the effective refractory period. Both may lead to atrial fibrillation through (b) re-entry mechanisms and (c) early and delayed afterdepolarizations. In support of this, Nielsen et al. showed that there is a J-shaped relationship between QTc interval and risk of atrial fibrillation (d). Graph reprinted with courtesy of Nielsen et al.45 EAD, early afterdepolarization; DAD, delayed afterdepolarization. A full color version of this figure is available at the Journal of Human Genetics journal online.
The other major repolarizing current in the myocardial cells is the rapidly repolarizing potassium current IKr, encoded by KCNH2. While analyzing a family with short QT syndrome and a high incidence of paroxysmal AF, Hong et al.38 identified a mutation N588K in KCNH2. Programmed electrical stimulation of the family members revealed short atrial and ventricular refractory period and inducibility of atrial and ventricular fibrillation, suggesting gain-of-function of IKr. In contrast, Mann et al.37 identified a mutation with a loss-of-function for IKr in whole-cell voltage clamp studies.
The ultra-rapid repolarizing current IKur is a particularly interesting potential candidate for AF, since it is largely atrial specific. The α-subunit is encoded by the gene KCNA5. A loss-of-function truncating mutation that eliminated the S4-S6 voltage sensor, pore region and C-terminus resulted in failure to generate an IKur current was reported by Olson et al.39 Several reports have described loss-of-function mutations,40, 41 but in contrast, both loss- and gain-of-function mutations were identified in 307 unrelated Danish individuals with early-onset lone AF.42 This implies that prolongation of the effective refractory period and the action potential duration can also cause AF. The predominant theory is that the resulting changes in Ca2+ homeostasis lead to early and/or delayed afterdepolarizations which can in turn trigger AF (Figures 1a and c).43, 44 These findings are also supported by a report from Nielsen et al.45 where they showed that both an increase or a decrease in the QTc interval duration is associated with an increased risk of AF (Figure 1d).
Sodium channel genes
The major depolarizing current in the cardiac cells is the INa, which is mediated by the sodium channel α-subunit Nav1.5 in complex with several regulatory β-subunits. Mutations in SCN5A were long known to cause Long QT syndrome type 3, but in 2005, Olson et al.46 reported a missense mutation that was associated with several cardiac phenotypes, including dilated cardiomyopathy, AF, impaired automaticity and conduction delay. Subsequently, several reports have established that mutations in SCN5A47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58 and genes encoding four regulatory β-subunits59, 60, 61, 62, 63 can cause AF.
The voltage-gated sodium channel Nav1.8, encoded by SCN10A, is expressed in sensory nerve fibers and has been thought to be primarily involved in pain mediation.64, 65 Recently, expression have also been detected in human cardiomyocytes and intracardiac neurons, especially in the conduction system,66, 67, 68, 69 and both gain- and loss-of-function mutations have been identified in two separate early-onset AF populations.70, 71 Functional studies suggested a role for Nav1.8 channels as a component of the late sodium current (INa-L) in cardiomyocytes67, 70 and SCN10A is also shown to modulate SCN5A expression.72
Genetic variation in other genes
The integrity of the conduction velocity in the cardiac tissue is largely maintained by the connexins. These proteins form gap junctions, which mediate the flow of ions between two neighboring cells, enabling the myocardium to depolarize and contract in an organized manner. Mouse models have shown that depletion of the atrial specific connexin 40, encoded by GJA5, can lead to atrial tachyarrhythmias. Gollob et al.73 described three somatic mutations in atrial cardiac tissue and one germline mutation in GJA5 in 15 individuals with sporadic, lone AF, and these were shown to impair cell–cell electrical coupling. These mutations may facilitate AF through a decrease in the conduction velocity resulting in shortening of the wavelength and increased risk of re-entry (Figure 1b). In support of this, the germline mutation has also been identified in an independent population with early-onset lone AF.74 Further, other mutations in GJA5 have been identified;75, 76, 77, 78, 79 however, atrial specific somatic mutations appear to be rare causes of AF.80
Atrial natriuretic peptide is a circulating hormone produced in the cardiac atria, important in the regulation of sodium homeostasis. A truncating frameshift mutation in NPPA, the gene encoding the precursor protein for atrial natriuretic peptide, was identified in a family with an autosomal dominant inheritance pattern of AF.18 The mutation was later shown to slow degradation of human atrial natriuretic peptide81 and to shorten the action potential duration in a rat Langendorff model.18 Both common and rare variants in NPPA have since been identified in a Chinese population.82
Although there have been a large number of mutations reported in patients with AF, there are many limitations pertaining to the findings from candidate gene studies. First, the number of genes considered is typically small. Given this limitation, candidate gene approaches are quickly being superseded by exome and genome sequencing. While such studies will provide a more comprehensive approach, they will require a robust sample size to have enough power to implicate any specific gene in the pathogenesis of AF.
Second, it remains difficult to know how often these apparent mutations may actually be natural genetic variation in the population. With the emergence of large-scale, publicly available databases such as the ExAC browser,83 which contains exome sequencing data on up to 60 000 individuals, several mutations that were thought to be rare and causal, are actually much more common in the general population than anticipated.84, 85, 86
Finally, in most cases, the functional effect of these mutations has not been evaluated. Even when a variant has been characterized, the cell lines or model systems may not faithfully recapitulate the complexity of human atrial cells.
Using Association Studies to Identify Common AF Variants
Genetic association studies involve testing whether the frequency of a genetic variant differs between cases and a control population. The most common type of genetic variation, or single-nucleotide polymorphism (SNP), is genotyped in cases and controls and tested for association with the desired phenotype. The first genome-wide association study (GWAS) was published in 2002 and described the association of a locus on chromosome 6p21 with myocardial infarction.87 Such an approach provides for a relatively unbiased method for testing SNPs throughout the whole genome. In the intervening years, GWAS have been performed on many traits and diseases. Table 2 shows all loci associated with AF through GWAS.
The first GWAS on AF was performed in an Icelandic population in 2006, analyzing ~300 000 SNPs throughout the genome. The most significantly associated SNPs were found in a region on chromosome 4q25. The SNP rs2200733 was most significantly associated with AF, a find that was replicated in populations of European and Asian ethnicity in the same study88 and later in independent European,89, 90, 91, 92, 93 African94 and Asian populations.92, 95 Similarly, association has been shown with cardioembolic stroke94, 96, 97 and PR interval,95, 98 which are both related to AF.
Fine mapping of the locus revealed two independent signals in addition to rs2200733, and the cumulative AF risk in carriers of all six risk alleles was sixfold increased.99 The surroundings of the 4q25 locus have been described as a ‘genetic desert’, with the closest gene (PITX2) located 150 000 base pairs upstream. PITX2 encodes the paired-like homeodomain transcription factor 2 and PITX2c is the major isoform expressed in the heart.100 It is active during the embryonic cardiogenesis, where it is crucial to right–left asymmetry, the suppression of the formation of a sinus node in the left atrium,101 and the formation of the myocardial sleeves in the pulmonary veins.102 The latter is interesting, because electrical impulses generated ectopically in the pulmonary myocardial sleeves are known triggers of AF. Isolation of the pulmonary veins using ablative techniques is one of the most common treatment strategies for AF.
