
Abstract
Marine cyanobacteria of the genus Acaryochloris are the only known organisms that use chlorophyll d as a photosynthetic pigment. However, based on chemical sediment analyses, chlorophyll d has been recognized to be widespread in oceanic and lacustrine environments. Therefore it is highly relevant to understand the genetic basis for different physiologies and possible niche adaptation in this genus. Here we show that unlike all other known isolates of Acaryochloris, the strain HICR111A, isolated from waters around Heron Island, Great Barrier Reef, possesses a unique genomic region containing all the genes for the structural and enzymatically active proteins of nitrogen fixation and cofactor biosynthesis. Their phylogenetic analysis suggests a close relation to nitrogen fixation genes from certain other marine cyanobacteria. We show that nitrogen fixation in Acaryochloris sp. HICR111A is regulated in a light–dark-dependent fashion. We conclude that nitrogen fixation, one of the most complex physiological traits known in bacteria, might be transferred among oceanic microbes by horizontal gene transfer more often than anticipated so far. Our data show that the two powerful processes of oxygenic photosynthesis and nitrogen fixation co-occur in one and the same cell also in this branch of marine microbes and characterize Acaryochloris as a physiologically versatile inhabitant of an ecological niche, which is primarily driven by the absorption of far-red light.
Similar content being viewed by others

Introduction
The fixation of dinitrogen is a major and crucial biogeochemical process in the ocean where it replaces the losses of bioavailable combined nitrogen due to denitrification and anaerobic ammonium oxidation (ANAMMOX) and allows new primary production (Zehr and Kudela, 2011). The capacity of fixing dinitrogen is widely distributed among Bacteria and a few Archaea, but not among Eukarya, and it gives the organisms that possess this capacity access to the largest pool of nitrogen, atmospheric N2. Nitrogenase, the enzyme complex that is responsible for the fixation of dinitrogen, is extremely oxygen sensitive and therefore most of the N2-fixing microorganisms are obligate or facultative anaerobes. A notable exception is the oxygenic phototrophic cyanobacteria, many of which possess the capacity of fixing dinitrogen (Zehr, 2011). Therefore, it is generally assumed that cyanobacteria are the main group of organisms that are responsible for the fixation of N2 in the ocean, although this viewpoint has been challenged (Riemann et al., 2010). Obviously, cyanobacteria need adaptations and follow certain strategies to provide nitrogenase with an anaerobic environment (Bergman et al., 1997; Berman-Frank et al., 2003). Heterocystous cyanobacteria confine nitrogenase to special differentiated anoxic cells that lack photosystem II, thereby separating it spatially from oxygenic photosynthesis. Many marine unicellular diazotrophic cyanobacteria, such as Crocosphaera or Cyanothece, follow another strategy and separate the two incompatible processes temporally, confining the fixation of dinitrogen to the dark period. A notable exception is the filamentous non-heterocystous cyanobacterium Trichodesmium, which is abundant in the tropical oceans and one of the most important diazotrophs (Karl et al., 2002). This organism may exhibit a combination of spatial and temporal separation, in which cells temporally switch off oxygenic photosynthesis and fix N2 (Berman-Frank et al., 2001). Another exception is the hitherto uncultivated putative unicellular diazotrophic cyanobacteria UCYN-A. The analysis of the metagenome of flow cytometrically sorted cells has shown that the genes for the oxygenic photosystem II and for CO2 fixation are absent (Tripp et al., 2010). The nitrogen fixation (nif) genes of this organism are expressed during the daytime and N2 fixation also occurs during the day but further nothing is known about the identity of this enigmatic cyanobacterium.
