
Abstract
In humans, narcolepsy with cataplexy (narcolepsy) is a sleep disorder that is characterized by sleepiness, cataplexy and rapid eye movement (REM) sleep abnormalities. Narcolepsy is caused by a reduction in the number of neurons that produce hypocretin (orexin) neuropeptide. Both genetic and environmental factors contribute to the development of narcolepsy.
Rare and large copy number variations (CNVs) reportedly play a role in the etiology of a number of neuropsychiatric disorders. Narcolepsy is considered a neurological disorder; therefore, we sought to investigate any possible association between rare and large CNVs and human narcolepsy. We used DNA microarray data and a CNV detection software application, PennCNV-Affy, to detect CNVs in 426 Japanese narcoleptic patients and 562 healthy individuals. Overall, we found a significant enrichment of rare and large CNVs (frequency ⩽1%, size ⩾100 kb) in the patients (case–control ratio of CNV count=1.54, P=5.00 × 10−4). Next, we extended a region-based association analysis by including CNVs with its size ⩾30 kb. Rare and large CNVs in PARK2 region showed a significant association with narcolepsy. Four patients were assessed to carry duplications of the gene region, whereas no controls carried the duplication, which was further confirmed by quantitative PCR assay. This duplication was also found in 2 essential hypersomnia (EHS) patients out of 171 patients. Furthermore, a pathway analysis revealed enrichments of gene disruptions by rare and large CNVs in immune response, acetyltransferase activity, cell cycle regulation and regulation of cell development. This study constitutes the first report on the risk association between multiple rare and large CNVs and the pathogenesis of narcolepsy. In the future, replication studies are needed to confirm the associations.
Similar content being viewed by others


Introduction
In humans, narcolepsy is characterized by excessive daytime sleepiness, cataplexy (sudden loss of muscle tone in response to strong emotion) and pathological manifestations of rapid eye movement (REM) sleep—including hypnagogic hallucinations, sleep paralysis and sleep-onset REM sleep. Narcolepsy usually begins during adolescence, and it affects 0.16–0.18% of the general population in Japan and 0.02–0.06% of the population in the United States and Europe; men and women are equally affected.1, 2 The relative risk of narcolepsy in first-degree family members of patients with narcolepsy is 10- to 40-fold higher than that in the general population.1
Narcolepsy is a multifactorial disease, and genetic variations at multiple loci are associated with the condition. Initially, narcolepsy was shown to be strongly associated with a human leukocyte antigen (HLA) class II allele, specifically HLA-DQB1*06:02.3, 4, 5, 6, 7 This HLA variant is thought to be necessary, but not sufficient, for the development of narcolepsy.7 Recent genome-wide association studies have identified several narcolepsy susceptibility loci. An association between narcolepsy in Japanese patients and one single-nucleotide polymorphism (SNP) located between CPT1B (carnitine palmitoyltransferase 1B) and CHKB (choline kinase-β) indicated a new pathogenic mechanism that is related to fatty acid oxidation.8 Further genome-wide association studies identified additional susceptibility genes: TRA@ (T cell receptor alpha), TRB @ (T cell receptor beta), P2RY11 (purinergic receptor P2Y, G-protein coupled, 11), CTSH (cathepsin H), TNFSF4 (tumor necrosis factor superfamily member 4), ZNF365 (zinc finger protein 365) and IL10RB (interleukin 10 receptor, beta).9, 10, 11, 12 These findings provided additional evidence that narcolepsy results from autoimmunity.
Narcolepsy is caused by the destruction of hypothalamic neurons that secrete a wake-promoting neuropeptide hypocretin (orexin).13, 14 Most of the patients with narcolepsy have low or undetectable levels of hypocretin-1 in the cerebrospinal fluid.15, 16 However, it has been reported that narcolepsy cannot be explained by mutations and polymorphisms in prepro-hypocretin and hypocretin-receptor genes, except for in rare cases.17, 18, 19, 20
Recent methodological developments make it possible to conduct genome-wide studies on copy number variations (CNVs). Based on several reports, patients with neuropsychiatric diseases (for example, autism, schizophrenia or severe neurodevelopmental disorders) bear a higher global burden of rare and large CNV than do control subjects.21, 22, 23, 24 Moreover, in several cases, a single CNV locus is associated with multiple neuropsychiatric diseases; this observation indicates that neuropsychiatric diseases may share some genetic origins. Here, we worked from the hypothesis that CNVs have an impact on narcolepsy because narcolepsy most likely has neuropsychiatric etiology.13, 16, 19 We conducted a genome-wide CNV study in order to identify novel genetic susceptibility loci for narcolepsy.
