
Abstract
Opsismodysplasia is an autosomal recessive skeletal disorder characterized by facial dysmorphism, micromelia, platyspondyly and retarded bone maturation. Recently, mutations in the gene encoding inositol polyphosphate phosphatase-like 1 (INPPL1) are found in several families with opsismodysplasia by a homozygosity mapping, followed by whole genome sequencing. We performed an exome sequencing in two unrelated Japanese families with opsismodysplasia and identified a novel INPPL1 mutation, c.1960_1962delGAG, in one family. The mutation is predicted to result in an in-frame deletion (p.E654del) within the central catalytic 5-phosphate domain. Our results further support that INPPL1 is the disease gene for opsismodysplasia and that opsismodysplasia has genetic heterogeneity.
Similar content being viewed by others


Introduction
Opsismodysplasia (OMIM 258480) is a rare skeletal dysplasia identifiable at birth. Its clinical features are rhizomelic micromelia and facial dysmorphism, including prominent brow, large fontanels, depressed nasal bridge and small anteverted nose with long philtrum, as well as short feet and hands with sausage-like fingers.1 Its main radiological features include retarded bone maturation, marked shortness of the bones of hands and feet with concave metaphyses and thin, lamellar vertebral bodies. Some patients show severe phosphate wasting. Autosomal recessive inheritance is the most likely mode of inheritance; to date, at least three consanguineous families with opsismodysplasia are reported.2, 3, 4
Recently, Below et al.5 performed a homozygosity mapping coupled with whole genome sequencing in a consanguineous family with opsismodysplasia, and identified INPPL1 (inositol polyphosphate phosphatase-like 1) as a causative gene for opsismodysplasia. They first identified a homozygous missense mutation, p.Pro659Leu, in the consanguineous family, and then found INPPL1 mutations in additional five unrelated families with opsismodysplasia. We performed a whole exome sequencing for two patients from two unrelated families and identified a homozygous in-frame deletion of INPPL1 in one family.
Subjects and methods
Subjects and DNA samples
Two families with clinical diagnosis of opsismodysplaisa were included in the study. Family 1 consisted of parents and affected sibs (Figure 1a), and Family 2 consisted of parents and a patient. Genomic DNA was extracted by standard procedures from peripheral blood of the patients and their family members after informed consent. The study was approved by the ethical committee of RIKEN, Yokohama City University, and participating institutions.

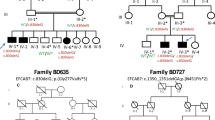
INPPL1 mutation in a Japanese family with opsismodysplasia. (a) Pedigree, (b) an in-frame deletion c.1960_1962delGAG (p.E654del) within exon 17 and (c) conservation of p.E654 in INPPL1 among different species.
Exome sequencing
Six individuals in the two families were analyzed by the whole exome sequence as described previously.6 Briefly, 3 μg of genomic DNA was sheared by Covaris 2S system (Covaris, Woburn, MA, USA) and partitioned using SureSelect Human All Exon V4 (Agilent technology, Santa Clara, CA, USA) according to the manufacturer’s instructions. The exon-enriched DNA libraries were sequenced using HiSeq2000 (Illumina, San Diego, CA, USA) with a 101-bp paired-end reads and a 7-bp index reads. Four samples (2.5 pM each, with different index) were run in one lane. HiSeq Control Software/Real-Time Analysis and CASAVA1.8.2 (Illumina) were used for image analysis and base calling. The mapping was performed to human genome hg19 using Novoalign (http://www.novocraft.com/main/page.php?s=novoalign). The aligned reads were processed by Picard to remove the polymerase chain reaction (PCR) duplicate (http://picard.sourceforge.net). The variants were called by Genome Analysis Toolkit 1.6-5 (GATK; http://www.broadinstitute.org/gsa/wiki/index.php/Main_Page) with the best practice variant detection with the GAKT v.3 (http://www.broadinstitute.org/gsa/wiki/index.php/Best_Practice_Variant_Detection_with_the_GATK_v3) and annotated by ANNOVAR (23 February 2012) (http://www.openbioinformatics.org/annovar/). Through this flow, common variants registered in dbSNP135 (minor allele frequeny ⩾0.01) (http://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=316787363&g=snp135Common&hgTracksConfigPage=configure) were removed.