Acaryochloris is a unicellular marine cyanobacterium that is atypical in using chlorophyll d for light harvesting (Miyashita et al., 1996). Based on chemical analyses, chlorophyll d has only recently been recognized to be widespread in oceanic and lacustrine environments (Kashiyama et al., 2008). Therefore it is highly relevant to understand the genetic basis for different physiologies and possible niche adaptation in this genus. At present, genome sequences are available for two Acaryochloris strains. Acaryochloris marina MBIC 11017 (from here A. marina) was isolated from waters around the Palau islands in the Pacific Ocean (Miyashita et al., 1996), Acaryochloris sp. CCMEE 5410 from the Salton Sea lake in California (Miller et al., 2005). Compared with other cyanobacteria, large and complex genomes were found for both isolates. A. marina harbors a total of 8.3 million base pairs with more than 25% of its 8462 genes being located on nine different plasmids, eight of which are larger than 100 kb (Swingley et al., 2008). A similarly complex genome was also reported for the Salton Sea strain (Miller et al., 2011). Despite this impressive coding capacity, no genes for dinitrogen fixation were found in either of these two well-studied isolates.
We recently described Acaryochloris sp. HICR111A, a new member of the Acaryochloris clade (Mohr et al., 2010). The strain was isolated from microbial mat-like material on a dead coral skeleton in shallow water on the southern reef flat of Heron Island, Great Barrier Reef (23°26′ 31.2′′ S, 151°54′ 50.4′′ E). The cells are rod shaped, between 1 and 2 μm long with a diameter of 0.75–1 μm, which is somewhat smaller than the 2–3 μm in length and the diameter of 1.5–2.0 μm reported for A. marina (Mohr et al., 2010). The presence of Chl d was monitored spectrophotometrically (Qy maxima of Chl d detected at 707 nm and a single fluorescence maximum at 750 nm after Chl d excitation at 470 nm) and by high-performance liquid chromatography repeatedly from the early cultivation phase onwards. Purified cultures use Chl d as main photopigment, as indicated by a 1:50 ratio of Chl a/Chl d, which changed only little with increasing light intensity (from 5–10 μmol to 75–100 μmol photons m−2 s−1) (Mohr et al., 2010). Acaryochloris sp. HICR111A is, according to 16S rRNA-based phylogeny, an early branching member of the genus Acaryochloris (Mohr et al., 2010). Its 16S ribosomal RNA is 2% divergent from the two previously described A. marina strains, which, together with several distinct genetic, morphological and physiological differences, led us to conclude that cyanobacteria of the genus Acaryochloris occur as distinct ecotypes. A preliminary sequence analysis of its genome based on Illumina technology yielded a genome with an accumulated length of 8 371 965 nt (Mohr et al., 2010). Here we report the existence of a complete nitrogenase gene cluster in Acaryochloris sp. HICR111A, we demonstrate that the nif genes are strongly induced upon removal of combined nitrogen, and we detected nitrogenase activity.
Materials and methods
Culture conditions
Cultures of Acaryochloris sp. HICR111A (Mohr et al., 2010) were grown in ASN medium (artificial seawater+BG-11 nutrients) (Rippka et al., 1979). For preliminary analysis of dinitrogen fixation genes by qPCR, cultures were grown at room temperature (summer: ∼25 °C) under naturally changing light conditions (from here: ‘summer cultures’) and later that year moved to an incubator at 26 °C under a 12-h/12-h light/dark cycle with warm white light (40 W fluorescent tubes, Sanyo, Munich, Germany) of 35 μmolquanta m−2 s−1. For measurement of nitrogenase activity, two cultures of Acaryochloris HICR111A were grown and used as biological replicates (the assays were done on subsequent days). Both cultures were initially grown under a constant 12-h/12-h light/dark cycle at 26 °C and 35 μmolquanta m−2 s−1. Nine days before the nitrogenase assay, half of one culture was shifted to nitrogen-depleted medium (ASN0) by centrifugation in a sterilized tube at 6000 × g for 20 min, washed once with ASN0 (a third of the original volume) and resuspended in the original volume of ASN0. The other half of this culture served as control and was also centrifuged, but resuspended in an equal volume of fresh ASN medium. Subsequently, both were incubated under a light/dark regime of 12-h/12-h at 50–60 μmol quanta m−2 s−1. The replicate culture had been incubated at 50–60 μmol quanta m−2 s−1 for 22 days and half of it was subsequently shifted to ASN0 7 days before the assay as described above. One day before the nitrogenase assay, all cultures were shifted to 150 μmol quanta m−2 s−1 and 29 °C. For the nitrogenase assay, six 15-ml samples of the ASN0 culture and one 15-ml sample of ASN culture were filtered onto GF-F filters and placed on a perforated support in contact with ASN0 (or ASN in case of the control) in a custom-made, temperature-controlled incubator. Each of the seven chambers was closed by a glass window, leaving 3 ml headspace. The cultures were incubated under a constant flow of N2+0.03% CO2. The light/dark cycle, light intensity and temperature were kept the same.