Knockout and haploinsufficient PITX2 mice, have been shown to have a propensity to atrial arrhythmias103 and AF.104 In addition, PITX2 knockout mice hearts display a substantial decrease in cardiac Nav1.5 protein expression, which may be involved in the atrioventricular block observed in these mice.105 In humans and mice, depletion of PITX2c leads to reduced levels of mRNA encoding sodium- and potassium channel protein subunits, suggesting that this gene is an upstream transcriptional regulator of atrial electrical function.105, 106
The CHARGE-AF (Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium), which now is known as the AFGen (AF Genetics) Consortium, was established to increase the power of GWAS to identify robust associations. In 2009, the AFGen consortium reported a locus associated with AF at 16q22 in a meta-analysis of GWAS from five populations of European ethnicity.93 The same association was simultaneously reported in the Icelandic population, replicated in European and Han Chinese populations,107 and later replicated in an African-American population.94 The top SNP at this locus is intronic to ZFHX3 which encodes the zinc finger homeobox 3, a transcription factor with unknown function in cardiac tissue.
Since then, the AFGen Consortium has reported many additional loci associated with AF.108, 109 One locus was identified intronic to the gene KCNN3 encoding the calcium-activated potassium channel SK3, which is active during the repolarization phase of the cardiac action potential. Blocking SK channels has been shown to reverse action potential duration shortening in a burst-pacing model in rabbits, suggesting a link to AF. Another locus was identified on chromosome 15q24, intronic to HCN4, which encodes the hyperpolarization-activated cyclic nucleotide-gated channel carrying the funny current (If) responsible for the pacemaker activity in the sinoatrial node in the left atrium. This gene has been related to sinus node dysfunction,110, 111, 112 tachy-brady syndrome and AF,113 and recently a mutation causing a channel trafficking defect was reported in a population of early-onset AF patients.114
More recently, Kolek et al.115 set out to identify genomic modulators of rate control therapy in patients with AF; however, no variant reached genome-wide significance, probably because of small sample size. Of note, SOX5, which previously has been associated with resting heart rate and PR interval, was among the top hits.
Identifying additional AF GWAS loci
The genome-wide significance level most commonly used in GWAS is 5x10−8, resulting from Bonferroni correction of a 5% significance level for the testing of ~1 million SNPs (0.05/1 000 000). Such a correction is conservative and comes at the expense of potentially missing SNPs that are truly associated with AF. The simplest way to identify novel AF-related loci would be to increase the study sample size. In 2014, Sinner et al.116 combined genotyping in additional cases in Europeans and Japanese, eQTL mapping, and functional characterization to identify five novel AF loci.
The most significantly associated variants in both Europeans and Japanese was at chromosome 10q24, intronic to NEURL. The protein product is an E3 ubiquitin ligase with a putative RING finger domain, and it was found to interact with PITX2 protein by co-immunoprecipitation. Further, knockdown of the NEURL ortholog in zebrafish resulted in significant prolongation of the atrial action potential duration, suggesting a possible link with AF.
Interestingly, one of the loci associated with AF was identified only in Japanese and is located intronic to CUX2 on chromosome 12q24. The top AF SNP at this locus was also associated with decreased risk of ischemic stroke in the Metastroke collaboration. A separate SNP at this locus, rs1265564, that is also intronic to CUX2 was associated with type 1 diabetes in the Wellcome Trust Consortium,117 but is monomorphic in the Asians. Recently, a variant located upstream of CUX2, rs2188380, was associated with gout in a Japanese population.118 Additional variants at this locus have been suggested as susceptibility variants for coronary artery disease in Koreans and Japanese, but failed to replicate.119 Currently, the mechanistic link between CUX2 and AF remains unclear.
Target Sequencing of Genes at GWAS loci
The AF GWAS loci have been identified using common genetic variants with either increased or decreased frequency among cases versus controls. A logical next step would be to ask if rare genetic variation in the coding region of any of the genes in the loci may also be related to AF. Two studies have taken an initial foray into this approach. First, Tsai et al.120 performed next generation sequencing of nine AF GWAS genes in 20 individuals with AF. They identified six novel mutations exonic to or in the 5′ untranslated regions of four genes (PITX2, SYNE2, ZFXH3, KCNN3), and a promoter variant in PITX2 reduced luciferase activity compared to a wild-type construct. In a second report, Lin et al. sequenced 77 target gene regions from GWAS loci of complex diseases or traits, including four AF genes (PRRX1, CAV1, CAV2 and ZFHX3), in 948 individuals with and 3330 without AF. One common variant intronic to the interleukin-6 receptor gene was identified, and in rare variant analysis, deleterious variants within the PRRX1 region had a significant association with AF.121
Figure 2 shows the main pathways for atrial fibrillation identified through genetic studies.

The genes and genetic loci found to be associated with atrial fibrillation can roughly be grouped according to three main biological pathways: variants influencing cardiogenesis, cell architecture and electrical coupling and ion channel dysfunction. Gene names in black font terms genes identified through resequencing, while gray font refers to genes identified in association studies.
Clinical Application of GWAS Findings
The fourteen AF loci discovered through GWAS have opened up new potential pathways for the development of AF, yet it is natural to wonder whether this knowledge can be transferred from the bench to the bedside. In an attempt to examine this, several studies have incorporated all of the GWAS SNPs into an AF-genetic risk score (GRS). Most recently, an AF-GRS including the 12 top AF SNPs was examined in a community-based study of 27 000 Swedish individuals.122 The AF-GRS was associated with incident AF and ischemic stroke when adjusting for clinical risk factors and it identified the upper quintile of the population, with >twofold increased risk for AF and ~25% increased risk for ischemic stroke. Interestingly, the AF-GRS displayed similar magnitude as a risk factor as hypertension.
In 2013, investigators in the Women’s Genome Health Study, a cohort of more than 20 000 women, found that the addition of a GRS including the same 12 risk alleles, to a clinical AF risk score improved discrimination (C-index=0.741) and improved category-less reclassification.123 A range of genotype risk scores have also been used to examine the risk of postoperative AF, yet they have failed to improve discrimination of AF risk beyond the usual clinical AF risk factors.124, 125, 126
These efforts show that an AF-GRS improves prediction of incident AF in asymptomatic individuals.
Prediction models of AF recurrence after treatment for AF have also been evaluated. The presence of any of the two SNPs, rs2200733 and rs10033464, at 4q25 was an independent predictor of AF recurrence in a study of 184 individuals undergoing an electrical cardioversion.127 Similarly, in 195 individuals with AF who underwent AF catheter ablation, the presence of any of the same two SNPs increased risk of early AF recurrence (⩽7 days) by twofold, and late AF recurrence (3–6 months) by fourfold.128 Parvez et al.129 have reported that the ancestral allele at rs10033464 at 4q25 was an independent predictor of successful rhythm control in 478 individuals with AF treated with antiarrhythmic drugs. This finding was replicated in 198 additional individuals, but in a follow-up study, the effect was attenuated when obstructive sleep apnea was present.130 None of the prediction models described above have been implemented in clinical practice thus far. Some studies are underpowered, and most lack generalizability to broader populations; however, the most prominent reason is that the clinical value of screening individuals for AF risk by using genetic markers depend on the availability of effective interventions that can prevent AF or diminish the AF burden. In contrast to many other conditions, we do not yet have any known preventive strategies against AF, although such interventions could include antihypertensive treatment, polysomnography and more extensive electrocardiogram-monitoring. Further studies are needed to evaluate the efficacy of these or other measures, before genetic screening with the aim to identify individuals at risk for AF can be implemented in clinical guidelines.