Priority scheme
On the basis of the hypothesis that opsismodysplasia is inherited in an autosomal recessive manner, variants were filtered by following conditions using the script created by BITS (Tokyo, Japan). For the homozygous mutation model: (1) variant allele frequency (variant alleles/total alleles) in probands ⩾0.8, (2) variant allele frequency in parents ⩽0.8, (3) excluding synonymous changes and (4) excluding the variants observed in our in-house database (n=429). For the compound heterozygous mutation model: (1) mutation allele frequency in probands: 0.2–0.8, (2) variant allele frequency in parents ⩽0.8, (3) excluding synonymous changes, (4) excluding the variants observed in our in-house database (n=429) and (5) selecting genes with compound heterozygous change. After combining variants selected by both models, genes commonly found in the two families were searched.
Sanger sequencing
We performed Sanger sequencing to confirm the deletion identified in the proband of Family 1 by the exome sequencing. We amplified exon 17 by PCR using primer sequences, 5′-AAGCACAAGGTCTTCCTTCGATTCA-3′ and 5′-CCATACCCTTGACCCAAATTCTTGAT-3′. We directly sequenced the PCR product using an Applied Biosystems 3730xl DNA analyzer (Life Technologies, Forster City, CA, USA). For the patient in Family 2, we screened 28 exons of INPPL1 and exon–intron boundaries by direct sequencing of PCR products from genome DNA. The primer sequences are available on request.
Evaluation of polymorphism
We used the invader assay coupled with PCR7 to exclude the possibility of polymorphism in 188 Japanese general populations. The deletion was evaluated by databases, PROVEAN v.1.1 (http://provean.jcvi.org/genome_submit.php), dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/) and 1000 genomes (http://www.1000genomes.org/). We used Evola website to investigate the conservation of p.E654 of INPPL1 (http://www.h-invitational.jp/hinv/ahg-db/index.jsp).
Results
Exome sequencing
By the whole exome sequencing, 3.8–5.1 Gb sequences uniquely mapped to all human RefSeq coding region were obtained. For all subjects, at least 95.9% of all coding regions were covered in five reads depth and more (Supplementary Table 1). No candidate genes that had mutations in the two families were identified.
Because INPPL1 mutations have recently been identified in opsismodysplasia,5 we checked INPPL1 mutations in the exome sequence data. Five or more reads covered 100% of its coding regions (Supplementary Table 1). A homozygous deletion, c.1960_1962 (p.E654del), was found in the proband of Family 1 (Figure 1a). However, this deletion had been excluded as a candidate mutation because no INPPL1 variant likely to be a mutation was detected in Family 2.
Confirmation and evaluation of c.1960_1962delGAG
We confirmed the deletion by direct sequence of PCR product from genomic DNA in the proband of Family 1 (Figure 1b). Next, we performed the invader assay coupled with PCR in the family. The parents were compound heterozygous for the deletion and the affected sibs were homozygous for it. The deletion was not found in 188 Japanese controls and in the public databases. The E654 is conserved between different species (Figure 1c). It is within the central catalytic 5-phosphate domain, but located at the position far from active site (25 amino acids) and within a loop region, which is thought to have structural flexibility in general. Inositol polyphosphate 5-phosphatase domain (ipp5c) of yeast synaptojanin in complex with inositol (1,4)-bisphosphate and calcium ion (PDB ID 1i9z) is the most analogous structure to the human INPPL1 catalytic domain among the currently available structures; however, its sequence identity with the human INPPL1 catalytic domain is low (26%). These make the structural assessment of the mutation equivocal. The PROVEAN database showed that p.E654del had a deleterious function against the gene product (score: −12.1).
Mutation screening of INPPL1 in Family 2
We screened the INPPL1 mutation in the patient of Family 2 by direct sequencing of the entire coding exons and their flanking regions. A total of nine SNPs were found, but no mutation was found in the patient.