Determination of nitrogenase activity
Nitrogenase activity was measured in custom-made incubators with separate chambers to hold filters of 45 mm diameter (one incubator containing six chambers and the other containing a single chamber that served as the control). The chambers were identical to the one described in Staal et al. (2001). The incubator was connected to a circulating cryostat water bath in order to thermostat the incubation chambers. The single chamber was temperature controlled by a computer-controlled Peltier element. The chambers were connected in series to a gas line of N2 with 0.03% CO2 premixed (Westfalen Gases, Münster, Germany) at 3 L h−1, controlled by a mass flow controller (5850S) and control unit (0152, both Brooks Instrument, Ede, The Netherlands). The incubator was covered with a hood containing the light source (two cool white fluorescent tubes) that was turned on and off by a timer.
Nitrogenase activity was measured using the acetylene reduction assay (ARA) (Steward et al., 1967). Nitrogenase activity was measured at six time points: (1) −2 to 0 h, (2) 0–2 h, (3) 3–5 h, (4) 6–8 h, (5) 10–12 h and (6) 12–14 h (where the beginning of the dark phase is taken as 0 h). The ASN culture (nitrate-supplemented) was taken as the control and measured along with the second and the fifth time points in the two replicate experiments, respectively. The samples had been incubated under anaerobic conditions for 7–8 h before the acetylene reduction assay was started to prevent a false-negative result due to oxygen inactivation.
For each time point, one chamber was disconnected from the gas flow and 1 ml of acetylene was injected into the chamber (∼30% of gas phase) while the gas was displaced into a second syringe connected to the gas outlet of the incubator. The gas in the headspace was mixed by ‘pumping’ using the two syringes. After 2 h of incubation the gas was mixed again, two 0.5-ml gas samples (technical duplicates) were withdrawn from the headspace and the filter with the culture was snap-frozen in liquid nitrogen for later RNA extraction. Acetylene and ethylene in the gas samples were determined gas chromatographically (Chrompack CP9001, Middelburg, The Netherlands). The gas chromatograph was equipped with a flame ionization detector (FID) and a wide-bore silica-fused (0.53 mm internal diameter) Porapak U column (Chrompack). The carrier gas was N2 at 10 ml min−1, and the flows of H2 and air for the FID were 30 and 300 ml min−1, respectively. The temperatures for injector, detector and oven were 90, 120, and 55 °C, respectively. The total amount of ethylene produced by the culture was calculated using acetylene as an internal standard (Stal, 1988) and normalized to chlorophyll d (measured in a separate 15-ml sample of the original culture).
Determination of chlorophyll d
A total of 15 ml of culture was transferred to a 50-ml Falcon tube and centrifuged for 15 min at 5000 g. 5 ml of 96% ethanol was added to the cell pellet vortexed, and the chlorophyll was extracted overnight in the dark at room temperature. The extract was centrifuged for 20 min at 6000 g and the absorption was read spectrophotometrically at 665 and 696 nm. The concentration of chlorophyll d was calculated using the formula Chl d (μg/ml)=A665 nm (−0.2006)+A696 nm (12.0995) (Ritchie, 2006).