Familial AF as a risk factor for AF; however, is a factor easily ascertained in a clinical setting without incremental risk for the patient, which has been found to increase discrimination in a prediction model for AF, most dominantly in early-onset familial AF (C-statistic 0.842–0.846).5 Familial AF is included as a clinical risk factor in the 2014 AHA/ACC/HRS Guidelines for the management of patients with AF and referral to genetic counseling at a tertiary center may be considered as a class IIb recommendation in families with multigenerational AF aggregation.131
Future Directions
The rare and common variants identified to date do not seem to fully account for the considerable heritability of AF observed in epidemiologic studies. There is a range of possible explanations for this missing heritability, including unidentified rare or common genetic variants, gene–gene interactions, gene–environment interactions or variations in copy number. For example, Ritchie et al. found a significant interaction between SNPs in the 4q25 region and rare genetic variants in known AF genes.132
In the upcoming years, it will be interesting to see the results of large-scale exome and genome sequencing for AF. As we await these results, recent findings on lipids and myocardial infarction provide a glimpse into the future. Exome sequencing of ~4000 participants in the Exome Sequencing Project identified variants in apolipoprotein C3 that reduced triglyceride levels with 39% and was protective against coronary heart disease.133 In the early-onset myocardial infarction consortium, exome sequencing revealed mutations in the low-density lipoprotein receptor and in apolipoprotein A-V that increased levels of low-density lipoprotein cholesterol and triglycerides, respectively, and increased risk of myocardial infarction.134 Although these are valuable findings in the search for new preventive treatments, these variants only explain a small fraction (0.48% and 0.28%, respectively) of the heritability of myocardial infarction.
Conclusion
Great strides have been made in the past decade in defining the heritability and genetic basis of AF. In the upcoming years, we expect the identification of further AF-related genes in larger association studies, exome sequencing and genome sequencing studies. The results will provide the foundation for further work needed to determine the role of these genes in AF and to define the molecular pathways leading to this common arrhythmia.
References
Chugh, S. S., Havmoeller, R., Narayanan, K., Singh, D., Rienstra, M., Benjamin, E. J. et al. Worldwide epidemiology of atrial fibrillation: a global burden of disease 2010 study. Circulation 129, 837–847 (2014).
Benjamin, E. J., Wolf, P. A., D’Agostino, R. B., Silbershatz, H., Kannel, W. B. & Levy, D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation 98, 946–952 (1998).
Kim, M. H., Johnston, S. S., Chu, B. -C., Dalal, M. R. & Schulman, K. L. Estimation of total incremental health care costs in patients with atrial fibrillation in the United States. Circ. Cardiovasc. Qual. Outcomes 4, 313–320 (2011).
Orgain, E. S. Uncomplicated auricular fibrillation and auricular flutter. Arch. Intern. Med. 57, 493 (1936).
Lubitz, S., Yin, X. & Fontes, J. Association between familial atrial fibrillation and risk of new-onset atrial fibrillation. JAMA 304, 2263–2269 (2010).
Arnar, D. O., Thorvaldsson, S., Manolio, T. A., Thorgeirsson, G., Kristjansson, K., Hakonarson, H. et al. Familial aggregation of atrial fibrillation in Iceland. Eur. Heart J. 27, 708–712 (2006).
Fox, C. S., Parise, H., D’Agostino, R. B. Sr ., Lloyd-Jones, D. M., Vasan, R. S., Wang, T. J. et al. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA 291, 2851–2855 (2004).
Ellinor, P. T., Yoerger, D. M., Ruskin, J. N. & MacRae, C. A. Familial aggregation in lone atrial fibrillation. Hum. Genet. 118, 179–184 (2005).
Darbar, D., Herron, K. J., Ballew, J. D., Jahangir, A., Gersh, B. J., Shen, W. K. et al. Familial atrial fibrillation is a genetically heterogeneous disorder. J. Am. Coll. Cardiol. 41, 2185–2192 (2003).
Oyen, N., Ranthe, M. F., Carstensen, L., Boyd, H. A., Olesen, M. S., Olesen, S. -P. et al. Familial Aggregation of lone atrial fibrillation in young persons. J. Am. Coll. Cardiol. 60, 917–921 (2012).
Yang, Y. -Q., Zhang, X. -L., Wang, X. -H., Tan, H. -W., Shi, H. -F., Fang, W. -Y. et al. Familial aggregation of lone atrial fibrillation in the Chinese population. Intern. Med. 49, 2385–2391 (2010).
Christophersen, I. E., Ravn, L. S., Budtz-Joergensen, E., Skytthe, A., Haunsoe, S., Svendsen, J. H. et al. Familial aggregation of atrial fibrillation: a study in Danish twins. Circ. Arrhythm. Electrophysiol. 2, 378–383 (2009).
Brugada, R., Tapscott, T., Czernuszewicz, G. Z., Marian, A. J., Iglesias, A., Mont, L. et al. Identification of a genetic locus for familial atrial fibrillation. N. Engl. J. Med. 336, 905–911 (1997).
Ellinor, P. T., Shin, J. T., Moore, R. K., Yoerger, D. M. & MacRae, C. A. Locus for atrial fibrillation maps to chromosome 6q14-16. Circulation 107, 2880–2883 (2003).
Oberti, C., Wang, L., Li, L., Dong, J., Rao, S., Du, W. et al. Genome-wide linkage scan identifies a novel genetic locus on chromosome 5p13 for neonatal atrial fibrillation associated with sudden death and variable cardiomyopathy. Circulation 110, 3753–3759 (2004).
Volders, P. G. A., Zhu, Q., Timmermans, C., Eurlings, P. M. H., Su, X., Arens, Y. H. et al. Mapping a novel locus for familial atrial fibrillation on chromosome 10p11-q21. Heart Rhythm. 4, 469–475 (2007).
Chen, Y. H., Xu, S. J., Bendahhou, S., Wang, X. L., Wang, Y., Xu, W. Y. et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science 299, 251–254 (2003).
Hodgson-Zingman, D. M., Karst, M. L., Zingman, L. V., Heublein, D. M., Darbar, D., Herron, K. J. et al. Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N. Engl. J. Med. 359, 158–165 (2008).
Restier, L., Cheng, L. & Sanguinetti, M. C. Mechanisms by which atrial fibrillation-associated mutations in the S1 domain of KCNQ1 slow deactivation of IKs channels. J. Physiol. 586, 4179–4191 (2008).
Moe, G. K. Evidence for reentry as a mechanism of cardiac arrhythmias. Rev. Physiol. Biochem. Pharmacol. 72, 55–81 (1975).
Bartos, D. C., Duchatelet, S., Burgess, D. E., Klug, D., Denjoy, I., Peat, R. et al. R231C mutation in KCNQ1 causes long QT syndrome type 1 and familial atrial fibrillation. Heart Rhythm. 8, 48–55 (2011).
Bartos, D. C., Anderson, J. B., Bastiaenen, R., Johnson, J. N., Gollob, M. H., Tester, D. J. et al. A KCNQ1 mutation causes a high penetrance for familial atrial fibrillation. J. Cardiovasc. Electrophysiol. 24, 562–569 (2013).
Das, S., Makino, S., Melman, Y. F., Shea, M. A., Goyal, S. B., Rosenzweig, A. et al. Mutation in the S3 segment of KCNQ1 results in familial lone atrial fibrillation. Heart Rhythm. 6, 1146–1153 (2009).
Otway, R., Vandenberg, J. I., Guo, G., Varghese, A., Castro, M. L., Liu, J. et al. Stretch-sensitive KCNQ1 mutation A link between genetic and environmental factors in the pathogenesis of atrial fibrillation? J. Am. Coll. Cardiol. 49, 578–586 (2007).