Materials and methods
Subjects
Japanese patients with narcolepsy–cataplexy (n=426) were unrelated individuals living in Tokyo or neighboring areas. Each patient had received a diagnosis at the Neuropsychiatric Research Institute; diagnoses were made according to the ICSD (International Classification of Sleep Disorders, 2nd edition-2). In our study, all patients were diagnosed by narcolepsy specialists. All patients with narcolepsy–cataplexy carried HLA-DQB1*06:02. It has been reported that most of the narcolepsy patients with low level of hypcocretin-1 carried HLA-DQB1*06:02.15, 25, 26 A total of 171 essential hypersomnia (EHS) patients were diagnosed based on the following three clinical items in central nervous system hypersomnias: (1) recurrent daytime sleep episodes that occur basically everyday over a period of at least 6 months; (2) absence of cataplexy; (3) the hypersomnia is not better explained by another sleep disorder, medical or neurological disorder, mental disorder, medication use or substance use disorder.27, 28, 29 The diagnostic criteria for EHS correspond to narcolepsy without cataplexy and part of idiopathic hypersomnia without long sleep time if we apply the criteria according to the ICSD-2. Cataplexy is absent in both disorders. Excessive daytime sleepiness of narcolepsy without cataplexy is typically associated with naps that are of refreshing nature. Meanwhile, idiopathic hypersomnia without long sleep time results in unintended naps with or without refreshing nature. EHS includes patients with short refreshing naps in idiopathic hypersomnia without long sleep time. The nocturnal sleep and total amount of sleep are basically normal in both disorders. Multiple sleep latency test (MSLT) is required for the diagnosis in the ICSD-2. Idiopathic hypersomnia without long sleep time is differentiated from narcolepsy without cataplexy by the absence of REM-related features, most notably two or more sleep-onset REM periods on MSLT. However, we consider that the nature of naps (short or long, refreshing or nonrefreshing) is more appropriate to distinguish a group of patients for study than MSLT, because MSLT is useful for the diagnosis of narcolepsy with cataplexy but is not optimized for the diagnosis of related hypersomnia patients using the number of sleep-onset REM periods and mean sleep latency on MSLT to distinguish between narcolepsy without cataplexy and idiopathic hypersomnia without long sleep time. In addition, MSLT requires a whole day and impose an economic burden on patients. Therefore, we focused on the symptoms themselves and adopted the definition of EHS. The control group comprised 562 unrelated healthy Japanese individuals living in Tokyo or neighboring areas. The control subjects did not have a history of narcolepsy and central nervous system hypersomnia. Age and gender were not matched between cases and controls. Ethical approval was obtained from the local institutional review boards of all participating organizations. All individuals gave written informed consent for their participation in the study.
Preparation of gene chip data
We conducted a genome-wide CNV analysis with DNA samples from 426 narcoleptic patients, 171 EHS patients and 562 healthy controls. The Affymetrix Genome-Wide Human SNP Array 6.0 (Affymetrix, Santa Clara, CA, USA) was used according to the manufacturer’s protocols (http://www.affymetrix.com). This array comprises about ∼906 600 SNP probes and ∼946 000 CNV probes. Samples with overall call rates <99% were excluded from the CNV analysis because low-quality genotyping is strongly correlated with inaccurate CNV detection.30
CNV detection
We made use of PennCNV-Affy (http://www.openbioinformatics.org/) in order to detect CNVs. This software uses called genotypes and normalized intensity to create reference cluster positions to compute relative differences in the signal from each sample in the form of B allele frequency (BAF) and log R ratio. BAF is a normalized measure of relative signal intensity ratio of the B and A alleles in the SNP array. Log R ratio is a measure of normalized total signal intensity relative to expected value (=log2(Robserved/Rexpected), where R is a sum of probe intensities). For normalization of intensity data, Affymertix Power Tools software (http://www.affymetrix.com/) was utilized before the CNV detection.