Clinical information of the patients with the INPPL1 mutation
The proband of Family 1 (II-1 in Figure 1a) was a 9-year-old girl born to non-consanguineous healthy parents. Family history was unremarkable. She was referred to one of us because fetal echogram revealed short extremities. She was born at 40 weeks’ of gestation. Her birth weight was 2119 g (<3 percentile), length 38.0 cm (<3 percentile) and head circumference 35.1 cm (<3 percentile). She had a wide fontanelle, widely patent sutures, frontal bossing, flat nasal bridge, low set ears, anteverted nostrils, micrognathia, narrow thorax and distended abdomen, and her extremities were remarkably short (Figure 2a). Her respiratory activity was weak and inspiratory wheezing was noted. Tracheal intubation became necessary 4 h after birth. Radiological investigations of her skeleton showed characteristics of opsisimodysplasia (Figures 2b–d). She was repeatedly admitted because of respiratory insufficiency due to infections. At 2 years of age, tracheotomy was performed to care for respiratory problems. She was noticed to show low serum phosphate levels at around 1 year and since then had been treated on phosphate supplements and/or alfacalcidol (1α-OH-D3). At age 9 years, her height was 65 cm (<–6 s.d.) and weight 9 kg (–4 s.d). Her intellectual development was normal and was attending an elementary school.

Phenotype of patients in Family 1. (a) Appearance of the proband in Family 1. Rhizomelic micromelia, frontal bossing, flat nasal bridge, low set ears, anteverted nostrils, micrognathia, narrow thorax and distended abdomen were noted. Radiographs of the proband (II-1) at birth (b–d) and the aborted fetus (II-2) (e). Characteristics of opsismodysplasia including retarded bone maturation, shortness of the bones of hands and feet, concave metaphyses and thin, lamellar vertebral bodies were noted.
In the second pregnancy, similar conditions were found by a fetal echogram. Artificial abortion was carried out. The post-mortem radiograph showed skeletal findings similar to the proband (Figure 2e).
Discussion
Below et al.5 examined INPPL1 in a total of 12 unrelated families with opsismodysplasia and found its mutations in seven families. The list of mutation includes missense, nonsense and splicing mutations; all are predicted to be loss of function mutations. In one family, we also found a deletion mutation in INPPL1 that is predicted to be a loss of function mutation, but in another family, we could not detect an INPPL1 mutation. These results further support the results of the previous study that INPPL1 is the disease gene for opsismodysplasia and that opsismodysplasia has genetic heterogeneity.5 In retrospect, the patient of Family 2 showed significant platyspondyly, yet some of the radiographic features for opsismodysplasia that include hypoplasia of the base of the skull on lateral views and lateral spikes of the acetabular roof were absent. Further, the fragmented epiphyses and coning of the distal femora are not characteristically seen in opsismodysplasia. This case is also different from the other cases with an opsismodysplasia phenotype that do not have INPPL1 mutations (Prof. Debora Krakow, personal communication). Further collection of INPPL1 mutation-proven cases would help in defining the phenotype of opsismodysplasia. While we were preparing the manuscript, another study reporting the identification of INPPL1 as the cause of opsismodysplasia was published.8 It reports identification of the INPPL1 mutation in all 10 families examined.
INPPL1 (also known as SHIP2) is a member of the inositol 5′-phosphatase family that hydrolyzes phosphatidlylinositol 3,4,5-triphosphate (PtdIns(3,4,5)P3) and generates phosphatidylinositol 3,4-bisphosphate (PtdIns(3,4)P2).9 INPPL1 encodes a 142-kDa protein with a variety of protein interaction domains, including an N-terminal SH2 domain, a central catalytic 5-phosphatase domain, a C-terminal proline-rich domain, an NPXY site and a sterile a motif domain in the C-terminal region.10 At least 12 proteins of binding partners for INPLL1, such as Shc, APS, filamin and EphA2, have been identified.10 The genes for these binding partners are good candidates for the disease gene for the opsismodysplasia-like phenotype.
Biological roles of INPPL1 remain unclear. INPPL1 expression is particularly high in heart, skeletal muscle and placenta.11, 12 Its proposed roles are cell adhesion and spreading, actin cytoskeletal remodeling and receptor internalization. INPPL1 negatively regulates insulin signaling through its catalytic PtdIns(3,4,5) P3 5-phosphatase activity.9 The INPPL1−/− mice show a shortened snout and grow more slowly than wild-type littermates.13 After 6 weeks of age, they showed a substantial reduced body length and body weight; however, radiographic analysis showed no gross skeletal deficit. Further studies are necessary to clarify the role of INPPL1 in skeletal development and homeostasis.
References
Cormier-Daire, V., Delezoide, A. L., Philip, N., Marcorelles, P., Casas, K., Hillion, Y. et al. Clinical, radiological, and chondro-osseous findings in opsismodysplasia: survey of a series of 12 unreported cases. J. Med. Genet. 40, 195–200 (2003).