Ki, C. S., Jung, C. L., Kim, H. J., Baek, K. H., Park, S. J., On, Y. K. et al. A KCNQ1 mutation causes age-dependant bradycardia and persistent atrial fibrillation. Pflugers Arch. 466, 529–540 (2014).
Wang, Q., Curran, M. E., Splawski, I., Burn, T. C., Millholland, J. M., VanRaay, T. J. et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 12, 17–23 (1996).
Lundby, A., Ravn, L. S., Svendsen, J. H., Olesen, S. P. & Schmitt, N. KCNQ1 mutation Q147R is associated with atrial fibrillation and prolonged QT interval. Heart Rhythm. 4, 1532–1541 (2007).
Abraham, R. L., Yang, T., Blair, M., Roden, D. M. & Darbar, D. Augmented potassium current is a shared phenotype for two genetic defects associated with familial atrial fibrillation. J. Mol. Cell Cardiol. 48, 181–190 (2010).
Hong, K., Piper, D. R., Diaz-Valdecantos, A., Brugada, J., Oliva, A., Burashnikov, E. et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc. Res. 68, 433–440 (2005).
Hasegawa, K., Ohno, S., Ashihara, T., Itoh, H., Ding, W. G., Toyoda, F. et al. A novel KCNQ1 missense mutation identified in a patient with juvenile-onset atrial fibrillation causes constitutively open IKs channels. Heart Rhythm. 11, 67–75 (2014).
Lai, L. P., Su, M. J., Yeh, H. M., Lin, J. L., Chiang, F. T., Hwang, J. J. et al. Association of the human minK gene 38G allele with atrial fibrillation: evidence of possible genetic control on the pathogenesis of atrial fibrillation. Am. Heart J. 144, 485–490 (2002).
Yang, Y., Xia, M., Jin, Q., Bendahhou, S., Shi, J., Chen, Y. et al. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am. J. Hum. Genet. 75, 899–905 (2004).
Lundby, A., Ravn, L. S., Svendsen, J. H., Haunsoe, S., Olesen, S. -P. & Schmitt, N. KCNE3 mutation V17M identified in a patient with lone atrial fibrillation. Cell Physiol. Biochem. 21, 47–54 (2008).
Ravn, L. S., Hofman-Bang, J., Dixen, U., Larsen, S. O., Jensen, G., Haunsoe, S. et al. Relation of 97T polymorphism in KCNE5 to risk of atrial fibrillation. Am. J. Cardiol. 96, 405–407 (2005).
Olesen, M. S., Bentzen, B. H., Nielsen, J. B., Steffensen, A. B., David, J. -P. P., Jabbari, J. et al. Mutations in the potassium channel subunit KCNE1 are associated with early-onset familial atrial fibrillation. BMC Med. Genet. 13, 24 (2012).
Nielsen, J. B., Bentzen, B. H., Olesen, M. S., David, J. -P., Olesen, S. -P., Haunsø, S. et al. Gain-of-function mutations in potassium channel subunit KCNE2 associated with early-onset lone atrial fibrillation. Biomark Med. 8, 557–570 (2014).
Mann, S. A., Otway, R., Guo, G., Soka, M., Karlsdotter, L., Trivedi, G. et al. Epistatic effects of potassium channel variation on cardiac repolarization and atrial fibrillation risk. J. Am. Coll. Cardiol. 59, 1017–1025 (2012).
Hong, K., Bjerregaard, P., Gussak, I. & Brugada, R. Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J. Cardiovasc. Electrophysiol. 16, 394–396 (2005).
Olson, T. M., Alekseev, A. E., Liu, X. K., Park, S., Zingman, L. V., Bienengraeber, M. et al. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum. Mol. Genet. 15, 2185–2191 (2006).
Yang, T., Yang, P., Roden, D. M. & Darbar, D. Novel KCNA5 mutation implicates tyrosine kinase signaling in human atrial fibrillation. Heart Rhythm. 7, 1246–1252 (2010).
Yang, Y., Li, J., Lin, X., Hong, K., Wang, L., Liu, J. et al. Novel KCNA5 loss-of-function mutations responsible for atrial fibrillation. J. Hum. Genet. 54, 277–283 (2009).
Christophersen, I. E., Olesen, M. S., Liang, B., Andersen, M. N., Larsen, A. P., Nielsen, J. B. et al. Genetic variation in KCNA5: impact on the atrial-specific potassium current IKur in patients with lone atrial fibrillation. Eur. Heart J. 34, 1517–1525 (2013).
Li, D., Fareh, S., Leung, T. K. & Nattel, S. Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation 100, 87–95 (1999).
Yeh, Y. H., Wakili, R., Qi, X. Y., Chartier, D., Boknik, P., Kaab, S. et al. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ. Arrhythm. Electrophysiol 1, 93–102 (2008).
Nielsen, J. B., Graff, C., Pietersen, A., Lind, B., Struijk, J. J., Olesen, M. S. et al. J-shaped association between QTc interval duration and the risk of atrial fibrillation: results from the Copenhagen ECG Study. J. Am. Coll. Cardiol. 61, 2557–2564 (2013).
Olson, T. M., Michels, V. V., Ballew, J. D., Reyna, S. P., Karst, M. L., Herron, K. J. et al. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA 293, 447–454 (2005).
Chen, L. Y., Ballew, J. D., Herron, K. J., Rodeheffer, R. J. & Olson, T. M. A common polymorphism in SCN5A is associated with lone atrial fibrillation. Clin. Pharmacol. Ther. 81, 35–41 (2007).
Makiyama, T., Akao, M., Shizuta, S., Doi, T., Nishiyama, K., Oka, Y. et al. A novel SCN5A gain-of-function mutation M1875T associated with familial atrial fibrillation. J. Am. Coll. Cardiol. 52, 1326–1334 (2008).
Watanabe, H., Yang, T., Stroud, D. M., Lowe, J. S., Harris, L., Atack, T. C. et al. Striking in vivo phenotype of a disease-associated human SCN5A mutation producing minimal changes in vitro. Circulation 124, 1001–1011 (2011).
Olesen, M. S., Yuan, L., Liang, B., Holst, A. G., Nielsen, N., Nielsen, J. B. et al. High prevalence of long QT syndrome associated scn5a variants in patients with early-onset lone atrial fibrillation. Circ. Cardiovasc. Genet. 5, 450–459 (2012).
Robyns, T., Nuyens, D., Van Casteren, L., Corveleyn, A., De Ravel, T., Heidbuchel, H. et al. Reduced penetrance and variable expression of SCN5A mutations and the importance of co-inherited genetic variants: Case Report and Review of the Literature. Indian Pacing Electrophysiol. J. 14, 133–149 (2014).
Amin, A. S., Boink, G. J. J., Atrafi, F., Spanjaart, A. M., Asghari-Roodsari, A., Molenaar, R. J. et al. Facilitatory and inhibitory effects of SCN5A mutations on atrial fibrillation in Brugada syndrome. Europace 13, 968–975 (2011).
Ziyadeh-Isleem, A., Clatot, J., Duchatelet, S., Gandjbakhch, E., Denjoy, I., Hidden-Lucet, F. et al. A truncating SCN5A mutation combined with genetic variability causes sick sinus syndrome and early atrial fibrillation. Heart Rhythm. 11, 1015–1023 (2014).