Quality control
Quality controls were done after the CNV detection (Supplementary Figure 1). In the raw detection data from PennCNV-Affy, samples with CNV call counts >100 were excluded because an unusually large number of CNVs indicates that a sample contains low-quality DNA.30 Then, CNVs that included <10 probes were removed in order to increase the reliability of the analysis because CNVs with lower numbers of probes cause false detection of CNVs.21, 23, 30
Statistical analysis
Statistical analyses of the CNV data were performed using PLINK ver1.0.7 (http://pngu.mgh.harvard.edu/~purcell/plink/). Gene information was based on glist-hg18 list provided by PLINK and NCBI Build 36. In this analysis, we focused on rare and large CNVs that were found in ⩽1% of the all samples and that were ⩾100 kb in size. In a global burden analysis, the average number of all rare and large CNVs within individual whole genome was assessed between cases and controls. A region-based analysis was performed in order to identify specific CNV-associated regions, using CNV with its size ⩾30 kb. A significant region identified in the region-based analysis was validated using a TaqMan assay (Supplementary Information).
A pathway analysis was designed to discover disease-related functional pathways and this design was based on those in previous studies, using CNV with its size ⩾100 kb.23, 31, 32 Briefly, gene sets were derived from the Gene Ontology (http://www.geneontology.org/) and from KEGG (http://www.genome.jp/kegg/). The analysis was conducted using Perl (http://www.perl.org/) and S (http://www.r-project.org/) programming (Supplementary Information). Functional clusters were created to interpret the relation of a large number of significant gene sets. Cytoscape (http://www.cytoscape.org/) was used to create functional clusters of significant gene sets. Edge indicates that two gene sets share ‘support-genes’ that are more frequently affected in the patients. Edge width is the proportion of shared support-genes versus the total number of genes within two gene sets. Node indicates a gene set. Node size is proportional to the number of genes in a gene set (Supplementary Information).
The global burden analysis and the region-based analysis were evaluated using one-tailed 100 000 permutation tests. False discovery rate was applied to correct for multiple testing in the pathway analysis. To estimate false discovery rate, 10 000 permutation tests were also used to correct for the dependency of gene sets because an individual gene could fall into more than one gene set. Corrections for multiple testing were not applied to the region-based analysis. All programming and file conversions were done with Perl or S script.
Results
SNP Gene chip data were used to detect CNVs within the genomes of 426 Japanese narcoleptic patients and 562 healthy controls. Each sample with an overall call rate of <99% was excluded from further analysis before detection of CNVs was performed; consequently, the samples from 345 cases and 470 controls were used for CNV detection (Supplementary Figure 1). We used PennCNV-Affy to detect CNVs; after the detection step, we eliminated whole samples or individual CNVs during the ensuing quality control steps. The CNV call count threshold was set to >100 for sample exclusion; this step left 327 patient and 459 healthy control samples (Supplementary Figure 1). Individual CNVs that included <10 probes were removed from subsequent analyses. Then, we eliminated all small CNVs and all common CNVs to target only those CNVs that were both rare and large (frequency: ⩽1%, CNV size: ⩾100 kb), leaving 1061 CNVs detected by PennCNV-Affy for statistical analyses.
The global burden of rare and large CNVs, which were stratified by size and CNV type, among narcoleptic patients was compared with that among control subjects (Table 1). Overall, we found a significant enrichment of rare and large CNVs in the patients (case–control ratio of CNV count=1.54, P=5.00 × 10−4). In particular, we observed a higher occurrence of duplications with sizes between 100 and 200 kb in the patients (case–control ratio of CNV count=1.93, P=2.00 × 10−5; Table 1). An excess of rare and large deletions with sizes between 500 kb and 1 Mb was evident in the patients (case–control ratio of CNV count=3.12, P=2.18 × 10−2; Table 1). The number of genes disrupted by rare and large CNVs was significantly higher in the patients than in control subjects (case–control ratio of gene count=2.18, P=5.75 × 10−3; Table 2). Especially, gene disruptions by duplications with sizes of 200 to 500 kb were more frequent in the patients in the same manner of Table 1 (case–control ratio of gene count=2.62, P=9.70 × 10−4; Table 2).