RNA extraction
Acaryochloris cells were collected by rapid filtration on hydrophilic polyethersulfone filters (Supor 800 Filter, 0.8 μm, Pall, New York, NY, USA) and transferred to 15-ml Sarstedt tubes containing 2 ml of PGTX RNA extraction solution (Pinto et al., 2009). Alternatively, the frozen GF-F filters with the samples from the acetylene reduction assays were quickly thawed, the cells were removed with forceps and immediately placed into a 15-ml Sarstedt plastic tube containing 2 ml of PGTX extraction solution.
Samples in Sarstedt tubes were vortexed together with the PGTX, frozen in liquid N2 and stored at −80 °C until extraction. Frozen Acaryochloris cells in PGTX were transferred from −80 °C immediately to a 95-°C water bath for 5 min with occasional vortexing and were subsequently put on ice for 5 min. For phase extraction, 700 μl of chloroform/isoamylalcohol (IAA) (v/v 24:1) per 1 ml PGTX was added, the mixture was shaken vigorously and left at 24 °C for 10 min with occasional shaking and vortexing. The phases were separated by centrifugation for 15 min (20 min for GF-F filter samples) at 3750 r.p.m. (Allegra 6 KR, Beckman Coulter, Krefeld, Germany). The aqueous phase containing the RNA was transferred to a new reaction tube and one volume chloroform/IAA (v/v 24:1) was added, followed by a second centrifugation step (for GF-F filter samples, the aqueous phase was first re-extracted with phenol (pH 4.5-5)/chloroform/IAA (v/v/v 25:24:1)). The aqueous phase was transferred to a 2-ml reaction tube (1 ml per tube) and the RNA precipitated overnight with 1 volume of isopropanol at −20 °C. Precipitated RNA was pelleted by centrifugation for 25 min at 13 000 g at 4 °C, the pellet was washed with 1 ml 70% ethanol and centrifuged for 10 min at 13 000 g at 4 °C. The pellet was subsequently air-dried and dissolved in 100 μl (30 μl for GF-F filter samples) nuclease-free Milli-Q H2O. RNA concentration and grade of contamination of a sample were measured with a spectrophotometer (NanoDrop ND-1000 or ND-2000, Peqlab, Erlangen, Germany). Quality of RNA was verified on 1.5% formaldehyde-agarose gels.
Northern blot analysis
Blots for the detection of nifD mRNA were prepared from the separation of 1 μg of total RNA on 1.5% denaturing formaldehyde-agarose gels. Separated RNA was transferred to Amersham Hybond-N nylon membranes (GE Healthcare, Munich, Germany) by capillary blotting with 20 × SSC buffer (3 M NaCl, 0.3 M C6H5Na3O7, pH 7.0). The membranes were hybridized with [α-32P]UTP-labeled transcript probes overnight at 62 °C in 50% deionized formamide, 7% SDS, 250 mM NaCl, 120 mM phosphate buffer (sodium salts), pH 7.2 in screw-cap hybridization tubes. The membranes were washed in 2 × SSC, 1% SDS for 10 min; 1 × SSC, 0.5% SDS for 10 min; and in 0.1 × SSC, 0.1% SDS for 10 min at 57 °C. Signals were detected and analyzed on a Personal Molecular Imager FX system with Quantity One software (Bio-Rad, Munich, Germany).