Shoemaker, M. B., Bollmann, A., Lubitz, S. A., Ueberham, L., Saini, H., Montgomery, J. et al. Common Genetic Variants and Response to Atrial Fibrillation Ablation. Circ. Arrhythm. Electrophysiol. 8, 296–302 (2015).
Li, Q., Huang, H., Liu, G., Lam, K., Rutberg, J., Green, M. S. et al. Gain-of-function mutation of Nav1.5 in atrial fibrillation enhances cellular excitability and lowers the threshold for action potential firing. Biochem. Biophys. Res. Commun. 380, 132–137 (2009).
Laitinen-Forsblom, P. J., Mäkynen, P., Mäkynen, H., Yli-Mäyry, S., Virtanen, V., Kontula, K. et al. SCN5A mutation associated with cardiac conduction defect and atrial arrhythmias. J. Cardiovasc. Electrophysiol. 17, 480–485 (2006).
Ellinor, P. T., Nam, E. G., Shea, M. A., Milan, D. J., Ruskin, J. N. & MacRae, C. A. Cardiac sodium channel mutation in atrial fibrillation. Heart Rhythm. 5, 99–105 (2008).
Darbar, D., Kannankeril, P. & Donahue, B. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation 117, 1927–1935 (2008).
Olesen, M. S., Holst, A. G., Svendsen, J. H., Haunsø, S. & Tfelt-Hansen, J. SCN1Bb R214Q found in 3 patients: 1 with Brugada syndrome and 2 with lone atrial fibrillation. Heart Rhythm. 9, 770–773 (2012).
Watanabe, H., Darbar, D., Kaiser, D. W., Jiramongkolchai, K., Chopra, S., Donahue, B. S. et al. Mutations in sodium channel beta1- and beta2-subunits associated with atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2, 268–275 (2009).
Olesen, M. S., Jespersen, T., Nielsen, J. B., Liang, B., Moller, D. V., Hedley, P. et al. Mutations in sodium channel beta-subunit SCN3B are associated with early-onset lone atrial fibrillation. Cardiovasc. Res. 89, 786–793 (2011).
Wang, P., Yang, Q., Wu, X., Yang, Y., Shi, L., Wang, C. et al. Functional dominant-negative mutation of sodium channel subunit gene SCN3B associated with atrial fibrillation in a Chinese GeneID population. Biochem. Biophys. Res. Commun. 398, 98–104 (2010).
Li, R. -G., Wang, Q., Xu, Y. -J., Zhang, M., Qu, X. -K., Liu, X. et al. Mutations of the SCN4B-encoded sodium channel β4 subunit in familial atrial fibrillation. Int. J. Mol. Med. 32, 144–150 (2013).
Souslova, V. A., Fox, M., Wood, J. N. & Akopian, A. N. Cloning and characterization of a mouse sensory neuron tetrodotoxin-resistant voltage-gated sodium channel gene, Scn10a. Genomics 41, 201–209 (1997).
Rabert, D. K., Koch, B. D., Ilnicka, M., Obernolte, R. A., Naylor, S. L., Herman, R. C. et al. A tetrodotoxin-resistant voltage-gated sodium channel from human dorsal root ganglia, hPN3/SCN10A. Pain 78, 107–114 (1998).
Facer, P., Punjabi, P. P., Abrari, A., Kaba, R. A., Severs, N. J., Chambers, J. et al. Localisation of SCN10A gene product Na(v)1.8 and novel pain-related ion channels in human heart. Int. Heart J. 52, 146–152 (2011).
Yang, T., Atack, T. C., Stroud, D. M., Zhang, W., Hall, L. & Roden, D. M. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ. Res. 111, 322–332 (2012).
Verkerk, A. O., Remme, C. A., Schumacher, C. A., Scicluna, B. P., Wolswinkel, R., De Jonge, B. et al. Functional NaV1.8 channels in intracardiac neurons: the link between SCN10A and cardiac electrophysiology. Circ. Res. 111, 333–343 (2012).
Chambers, J. C., Zhao, J., Terracciano, C. M., Bezzina, C. R., Zhang, W., Kaba, R. et al. Genetic variation in SCN10A influences cardiac conduction. Nat. Genet. 42, 149–152 (2010).
Savio-Galimberti, E., Weeke, P., Muhammad, R., Blair, M., Ansari, S., Short, L. et al. SCN10A/Nav1.8 modulation of peak and late sodium currents in patients with early onset atrial fibrillation. Cardiovasc. Res. 104, 355–363 cvu170– (2014).
Jabbari, J., Olesen, M. S., Yuan, L., Nielsen, J. B., Liang, B., Macri, V. et al. Common and rare variants in SCN10A modulate the risk of atrial fibrillation. Circ. Cardiovasc. Genet. 8, 64–73 (2014).
Van Den Boogaard, M., Smemo, S., Burnicka-Turek, O., Arnolds, D. E., Van De Werken, H. J. G., Klous, P. et al. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J. Clin. Invest. 124, 1844–1852 (2014).
Gollob, M. H., Jones, D. L., Krahn, A. D., Danis, L., Gong, X. Q., Shao, Q. et al. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N. Engl. J. Med. 354, 2677–2688 (2006).
Christophersen, I. E., Holmegard, H. N., Jabbari, J., Sajadieh, A., Haunso, S., Tveit, A. et al. Rare variants in GJA5 are associated with early-onset lone atrial fibrillation. Can. J. Cardiol. 29, 111–116 (2013).
Yang, Y. Q., Zhang, X. L., Wang, X. H., Tan, H. W., Shi, H. F., Jiang, W. F. et al. Connexin40 nonsense mutation in familial atrial fibrillation. Int. J. Mol. Med. 26, 605–610 (2010).
Yang, Y. -Q., Liu, X., Zhang, X. -L., Wang, X. -H., Tan, H. -W., Shi, H. -F. et al. Novel connexin40 missense mutations in patients with familial atrial fibrillation. Europace 12, 1421–1427 (2010).
Sun, Y., Yang, Y. Q., Gong, X. Q., Wang, X. H., Li, R. G., Tan, H. W. et al. Novel Germline GJA5/Connexin40 mutations associated with lone atrial fibrillation impair gap junctional intercellular communication. Hum. Mutat. 34, 603–609 (2013).
Sun, Y., Tong, X., Chen, H., Huang, T., Shao, Q., Huang, W. et al. An atrial-fibrillation-linked connexin40 mutant is retained in the endoplasmic reticulum and impairs the function of atrial gap-junction channels. Dis. Model Mech. 7, 561–569 (2014).
Shi, H. F., Yang, J. F., Wang, Q., Li, R. G., Xu, Y. J., Qu, X. K. et al. Prevalence and spectrum of GJA5 mutations associated with lone atrial fibrillation. Mol. Med. Rep. 7, 767–774 (2013).
Roberts, J. D., Longoria, J., Poon, A., Gollob, M. H., Dewland, T. A., Kwok, P. -Y. et al. Targeted deep sequencing reveals no evidence for somatic mosaicism in atrial fibrillation. Circ. Cardiovasc. Genet. 8, 50–57 (2014).
Dickey, D. M., Yoder, A. R. & Potter, L. R. A familial mutation renders atrial natriuretic peptide resistant to proteolytic degradation. J. Biol. Chem. 284, 19196–19202 (2009).
Ren, X., Xu, C., Zhan, C., Yang, Y., Shi, L., Wang, F. et al. Identification of NPPA variants associated with atrial fibrillation in a Chinese GeneID population. Clin. Chim. Acta 411, 481–485 (2010).