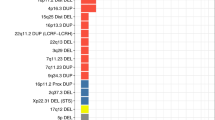
A region-based analysis was performed to identify regions that may be associated with narcolepsy susceptibility. We extended the definition of rare and large CNVs by including CNVs with its size ⩾30 kb and with its frequency ⩽1%, so that we reduce the possibility to miss true associations. The PARK2 (parkinson protein 2, E3 ubiquitin protein ligase) region (Chr 6: 162 685 167–162 762 467, P=3.07 × 10−2) was detected as a region of significant association. Four patients carried duplications with copy numbers=3 within the PARK2 region, but no controls carried duplications in this region (Figure 1). In order to ensure reliability of the duplications in the PARK2 region in these four patients, we validated these CNVs using quantitative PCR (Figure 1, Supplementary Figure 2 and Supplementary Information).

The significant region in PARK2 (parkinson protein 2, E3 ubiquitin protein ligase) gene (Chr 6: 162 685 167–162 762 467).
Next, we searched whether other hypersomnia patients such as EHS might have the same duplication in PARK2 region as narcolepsy. First, we used Affymetrix 6.0 chip data of 171 Japanese EHS samples and detected the two same duplications in PARK2 (Figure 1), using Birdsuite software (http://www.broadinstitute.org/scientific-community/software). These duplications were validated by quantitative PCR (Supplementary Figure 3 and Supplementary Information).
To assess the overall influence of rare and large CNVs in the pathogenic narcolepsy, we conducted a pathway analysis because association studies that involve only individual rare CNVs and a limited number of samples often have insufficient power to detect disease-causing variants. We compiled comprehensive collections of gene sets using KEGG and the Gene Ontology, and then we assessed which gene sets were more frequently affected by rare and large CNV in narcoleptic patients than in the control. We found that 32 gene sets showed significant associations when the false discovery rate was set to 5% (Supplementary Table 1). Functional clusters were created using these 32 gene sets; four clusters—immune responses, acetyltransferase activity, cell cycle regulation and regulation of cell development (Figure 2). The results indicated that these biological pathways might be involved in the pathogenesis of narcolepsy.

Functional clusters of the 32 gene sets that were found to be significantly associated with narcolepsy. The figure shows a network of gene sets (nodes) that are related by mutual overlap (edges). Node size is proportional to the total number of genes in each gene set. The number inside a node is the index used in the first column in Supplementary Table 1. Edge width represents the number of overlapping genes between two gene sets (see Supporting Information for details).
Discussion
In this study, we conducted a genome-wide CNV analysis using DNA samples from 426 narcoleptic patients and 562 healthy controls. To our knowledge, no previously reported genome-wide CNV study has focused on associations between narcolepsy and CNVs. Narcoleptic patients were found to carry a larger global burden of rare and large CNVs than did controls (Table 1). The number of genes disrupted by rare and large CNVs was also higher in the patients than in the controls (Table 2). At this moment, a further replication study is needed to confirm these results because the sample size of this study was relatively small.
To identify the influence of rare and large CNVs at specific loci on narcolepsy, a region-based analysis was conducted. Rare and large CNVs in the PARK2 region were found to be significantly associated with narcolepsy (Figure 1 and Supplementary Figure 2). Furthermore, we found that two EHS patients had the same duplication in PARK2 region (Figure 1 and Supplementary Figure 3). EHS is a sleep disorder characterized by excessive daytime sleepiness without cataplexy. Previous reports have reported that EHS and narcolepsy are associated with the same susceptibility genes. Approximately 30–50% of EHS patients carry HLA-DQB1*06:02.27, 33, 34 Narcolepsy-related SNPs in CPT1B and TRA@ reported by genome-wide association studies were also associated with EHS.33, 35 The pathogenesis of EHS is thought to be partially similar to that of narcolepsy.27, 33, 35, 36 The finding of the same duplication in PARK2 among EHS patients could be a supportive evidence for the association between narcolepsy and PARK2; however, the replication study will be necessary to validate the result.