Quantitative real-time reverse transcription PCR (RT-qPCR)
For elimination of contamination with genomic DNA, 2 or 8 μg (for first and second qPCR experiment, respectively) of each RNA sample was incubated twice with 2 units of TURBO DNase (TURBO DNA-free Kit, Ambion, Austin, TX, USA) for 20 min at 37 °C. The DNase was inactivated following the manufacturer's instructions. RNA was re-extracted by phenol (pH 4.5–5)/chloroform and precipitated with 3 volumes of NaOAc/ethanol. RNA samples were tested for residual contamination with DNA by PCR (30 amplification cycles) using the nifD primers of the qPCR reactions. All tests were negative. cDNA synthesis was done with 350 ng RNA of each sample using the Quantitect kit (Qiagen, Hilden, Germany) with a mix of nine primers (the reverse primer for each gene to be tested), each at a final concentration of 0.5 μM, and following the manufacturer's instructions. A 7500 Fast system (Applied Biosystems, Foster City, CA, USA) was used for qPCR. The modified ‘Fast Mode’ (10 min at 95 °C, 40 × (3 s at 95 °C, 30 s at 60 °C)) was used for the preliminary analysis of the ‘summer culture’ samples. For analysis of the samples from the acetylene reduction assays, the longer ‘Standard Mode’ (2 min at 50 °C, 10 min at 95 °C, 40 × (15 s at 95 °C, 60 s at 60 °C)) was used. Each 15-μl reaction contained 7.5 μl of Power SYBR Green Mix (Applied Biosystems), 4.5 μl of diluted cDNA (1:1280 for nifB, nifD, fdxH, psbA, psaA and coxA; 1:80 for pdhB; and 1: 5 242 880 for 16s rRNA), and 1.5 μl each of forward and reverse primers. Primers were designed manually to contain similar GC content and have an equal length of 24 nt. The ‘PrimersList’ tool on http://primerdigital.com/tools/ was used for all calculations; for melting temperature calculation, the option ‘using Allawi's thermodynamics parameters’ was chosen (Kalendar et al., 2011). Before the final experiment, a primer matrix was tested (amplifying genomic DNA) to determine the most efficient primer concentration for each primer pair and to confirm if primer efficiencies were similar. Primer sequences, concentrations, and efficiencies are shown in Table 1. Primers were synthesized by Sigma-Aldrich, Hamburg, Germany. All reactions were done in triplicate and for each reaction a ‘no RT’ control was included. 16s rRNA was amplified as reference. Melt curves for all the amplifications showed only one product (no product for ‘no RT’ control). Data were analyzed with the 7500 Fast System SDS software (Applied Biosystems), with manual Ct and automatic baseline setting (followed by manual control). Relative transcript quantities were calculated with the ΔΔCt method (Livak and Schmittgen, 2001). As calibrator conditions, samples of the nitrate-supplemented culture were used, namely the one harvested at 1200 and 2200 hours for the first and second experiments (replicate), respectively.
Software
The contig described here was fully annotated and submitted to Genbank (accession JN585763). Artemis Release 12 (Rutherford et al., 2000) was used for visualization and annotation of genome information stored in Genbank or text files. For multiple-sequence alignments and BLAST searches in local databases, BioEdit version 7.0.5.3 (Hall, 1999) was used. Phylogenetic analyses were done with MEGA version 5 (Tamura et al., 2011) and with Geneious Pro version 5.4.6 (Drummond et al., 2011) using the Mr. Bayes PlugIn (Huelsenbeck and Ronquist, 2001). The BLAST algorithm (Altschul et al., 1990) was used with the BLOSUM62 matrix to find putative homologs of genes (BLASTN) or proteins (BLASTP) in the NCBI database and to determine conservation between homologous sequences.
Results and discussion
A cassette for nitrogen fixation genes in Acaryochloris sp. HICR111A
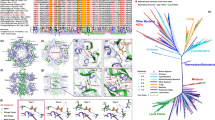
During annotation of the Acaryochloris sp. HICR111A genome, we encountered a possible cassette for nitrogen fixation (nif) genes on a 63 005-bp-long contig (GenBank accession JN585763). This contig harbors 60 protein-coding genes, from which 22 physically clustered genes are related to dinitrogen fixation. With a few exceptions like cysteine synthase A and serine-O-acetyltransferase, these have no homologs in the genomes of the two other sequenced Acaryochloris (Swingley et al., 2008; Miller et al., 2011) but several flanking genes on both sides of the nif gene cluster have homologs in A. marina (Figure 1), albeit at different positions in the genome. It should be noted that the genome was assembled from DNA of a non-axenic culture containing one strain of an α-proteobacterium and possibly another bacterium. However, these were the only nif genes found in the whole metagenome.