Exome Aggregation Consortium (ExAC), Cambridge, MA. URL http://exac.broadinstitute.org.
Andreasen, C., Nielsen, J. B., Refsgaard, L., Holst, A. G., Christensen, A. H., Andreasen, L. et al. New population-based exome data are questioning the pathogenicity of previously cardiomyopathy-associated genetic variants. Eur. J. Hum. Genet. 21, 918–928 (2013).
Andreasen, L., Nielsen, J. B., Darkner, S., Christophersen, I. E., Jabbari, J., Refsgaard, L. et al. Brugada syndrome risk loci seem protective against atrial fibrillation. Eur. J. Hum. Genet. 22, 1357–1361 (2014).
Risgaard, B., Jabbari, R., Refsgaard, L., Holst, A., Haunsø, S., Sadjadieh, A. et al. High prevalence of genetic variants previously associated with brugada syndrome in new exome data. Clin. Genet. 84, 489–495 (2013).
Ozaki, K., Ohnishi, Y., Iida, A., Sekine, A., Yamada, R., Tsunoda, T. et al. Functional SNPs in the lymphotoxin-alpha gene that are associated with susceptibility to myocardial infarction. Nat. Genet. 32, 650–654 (2002).
Gudbjartsson, D. F., Arnar, D. O., Helgadottir, A., Gretarsdottir, S., Holm, H., Sigurdsson, A. et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature 448, 353–357 (2007).
Olesen, M. S., Holst, A. G., Jabbari, J., Nielsen, J. B., Christophersen, I. E., Sajadieh, A. et al. Genetic loci on chromosomes 4q25, 7p31, and 12p12 are associated with onset of lone atrial fibrillation before the age of 40 years. Can. J. Cardiol. 28, 191–195 (2012).
Viviani Anselmi, C., Novelli, V., Roncarati, R., Malovini, A., Bellazzi, R., Bronzini, R. et al. Association of rs2200733 at 4q25 with atrial flutter/fibrillation diseases in an Italian population. Heart 94, 1394–1396 (2008).
Kääb, S., Darbar, D., van Noord, C., Dupuis, J., Pfeufer, A., Newton-Cheh, C. et al. Large scale replication and meta-analysis of variants on chromosome 4q25 associated with atrial fibrillation. Eur. Heart J. 30, 813–819 (2009).
Lubitz, S. A., Lunetta, K. L., Lin, H., Arking, D. E., Trompet, S., Li, G. et al. Novel genetic markers associate with atrial fibrillation risk in Europeans and Japanese. J. Am. Coll. Cardiol. 63, 1200–1210 (2014).
Benjamin, E. J., Rice, K. M., Arking, D. E., Pfeufer, A., Noord, C., van, Smith, A. V. et al. Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry. Nat. Genet. 41, 879–881 (2009).
Delaney, J. T., Jeff, J. M., Brown, N. J., Pretorius, M., Okafor, H. E., Darbar, D. et al. Characterization of genome-wide association-identified variants for atrial fibrillation in African Americans. PLoS One 7, e32338 (2012).
Goodloe, A. H., Herron, K. J. & Olson, T. M. Uncovering an intermediate phenotype associated with rs2200733 at 4q25 in lone atrial fibrillation. Am. J. Cardiol. 107, 1802–1805 (2011).
Shi, L., Li, C., Wang, C., Xia, Y., Wu, G., Wang, F. et al. Assessment of association of rs2200733 on chromosome 4q25 with atrial fibrillation and ischemic stroke in a Chinese Han population. Hum. Genet. 126, 843–849 (2009).
Gretarsdottir, S., Thorleifsson, G., Manolescu, A., Styrkarsdottir, U., Helgadottir, A., Gschwendtner, A. et al. Risk variants for atrial fibrillation on chromosome 4q25 associate with ischemic stroke. Ann. Neurol. 64, 402–409 (2008).
Kolek, M. J., Parvez, B., Muhammad, R., Shoemaker, M. B., Blair, M. A., Stubblefield, T. et al. A common variant on chromosome 4q25 is associated with prolonged PR interval in subjects with and without atrial fibrillation. Am. J. Cardiol. 113, 309–313 (2014).
Lubitz, S. A., Sinner, M. F., Lunetta, K. L., Makino, S., Pfeufer, A., Rahman, R. et al. Independent susceptibility markers for atrial fibrillation on chromosome 4q25. Circulation 122, 976–984 (2010).
Franco, D., Chinchilla, A. & Aranega, A. E. Transgenic insights linking pitx2 and atrial arrhythmias. Front. Physiol. 3, 206 (2012).
Mommersteeg, M. T. M., Hoogaars, W. M. H., Prall, O. W. J., de Gier-de Vries, C., Wiese, C., Clout, D. E. W. et al. Molecular pathway for the localized formation of the sinoatrial node. Circ. Res. 100, 354–362 (2007).
Mommersteeg, M. T. M., Brown, N. A., Prall, O. W. J., de Gier-de, V. C., Harvey, R. P., Moorman, A. F. M. et al. Pitx2c and Nkx2-5 are required for the formation and identity of the pulmonary myocardium. Circ. Res. 101, 902–909 (2007).
Wang, J., Klysik, E., Sood, S., Johnson, R. L., Wehrens, X. H. & Martin, J. F. Pitx2 prevents susceptibility to atrial arrhythmias by inhibiting left-sided pacemaker specification. Proc. Natl Acad. Sci. USA 107, 9753–9758 (2010).
Kirchhof, P., Kahr, P. C., Kaese, S., Piccini, I., Vokshi, I., Scheld, H. H. et al. PITX2c is expressed in the adult left atrium, and reducing Pitx2c expression promotes atrial fibrillation inducibility and complex changes in gene expression. Circ. Cardiovasc. Genet. 4, 123–133 (2011).
Chinchilla, A., Daimi, H., Lozano-Velasco, E., Dominguez, J. N., Caballero, R., Delpon, E. et al. PITX2 insufficiency leads to atrial electrical and structural remodeling linked to arrhythmogenesis. Circ. Cardiovasc. Genet. 4, 269–279 (2011).
Tao, Y., Zhang, M., Li, L., Bai, Y., Zhou, Y., Moon, A. M. et al. Pitx2, an atrial fibrillation predisposition gene, directly regulates ion transport and intercalated disc genes. Circ. Cardiovasc. Genet. 7, 23–32 (2014).
Gudbjartsson, D. F., Holm, H., Gretarsdottir, S., Thorleifsson, G., Walters, G. B., Thorgeirsson, G. et al. A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat. Genet. 41, 876–878 (2009).
Ellinor, P. T., Lunetta, K. L., Glazer, N. L., Pfeufer, A., Alonso, A., Chung, M. K. et al. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat. Genet. 42, 240–244 (2010).
Ellinor, P. T., Lunetta, K. L., Albert, C. M., Glazer, N. L., Ritchie, M. D., Smith, A. V. et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat. Genet. 44, 670–675 (2012).
Schulze-Bahr, E., Neu, A., Friederich, P., Kaupp, U. B., Breithardt, G., Pongs, O. et al. Pacemaker channel dysfunction in a patient with sinus node disease. J. Clin. Invest. 111, 1537–1545 (2003).
Ueda, K., Nakamura, K., Hayashi, T., Inagaki, N., Takahashi, M., Arimura, T. et al. Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J. Biol. Chem. 279, 27194–27198 (2004).
Milanesi, R., Baruscotti, M., Gnecchi-Ruscone, T. & DiFrancesco, D. Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N. Engl. J. Med. 354, 151–157 (2006).