Interestingly, the PARK2 region is associated with other neuropsychiatric diseases such as Parkinson disease, autism and attention deficit hyperactivity disorder. A large number of PARK2 mutations have been reported in patients with Parkinson disease; these mutations include point mutations, small insertions or deletions (indels) and single or multiple exon CNVs.37 Reportedly, eight patients with autism were found to carry PARK2 deletions, and another patient with autism carries a duplication of PARK2.22, 38 A recent publication reported the same PARK2 region as the candidate region for attention deficit hyperactivity disorder.39 They found 3 deletions and 9 duplications, validating 11 of them by quantitative PCR analysis. Taken together, these findings indicate that variations in the PARK2 region may result in genetic susceptibility to multiple neuropsychiatric diseases.
We conducted a pathway analysis to assess the influence of rare and large genic CNVs throughout the genome on narcolepsy. Using a false discovery rate of 5%, we found that 32 gene sets were significantly associated with narcolepsy (Supplementary Table 1 and Figure 2). Functional clustering of these 32 gene sets revealed four groups of gene sets: immune response, acetyltransferase activity, cell cycle regulation and regulation of cell development. Narcolepsy is well known to be associated with HLA and TRA@,3, 4, 5, 6, 9 and autoantibodies against Tribbles homolog 2 (Trib2) have been recently detected in some patients with narcolepsy.40, 41, 42 We also found an enrichment of CNVs disrupting functional gene sets involved in immune response, supporting the hypothesis that narcolepsy is caused by an immune attack on hypocretin-producing neurons. The largest group of gene sets in the functional map contained genes within the acetyltransferase activity pathways. A study of schizophrenia in a Korean population reported an association between schizophrenia and histone deacetylase genes.43 To our knowledge, no published report documents a relationship between narcolepsy and alterations in acetyltransferase activity; nevertheless, modified acetyltransferase activity might be an unrecognized component in the pathogenesis of narcolepsy. Novel observations included gene sets involved in cell cycle arrest, cell development and cell morphogenesis functional groups. Further studies will be needed to interpret the relation between narcolepsy and these pathways.
The finding of this study provided the first insight of the involvement of multiple rare and large CNVs, both genome-wide and at specific loci, in the pathogenesis of narcolepsy, even though the replication study would be necessary. Moreover, these findings revealed new genetic and functional targets in narcolepsy that may lead to an integrated understanding of the pathogenic mechanism of narcolepsy. Although some genetic factors leading or contributing to narcolepsy have been identified, we need to develop a more comprehensive understanding of the genetic underpinnings, especially the role of CNVs, in this disease because few studies on narcolepsy have been conducted.
References
Mignot, E Genetic and familial aspects of narcolepsy. Neurology 50 (2 Suppl 1), S16–S22 (1998).
Overeem, S, Mignot, E, van Dijk, JG & Lammers, GJ Narcolepsy: clinical features, new pathophysiologic insights, and future perspectives. J Clin. Neurophysiol. 18, 78–105 (2001).
Juji, T, Satake, M, Honda, Y & Doi, Y HLA antigens in Japanese patients with narcolepsy. All the patients were DR2 positive. Tissue Antigens 24, 316–319 (1984).
Langdon, N, Welsh, KI, van Dam, M, Vaughan, RW & Parkes, D Genetic markers in narcolepsy. Lancet 2, 1178–1180 (1984).
Matsuki, K, Juji, T, Tokunaga, K, Naohara, T, Satake, M & Honda, Y Human histocompatibility leukocyte antigen (HLA) haplotype frequencies estimated from the data on HLA class I, II, and III antigens in 111 Japanese narcoleptics. J. Clin. Invest. 76, 2078–2083 (1985).
Miyagawa, T, Hohjoh, H, Honda, Y, Juji, T & Tokunaga, K Identification of a telomeric boundary of the HLA region with potential for predisposition to human narcolepsy. Immunogenetics 52, 12–18 (2000).
Mignot, E, Lin, L, Rogers, W, Honda, Y, Qiu, X, Lin, X et al. Complex HLA-DR and -DQ interactions confer risk of narcolepsy-cataplexy in three ethnic groups. Am. J. Hum. Genet. 68, 686–699 (2001).
Miyagawa, T, Kawashima, M, Nishida, N, Ohashi, J, Kimura, R, Fujimoto, A et al. Variant between CPT1B and CHKB associated with susceptibility to narcolepsy. Nat. Genet. 40, 1324–1328 (2008).
Hallmayer, J, Faraco, J, Lin, L, Hesselson, S, Winkelmann, J, Kawashima, M et al. Narcolepsy is strongly associated with the T-cell receptor alpha locus. Nat. Genet. 41, 708–711 (2009).