Beemer, F. A. & Kozlowski, K. S Additional case of opsismodysplasia supporting autosomal recessive inheritance. Am. J. Med. Genet. 49, 344–347 (1994).
Santos, H. G. & Saraiva, J. M Opsismodysplasia: another case and literature review. Clin. Dysmorphol. 4, 222–226 (1995).
Tyler, K., Sarioglu, N. & Kunze, J Five familial cases of opsismodysplasia substantiate the hypothesis of autosomal recessive inheritance. Am. J. Med. Genet. 83, 47–52 (1999).
Below, J. E., Earl, D. L., Shively, K. M., McMillin, M. J., Smith, J. D., Turner, E. H. et al. Whole-genome analysis reveals that mutations in inositol polyphosphate phosphatase-like 1 cause opsismodysplasia. Am. J. Hum. Genet. 92, 137–143 (2013).
Miyake, N., Elcioglu, N. H., Iida, A., Isguven, P., Dai, J., Murakami, N. et al. PAPSS2 mutations cause autosomal recessive brachyolmia. J. Med. Genet. 49, 533–538 (2012).
Ohnishi, Y., Tanaka, T., Ozaki, K., Yamada, R., Suzuki, H. & Nakamura, Y A high-throughput SNP typing system for genome-wide association studies. J. Hum. Genet. 46, 471–477 (2001).
Huber, C., Faqeih, E. A., Bartholdi, D., Bole-Feysot, C., Borochowitz, Z., Cavalcanti, D. P. et al. Exome sequencing identifies INPPL1 mutations as a cause of opsismodysplasia. Am. J. Hum. Genet. 92, 144–149 (2013).
Dyson, J. M., Kong, A. M., Wiradjaja, F., Astle, M. V., Gurung, R. & Mitchell, C. A The SH2 domain containing inositol polyphosphate 5-phosphatase-2: SHIP2. Int. J. Biochem. Cell. Biol. 37, 2260–2265 (2005).
Suwa, A., Kurama, T. & Shimokawa, T. SHIP2 and its involvement in various diseases. Expert Opin. Ther. Targets 14, 727–737 (2010).
Hejna, J. A., Saito, H., Merkens, L. S., Tittle, T. V., Jakobs, P. M., Whitney, M. A. et al. Cloning and characterization of a human cDNA (INPPL1) sharing homology with inositol polyphosphate phosphatases. Genomics 29, 285–287 (1995).
Pesesse, X., Deleu, S., De Smedt, F., Drayer, L. & Erneux, C Identification of a second SH2-domain-containing protein closely related to the phosphatidylinositol polyphosphate 5-phosphatase SHIP. Biochem. Biophys. Res. Commun. 239, 697–700 (1997).
Sleeman, M. W., Wortley, K. E., Lai, K. M., Gowen, L. C., Kintner, J., Kline, W. O. et al. Absence of the lipid phosphatase SHIP2 confers resistance to dietary obesity. Nat. Med. 11, 199–205 (2005).
Acknowledgements
We thank the patients and their family for their help to the study. We also thank the Japanese Skeletal Dysplasia Consortium. This study is supported by research grants from the Ministry of Health, Labor and Welfare (23300101 to SI and NMat.; 23300201 to SI), by Grants-in-Aid for Young Scientists (23689052 to NMiy.) from the Japan Society for the Promotion of Science; by Research on intractable diseases, Health and Labour Sciences Research Grants, H23-Nanchi-Ippan-123 (SI) and by grants from the Japan Science and Technology Agency, the Strategic Research Program for Brain Sciences (11105137 to NMat.), a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (12024421 to NMat.), a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (12020465 to NMat.) and the Takeda Science Foundation (to N Miy. and N Mat.). We thank Professors Debora Krakow and Michael Bamshad for their comments on the patients’ phenotypes. We also thank Ms Tomoko Kusadokoro for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Iida, A., Okamoto, N., Miyake, N. et al. Exome sequencing identifies a novel INPPL1 mutation in opsismodysplasia. J Hum Genet 58, 391–394 (2013). https://doi.org/10.1038/jhg.2013.25
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2013.25
Keywords
This article is cited by
-
INPPL1 gene mutations in opsismodysplasia
Journal of Human Genetics (2017)
-
Japanese founder duplications/triplications involving BHLHA9 are associated with split-hand/foot malformation with or without long bone deficiency and Gollop-Wolfgang complex
Orphanet Journal of Rare Diseases (2014)