Duhme, N., Schweizer, P. A., Thomas, D., Becker, R., Schröter, J., Barends, T. R. M. et al. Altered HCN4 channel C-linker interaction is associated with familial tachycardia-bradycardia syndrome and atrial fibrillation. Eur. Heart J. 34, 2768–2775 (2013).
Macri, V., Mahida, S. N., Zhang, M. L., Sinner, M. F., Dolmatova, E. V., Tucker, N. R. et al. A novel trafficking-defective HCN4 mutation is associated with early-onset atrial fibrillation. Heart Rhythm. 11, 1055–1062 (2014).
Kolek, M. J., Edwards, T. L., Muhammad, R., Balouch, A., Shoemaker, M. B., Blair, M. A. et al. A genome-wide association study to identify genomic modulators of rate control therapy in patients with atrial fibrillation. Am. J. Cardiol. 114, 593–600 (2014).
Sinner, M. F., Tucker, N. R., Lunetta, K. L., Ozaki, K., Smith, J. G., Trompet, S. et al. Integrating genetic, transcriptional, and functional analyses to identify 5 novel genes for atrial fibrillation. Circulation 130, 1225–1235 (2014).
Huang, J., Ellinghaus, D., Franke, A., Howie, B. & Li, Y. 1000 Genomes-based imputation identifies novel and refined associations for the Wellcome Trust Case Control Consortium phase 1 Data. Eur. J. Hum. Genet. 20, 801–805 (2012).
Matsuo, H., Yamamoto, K., Nakaoka, H., Nakayama, A., Sakiyama, M., Chiba, T. et al. Genome-wide association study of clinically defined gout identifies multiple risk loci and its association with clinical subtypes. Ann Rheum. Dis. pii:annrheumdis-2014-206191. doi:10.1136/annrheumdis-2014-206191 (2015).
Lee, J. -Y., Lee, B. -S., Shin, D. -J., Woo Park, K., Shin, Y. -A., Joong Kim, K. et al. A genome-wide association study of a coronary artery disease risk variant. J. Hum. Genet. 58, 120–126 (2013).
Tsai, C. -T., Hsieh, C. -S., Chang, S. -N., Chuang, E. Y., Juang, J. -M. J., Lin, L. -Y. et al. Next-generation sequencing of nine atrial fibrillation candidate genes identified novel de novo mutations in patients with extreme trait of atrial fibrillation. J. Med. Genet. 52, 28–36 (2015).
Lin, H., Sinner, M. F., Brody, J. A., Arking, D. E., Lunetta, K. L., Rienstra, M. et al. Targeted sequencing in candidate genes for atrial fibrillation: the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Targeted Sequencing Study. Heart Rhythm. 11, 452–457 (2014).
Tada, H., Shiffman, D., Smith, J. G., Sjögren, M., Lubitz, S. A., Ellinor, P. T. et al. Twelve-single nucleotide polymorphism genetic risk score identifies individuals at increased risk for future atrial fibrillation and stroke. Stroke 45, 2856–2862 (2014).
Everett, B. M., Cook, N. R., Conen, D., Chasman, D. I., Ridker, P. M. & Albert, C. M. Novel genetic markers improve measures of atrial fibrillation risk prediction. Eur Heart J. 34, 2243–2251 (2013).
Kolek, M. J., Muehlschlegel, J. D., Bush, W. S., Parvez, B., Murray, K. T., Stein, C. M. et al. A Combined Genetic and Clinical Risk Prediction Model for Postoperative Atrial Fibrillation. Circ. Arrhythm. Electrophysiol 8, 25–31 (2015).
Body, S. C., Collard, C. D., Shernan, S. K., Fox, A. A., Liu, K. -Y., Ritchie, M. D. et al. Variation in the 4q25 chromosomal locus predicts atrial fibrillation after coronary artery bypass graft surgery. Circ. Cardiovasc. Genet. 2, 499–506 (2009).
Virani, S. S., Brautbar, A., Lee, V. V., Elayda, M., Sami, S., Nambi, V. et al. Usefulness of single nucleotide polymorphism in chromosome 4q25 to predict in-hospital and long-term development of atrial fibrillation and survival in patients undergoing coronary artery bypass grafting. Am. J. Cardiol. 107, 1504–1509 (2011).
Parvez, B., Shoemaker, M. B., Muhammad, R., Richardson, R., Jiang, L., Blair, M. A. et al. Common genetic polymorphism at 4q25 locus predicts atrial fibrillation recurrence after successful cardioversion. Heart Rhythm. 10, 849–855 (2013).
Husser, D., Adams, V., Piorkowski, C., Hindricks, G. & Bollmann, A. Chromosome 4q25 variants and atrial fibrillation recurrence after catheter ablation. J. Am. Coll. Cardiol. 55, 747–753 (2010).
Parvez, B., Vaglio, J., Rowan, S., Muhammad, R., Kucera, G., Stubblefield, T. et al. Symptomatic response to antiarrhythmic drug therapy is modulated by a common single nucleotide polymorphism in atrial fibrillation. J. Am. Coll. Cardiol. 60, 539–545 (2012).
Goyal, S. K., Wang, L., Upender, R., Darbar, D. & Monahan, K. Severity of obstructive sleep apnea influences the effect of genotype on response to anti-arrhythmic drug therapy for atrial fibrillation. J. Clin. Sleep Med. 10, 503–507 (2014).
January, C. T., Wann, L. S., Alpert, J. S., Calkins, H., Cleveland, J. C., Cigarroa, J. E. et al. 2014 AHA/ACC/HRS Guideline for the Management of Patients with Atrial Fibrillation: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J. Am. Coll. Cardiol. 64, 2246–2280 (2014).
Ritchie, M. D. M., Rowan, S., Kucera, G., Stubblefield, T., Blair, M., Carter, S. et al. Chromosome 4q25 variants are genetic modifiers of rare ion channel mutations associated with familial atrial fibrillation. J Am Coll Cardiol. 60, 1173–1181 (2012).
Crosby, J., Peloso, G. M., Auer, P. L., Crosslin, D. R., Stitziel, N. O., Lange, L. A. et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 371, 22–31 (2014).
Do, R., Stitziel, N. O., Won, H. -H., Jørgensen, A. B., Duga, S., Angelica Merlini, P. et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature 518, 102–106 (2014).
Olson, T. M., Alekseev, A. E., Moreau, C., Liu, X. K., Zingman, L. V., Miki, T. et al. KATP channel mutation confers risk for vein of Marshall adrenergic atrial fibrillation. Nat Clin Pract Cardiovasc Med. 4, 110–116 (2007).
Olesen, M. S., Refsgaard, L., Holst, A. G., Larsen, A. P., Grubb, S., Haunsø, S. et al. A novel KCND3 gain-of-function mutation associated with early-onset of persistent lone atrial fibrillation. Cardiovasc Res. 98, 488–495 (2013).
Sinner, M. F., Pfeufer, A., Akyol, M., Beckmann, B. M., Hinterseer, M., Wacker, A. et al. The non-synonymous coding IKr-channel variant KCNH2-K897T is associated with atrial fibrillation: results from a systematic candidate gene-based analysis of KCNH2 (HERG). Eur. Heart J. 29, 907–914 (2008).
Deo, M., Ruan, Y., Pandit, S. V., Shah, K., Berenfeld, O., Blaufox, A. et al. KCNJ2 mutation in short QT syndrome 3 results in atrial fibrillation and ventricular proarrhythmia. Proc. Natl Acad. Sci. USA. 110, 4291–4296 (2013).
