
Abstract
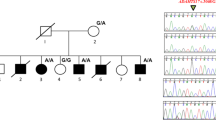
Acromesomelic dysplasia, PRKG2 type (AMDP, MIM 619636), is an extremely rare autosomal recessive skeletal dysplasia characterized by severe disproportionate short stature presenting with acromesomelia, mild metaphyseal widening of the long bones and mild spondylar dysplasia. To date, only four variants have been reported; one nonsense, one splice-site, and two frameshifts in five AMDP families. Here, we report the first missense variant and a second splice-site variant in PRKG2 in two patients with clinical and radiological features of acromesomelic dysplasia. Furthermore, functional studies of the novel missense variant, p.Val470Gly, revealed that it was unable to down-regulate FGF2-induced MAPK signaling and, thus, would be predicted to cause growth delay. Hence, this report expands the mutational spectrum in skeletal dysplasias associated with PRKG2 variants. In addition, we propose recognizable facial features with acromesomelic dysplasia, PRKG2 type.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Diaz-Gonzalez F, Wadhwa S, Rodriguez-Zabala M, Kumar S, Aza-Carmona M, Sentchordi-Montane L, et al. Biallelic cGMP-dependent type II protein kinase gene (PRKG2) variants cause a novel acromesomelic dysplasia. J Med Genet. 2022;59:28–38.
Pagnamenta AT, Diaz-Gonzalez F, Banos-Pinero B, Ferla MP, Toosi MB, Calder AD, et al. Variable skeletal phenotypes associated with biallelic variants in PRKG2. J Med Genet. 2022;59:947–50.
Mollaoglu E, Uludag Alkaya D, Yildiz CA, Kasap B, Tuysuz B. Natural history of clinical features in two brothers with acromesomelic dysplasia related to PRKG2. Clin Genet. 2023;103:574–9.
Pfeifer A, Aszodi A, Seidler U, Ruth P, Hofmann F, Fassler R. Intestinal secretory defects and dwarfism in mice lacking cGMP-dependent protein kinase II. Science. 1996;274:2082–6.
Chikuda H, Kugimiya F, Hoshi K, Ikeda T, Ogasawara T, Shimoaka T, et al. Cyclic GMP-dependent protein kinase II is a molecular switch from proliferation to hypertrophic differentiation of chondrocytes. Genes Dev. 2004;18:2418–29.
Kawasaki Y, Kugimiya F, Chikuda H, Kamekura S, Ikeda T, Kawamura N, et al. Phosphorylation of GSK-3beta by cGMP-dependent protein kinase II promotes hypertrophic differentiation of murine chondrocytes. J Clin Invest. 2008;118:2506–15.
Bartels CF, Bukulmez H, Padayatti P, Rhee DK, van Ravenswaaij-Arts C, Pauli RM, et al. Mutations in the transmembrane natriuretic peptide receptor NPR-B impair skeletal growth and cause acromesomelic dysplasia, type Maroteaux. Am J Hum Genet. 2004;75:27–34.
Vasques GA, Amano N, Docko AJ, Funari MF, Quedas EP, Nishi MY, et al. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature in patients initially classified as idiopathic short stature. J Clin Endocrinol Metab. 2013;98:E1636–44.
Wang SR, Jacobsen CM, Carmichael H, Edmund AB, Robinson JW, Olney RC, et al. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature. Hum Mutat. 2015;36:474–81.
Hisado-Oliva A, Ruzafa-Martin A, Sentchordi L, Funari MFA, Bezanilla-Lopez C, Alonso-Bernaldez M, et al. Mutations in C-natriuretic peptide (NPPC): a novel cause of autosomal dominant short stature. Genet Med. 2018;20:91–7.
Hisado-Oliva A, Garre-Vazquez AI, Santaolalla-Caballero F, Belinchon A, Barreda-Bonis AC, Vasques GA, et al. Heterozygous NPR2 mutations cause disproportionate short stature, similar to Leri-Weill dyschondrosteosis. J Clin Endocrinol Metab. 2015;100:E1133–42.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Campbell JC, Kim JJ, Li KY, Huang GY, Reger AS, Matsuda S, et al. Structural basis of cyclic nucleotide selectivity in cGMP-dependent protein kinase II. J Biol Chem. 2016;291:5623–33.
Vaandrager AB, Edixhoven M, Bot AG, Kroos MA, Jarchau T, Lohmann S, et al. Endogenous type II cGMP-dependent protein kinase exists as a dimer in membranes and can Be functionally distinguished from the type I isoforms. J Biol Chem. 1997;272:11816–23.
Vaandrager AB, Hogema BM, de Jonge HR. Molecular properties and biological functions of cGMP-dependent protein kinase II. Front Biosci. 2005;10:2150–64.
Koltes JE, Mishra BP, Kumar D, Kataria RS, Totir LR, Fernando RL, et al. A nonsense mutation in cGMP-dependent type II protein kinase (PRKG2) causes dwarfism in American Angus cattle. Proc Natl Acad Sci USA. 2009;106:19250–5.
Rudd Garces G, Turba ME, Muracchini M, Diana A, Jagannathan V, Gentilini F, et al. PRKG2 splice site variant in dogo argentino dogs with disproportionate Dwarfism. Genes (Basel). 2021;12:1489.
Acknowledgements
We would like to express our sincere appreciation to Prof. Gen Nishimura for his support in the radiographic analysis of Patient 1. We would also thank patients and families who have participated in the study.
Funding
This work was partly supported by a grant (PID2020-116263RB-I00) from the Ministerio de Economia, Industria Competividad (to KEH). Francisca Díaz-González was a recipient of a FPU Ph.D. studentship from the Ministerio de Ciencia, Innovación y Universidades; Grant 303294/2020-5 (to AALJ) from the National Council for Scientific and Technological Development (CNPq); and by Coordination of Superior Level Staff Improvement (CAPES; Finance Code 001 to NLMA and LPC).
Author information
Authors and Affiliations
Contributions
OAD, FG-D YA, and KEH generated the outline of the manuscript, and OAD, FD-G, AALJ, YA, and KEH wrote the first draft. OAD, YA, AALJ, FD-G, and KEH prepared the tables and figures. OAD, FD-G, AALJ, NOM, NLMA, LPC, SC, MBTM, YA, and KEH revised the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
AALJ received an independent research grant from BioMarin and consulting fees from Novo Nordisk. The other authors declare that they have no competing financial interests to declare.
Ethical approval
The Comite de Etica e Pesquisa do Hospital das Clínicas da Faculdade de Medicina da Universidade de Sao Paulo (CAAE, 06425812.4.0000.0068) provided approval for this study. Written informed consent for genetics studies and publication of individual details and images was obtained.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Akgun-Dogan, O., Díaz-González, F., de Lima Jorge, A.A. et al. Two new patients with acromesomelic dysplasia, PRKG2 type—identification and characterization of the first missense variant. Eur J Hum Genet (2023). https://doi.org/10.1038/s41431-023-01472-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41431-023-01472-z