Kornum, BR, Kawashima, M, Faraco, J, Lin, L, Rico, TJ, Hesselson, S et al. Common variants in P2RY11 are associated with narcolepsy. Nat. Genet. 43, 66–71 (2011).
Faraco, J, Lin, L, Kornum, BR, Kenny, EE, Trynka, G, Einen, M et al. ImmunoChip study implicates antigen presentation to T cells in narcolepsy. PLoS Genet. 9, e1003270 (2013).
Han, F, Faraco, J, Dong, XS, Ollila, HM, Lin, L, Li, J et al. Genome wide analysis of narcolepsy in China implicates novel immune loci and reveals changes in association prior to versus after the 2009 H1N1 influenza pandemic. PLoS Genet. 9, e1003880 (2013).
Lin, L, Faraco, J, Li, R, Kadotani, H, Rogers, W, Lin, X et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell 98, 365–376 (1999).
Chemelli, RM, Willie, JT, Sinton, CM, Elmquist, JK, Scammell, T, Lee, C et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 98, 437–451 (1999).
Mignot, E, Lammers, GJ, Ripley, B, Okun, M, Nevsimalova, S, Overeem, S et al. The role of cerebrospinal fluid hypocretin measurement in the diagnosis of narcolepsy and other hypersomnias. Arch. Neurol. 59, 1553–1562 (2002).
Nishino, S, Ripley, B, Overeem, S, Lammers, GJ & Mignot, E Hypocretin (orexin) deficiency in human narcolepsy. Lancet 355, 39–40 (2000).
Gencik, M, Dahmen, N, Wieczorek, S, Kasten, M, Bierbrauer, J, Anghelescu, I et al. A prepro-orexin gene polymorphism is associated with narcolepsy. Neurology 56, 115–117 (2001).
Olafsdottir, BR, Rye, DB, Scammell, TE, Matheson, JK, Stefansson, K & Gulcher, JR Polymorphisms in hypocretin/orexin pathway genes and narcolepsy. Neurology 57, 1896–1899 (2001).
Peyron, C, Faraco, J, Rogers, W, Ripley, B, Overeem, S, Charnay, Y et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat. Med. 6, 991–997 (2000).
Hungs, M, Lin, L, Okun, M & Mignot, E Polymorphisms in the vicinity of the hypocretin/orexin are not associated with human narcolepsy. Neurology 57, 1893–1895 (2001).
Consortium TIS. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455, 237–241 (2008).
Glessner, JT, Wang, K, Cai, G, Korvatska, O, Kim, CE, Wood, S et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573 (2009).
Pinto, D, Pagnamenta, AT, Klei, L, Anney, R, Merico, D, Regan, R et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372 (2010).
Williams, NM, Zaharieva, I, Martin, A, Langley, K, Mantripragada, K, Fossdal, R et al. Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet 376, 1401–1408 (2010).
Krahn, LE, Pankratz, VS, Oliver, L, Boeve, BF & Silber, MH Hypocretin (orexin) levels in cerebrospinal fluid of patients with narcolepsy: relationship to cataplexy and HLA DQB1*0602 status. Sleep 25, 733–736 (2002).
Kanbayashi, T, Yano, T, Ishiguro, H, Kawanishi, K, Chiba, S, Aizawa, R et al. Hypocretin-1 (orexin-A) levels in human lumbar CSF in different age groups: infants to elderly persons. Sleep 25, 337–339 (2002).
Honda, Y, Juji, T, Matsuki, K, Naohara, T, Satake, M, Inoko, H et al. HLA-DR2 and Dw2 in narcolepsy and in other disorders of excessive somnolence without cataplexy. Sleep 9 (1 Pt 2), 133–142 (1986).
Mukai, J, Inoue, Y, Honda, Y, Takahashi, Y, Ishii, A, Saitoh, K et al. Clinical characteristics of essential hypersomnia syndrome. Sleep. Biol. Rhythms 1, 229–231 (2003).
Komada, Y, Inoue, Y, Mukai, J, Shirakawa, S, Takahashi, K & Honda, Y Difference in the characteristics of subjective and objective sleepiness between narcolepsy and essential hypersomnia. Psychiatry Clin. Neurosci. 59, 194–199 (2005).