Xia, M., Jin, Q., Bendahhou, S., He, Y., Larroque, M. M., Chen, Y. et al. A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem. Biophys. Res. Commun. 332, 1012–1019 (2005).
Calloe, K., Ravn, L. S., Schmitt, N., Sui, J. L., Duno, M., Haunso, S. et al. Characterizations of a loss-of-function mutation in the Kir3.4 channel subunit. Biochem. Biophys. Res. Commun. 364, 889–895 (2007).
Delaney, J. T., Muhammad, R., Blair, M. A., Kor, K., Fish, F. A., Roden, D. M. et al. A KCNJ8 mutation associated with early repolarization and atrial fibrillation. Europace 14, 1428–1432 (2012).
Jiang, J. Q., Shen, F. F., Fang, W. Y., Liu, X. & Yang, Y. Q. Novel GATA4 mutations in lone atrial fibrillation. Int. J. Mol. Med. 28, 1025–1032 (2011).
Wang, J., Sun, Y. -M. & Yang, Y. -Q. Mutation spectrum of the GATA4 gene in patients with idiopathic atrial fibrillation. Mol. Biol. Rep. 39, 8127–8135 (2012).
Yang, Y. -Q., Wang, M. -Y., Zhang, X. -L., Tan, H. -W., Shi, H. -F., Jiang, W. -F. et al. GATA4 loss-of-function mutations in familial atrial fibrillation. Clin. Chim. Acta 412, 1825–1830 (2011).
Posch, M. G., Boldt, L. H., Polotzki, M., Richter, S., Rolf, S., Perrot, A. et al. Mutations in the cardiac transcription factor GATA4 in patients with lone atrial fibrillation. Eur. J. Med. Genet. 53, 201–203 (2010).
Gu, J. -Y., Xu, J. -H., Yu, H. & Yang, Y. -Q. Novel GATA5 loss-of-function mutations underlie familial atrial fibrillation. Clinics (Sao Paulo) 67, 1393–1399 (2012).
Wang, X. H., Huang, C. X., Wang, Q., Li, R. G., Xu, Y. J., Liu, X. et al. A novel GATA5 loss-of-function mutation underlies lone atrial fibrillation. Int. J. Mol. Med. 31, 43–50 (2013).
Yang, Y. -Q., Wang, J., Wang, X. -H., Wang, Q., Tan, H. -W., Zhang, M. et al. Mutational spectrum of the GATA5 gene associated with familial atrial fibrillation. Int. J. Cardiol. 157, 305–307 (2012).
Yang, Y. Q., Li, L., Wang, J., Zhang, X. L., Li, R. G., Xu, Y. J. et al. GATA6 loss-of-function mutation in atrial fibrillation. Eur. J. Med. Genet. 55, 520–526 (2012).
Li, J., Liu, W. -D., Yang, Z. -L. & Yang, Y. -Q. Novel GATA6 loss-of-function mutation responsible for familial atrial fibrillation. Int. J. Mol. Med. 30, 783–790 (2012).
Yang, Y. -Q., Wang, X. -H., Tan, H. -W., Jiang, W. -F., Fang, W. -Y. & Liu, X. Prevalence and spectrum of GATA6 mutations associated with familial atrial fibrillation. Int. J. Cardiol. 155, 494–496 (2012).
Thibodeau, I. L., Xu, J., Li, Q., Liu, G., Lam, K., Veinot, J. P. et al. Paradigm of genetic mosaicism and lone atrial fibrillation: physiological characterization of a connexin 43-deletion mutant identified from atrial tissue. Circulation 122, 236–244 (2010).
Müller, I. I., Melville, D. B., Tanwar, V., Rybski, W. M., Mukherjee, A., Shoemaker, M. B. et al. Functional modeling in zebrafish demonstrates that the atrial-fibrillation-associated gene GREM2 regulates cardiac laterality, cardiomyocyte differentiation and atrial rhythm. Dis. Model. Mech. 6, 332–341 (2013).
Beavers, D. L., Wang, W., Ather, S., Voigt, N., Garbino, A., Dixit, S. S. et al. Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization. J. Am. Coll. Cardiol. 62, 2010–2019 (2013).
Saj, M., Dabrowski, R., Labib, S., Jankowska, A., Szperl, M., Broda, G. et al. Variants of the lamin A/C (LMNA) gene in non-valvular atrial fibrillation patients: a possible pathogenic role of the Thr528Met mutation. Mol. Diagn. Ther. 16, 99–107 (2012).
Brauch, K. M., Chen, L. Y. & Olson, T. M. Comprehensive Mutation Scanning of LMNA in 268 Patients With Lone Atrial Fibrillation. Am. J. Cardiol. 103, 1426–1428 (2009).
Huang, R. -T., Xue, S., Xu, Y. -J., Zhou, M. & Yang, Y. -Q. A novel NKX2.5 loss-of-function mutation responsible for familial atrial fibrillation. Int. J. Mol. Med. 31, 1119–1126 (2013).
Xie, W. -H., Chang, C., Xu, Y. -J., Li, R. -G., Qu, X. -K., Fang, W. -Y. et al. Prevalence and spectrum of Nkx2.5 mutations associated with idiopathic atrial fibrillation. Clinics (Sao Paulo) 68, 777–784 (2013).
Wang, J., Zhang, D. -F., Sun, Y. -M., Li, R. -G., Qiu, X. -B., Qu, X. -K. et al. NKX2-6 mutation predisposes to familial atrial fibrillation. Int. J. Mol. Med. 34, 1581–1590 (2014).
Zhang, X., Chen, S., Yoo, S., Chakrabarti, S., Zhang, T., Ke, T. et al. Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell 135, 1017–1027 (2008).
Zhou, Y. -M., Zheng, P. -X., Yang, Y. -Q., Ge, Z. -M. & Kang, W. -Q. A novel PITX2c loss-of-function mutation underlies lone atrial fibrillation. Int. J. Mol. Med. 32, 827–834 (2013).
Zhabyeyev, P., Hiess, F., Wang, R., Liu, Y., Wayne Chen, S. R. & Oudit, G. Y. S4153R is a gain-of-function mutation in the cardiac Ca(2+) release channel ryanodine receptor associated with catecholaminergic polymorphic ventricular tachycardia and paroxysmal atrial fibrillation. Can. J. Cardiol. 29, 993–996 (2013).
Acknowledgements
IEC is supported by a mobility grant from the Research Council of Norway. PTE is supported by grants from the National Institutes of Health (1RO1HL092577, 1K24HL105780), an Established Investigator Award from the American Heart Association (13EIA14220013) and the Fondation Leducq (14CVD01).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Christophersen, I., Ellinor, P. Genetics of atrial fibrillation: from families to genomes. J Hum Genet 61, 61–70 (2016). https://doi.org/10.1038/jhg.2015.44
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.44
This article is cited by
-
Pathophysiology and clinical relevance of atrial myopathy
Basic Research in Cardiology (2024)
-
The network of cardiac KIR2.1: its function, cellular regulation, electrical signaling, diseases and new drug avenues
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
Pedigree analysis of atrial fibrillation in Irish wolfhounds supports a high heritability with a dominant mode of inheritance
Canine Genetics and Epidemiology (2019)
-
Identification of atrial fibrillation associated genes and functional non-coding variants
Nature Communications (2019)
-
Using iPSC Models to Probe Regulation of Cardiac Ion Channel Function
Current Cardiology Reports (2018)