Koike, A, Nishida, N, Yamashita, D & Tokunaga, K Comparative analysis of copy number variation detection methods and database construction. BMC Genet. 12, 29 (2011).
Wang, K, Zhang, H, Kugathasan, S, Annese, V, Bradfield, JP, Russell, RK et al. Diverse genome-wide association studies associate the IL12/IL23 pathway with Crohn Disease. Am. J. Hum. Genet. 84, 399–405 (2009).
Subramanian, A, Tamayo, P, Mootha, VK, Mukherjee, S, Ebert, BL, Gillette, MA et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Miyagawa, T, Honda, M, Kawashima, M, Shimada, M, Tanaka, S, Honda, Y et al. Polymorphism located between CPT1B and CHKB, and HLA-DRB1*1501-DQB1*0602 haplotype confer susceptibility to CNS hypersomnias (essential hypersomnia). PLoS One 4, e5394 (2009).
Honda, Y, Takahashi, Y, Honda, M, Watanabe, Y & Sato, T Genetic aspects of narcolepsy. Sleep and Sleep Disorders: From Molecule to Behavior (eds Hayaishi O. and Inoue S.) 341–358 Academic Press, 1998).
Miyagawa, T, Honda, M, Kawashima, M, Shimada, M, Tanaka, S, Honda, Y et al. Polymorphism located in TCRA locus confers susceptibility to essential hypersomnia with HLA-DRB1*1501-DQB1*0602 haplotype. J. Hum. Genet. 55, 63–65 (2010).
Khor, SS, Miyagawa, T, Toyoda, H, Yamasaki, M, Kawamura, Y, Tanii, H et al. Genome-wide association study of HLA-DQB1*06:02 negative essential hypersomnia. PeerJ 1, e66 (2013).
Crosiers, D, Theuns, J, Cras, P & Van Broeckhoven, C Parkinson disease: insights in clinical, genetic and pathological features of monogenic disease subtypes. J. Chem. Neuroanat. 42, 131–141 (2011).
Scheuerle, A & Wilson, K PARK2 copy number aberrations in two children presenting with autism spectrum disorder: further support of an association and possible evidence for a new microdeletion/microduplication syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156B, 413–420 (2011).
Jarick, I, Volckmar, AL, Putter, C, Pechlivanis, S, Nguyen, TT, Dauvermann, MR et al. Genome-wide analysis of rare copy number variations reveals PARK2 as a candidate gene for attention-deficit/hyperactivity disorder. Mol. Psychiatry 19, 115–121 (2012).
Cvetkovic-Lopes, V, Bayer, L, Dorsaz, S, Maret, S, Pradervand, S, Dauvilliers, Y et al. Elevated Tribbles homolog 2-specific antibody levels in narcolepsy patients. J. Clin. Invest. 120, 713–719 (2010).
Kawashima, M, Lin, L, Tanaka, S, Jennum, P, Knudsen, S, Nevsimalova, S et al. Anti-Tribbles homolog 2 (TRIB2) autoantibodies in narcolepsy are associated with recent onset of cataplexy. Sleep 33, 869–874 (2010).
Toyoda, H, Tanaka, S, Miyagawa, T, Honda, Y, Tokunaga, K & Honda, M Anti-Tribbles homolog 2 autoantibodies in Japanese patients with narcolepsy. Sleep 33, 875–878 (2010).
Kim, T, Park, JK, Kim, HJ, Chung, JH & Kim, JW Association of histone deacetylase genes with schizophrenia in Korean population. Psychiatry Res. 178, 266–269 (2010).
Acknowledgements
We thank all the individuals who participated in this study. This study was supported by Grants-in-Aid for Young Scientists (A) (23689022) and Scientific Research on Innovative Areas (22133008) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and Grants-in-Aid from ‘Takeda Science Foundation’ and ‘SENSHIN Medical Research Foundation’.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Yamasaki, M., Miyagawa, T., Toyoda, H. et al. Genome-wide analysis of CNV (copy number variation) and their associations with narcolepsy in a Japanese population. J Hum Genet 59, 235–240 (2014). https://doi.org/10.1038/jhg.2014.13
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.13

{kind=link}
{kind=link}
{kind=link}