Comparison of the Acaryochloris HICR111A and Synechococcus PCC 7335 nif clusters. (a) GC content aligned with the scaled map of the Acaryochloris nif cluster and surrounding genes. Numbers in the top right corner are %GC. Two intergenic regions exhibiting a characteristic drop in GC content are boxed in red. (b) Map and GC content of the Synechococcus PCC 7335 nif cluster. Orange block arrows indicate genes associated with dinitrogen fixation, gray block arrows indicate genes with known products, but not known to be associated with dinitrogen fixation, white block arrows indicate genes encoding hypothetical proteins. Gray triangles indicate stretches of sequence containing multiple genes not explicitly associated with dinitrogen fixation.
Besides the nitrogen fixation cassette, the genome of Acaryochloris sp. HICR111A contains genes for several other nitrogen assimilation pathways, including nitrate/nitrite-, ammonia, urea, and cyanate assimilation. It is missing the nitrate/nitrite transporters encoded by nrtABCD and focA, but contains the nrtP gene that also codes for a nitrate/nitrite transporter. A similar situation can be found in the reference strain A. marina.
Genome assemblies solely based on Illumina technology may be erroneous, in particular, in case of the Acaryochloris genomes which are among the biggest (Swingley et al., 2008; Miller et al., 2011) and most complex genomes in the phylum cyanobacteria (Hess, 2011). Therefore, the sequence assembly linking this nitrogenase gene cluster to the neighboring genes with homologs in A. marina was verified by PCR yielding three amplicons of 365, 2802 and 2182 nt (Figure 2). Among the 22 N2 fixation-related genes are the nifHDK genes encoding the three different nitrogenase subunits, genes related to molybdenum cofactor biosynthesis or transport (hesAB, nifBENX, modABC), Fe-S cluster assembly (nifSU), nif-specific ferredoxins (fdxHN) and a flavodoxin. On the left side, this cluster is flanked by a glgP gene for a putative glycogen/starch/alpha-glucan phosphorylase, closely related to A. marina gene AM1_2114, and on the right side by several genes of different phylogenetic matches, including four restriction/modification (RM) genes, followed by 12 homologs of A. marina genes AM1_4104 to AM1_4121 (Figure 1). Very surprisingly, the RM genes are interspersed by a gene encoding a protein of the XisH super family. The only known function of these proteins is the cell-type specific excision of a 55-kb element interrupting the fdxN gene during heterocyst differentiation in nitrogen-fixing filamentous cyanobacteria of the Nostocales group (Ramaswamy et al., 1997). The function of this XisH homolog in a unicellular non-heterocystous cyanobacterium appears enigmatic: the fdxN gene in Acaryochloris sp. HICR111A is not interrupted by a genetic element, moreover, the sequence comparison of XisH yielded mainly matches in the Nostocales, but also in some unicellular dinitrogen fixers such as Cyanothece and Crocosphaera, as well as in some cyanobacteria lacking this ability, such as Microcystis aeruginosa and, most interestingly, A. marina (gene AM1_4785).

Three linking PCRs were performed to validate the sequence assembly of contig 62 of the Acaryochloris sp. HICR111A sequencing project containing the nif gene cluster (for a detailed map of the cluster see Figure 1). The positions of forward and reverse primers are indicated by red arrows. Genes with the closest orthologs found in Acaryochloris marina are colored green; genes with the closest orthologs found in the Synechococcus sp. PCC 7335 nif gene cluster in the same order are colored orange; other genes in gray. Genes are clustered into boxes with only those important for the PCRs shown as single arrows. Arrows and boxes are not drawn to scale. The inset shows a 0.8% agarose gel with the three PCR products compared to a λ (PstI) ladder.
When the 22 core nif genes were compared against sequence databases, we noticed a remarkable co-linearity with a similar gene cluster in Synechococcus PCC 7335 including the same peculiar drop in GC content upstream of nifB (Figure 1). The average blastP amino-acid identity of all nif genes is 80%, showing the highest similarity between the homologs of the structural genes nifHDK (90% average identity) and the lowest similarity between the homologs of the nif related genes from cysE to nifT (73% average identity, compare Figure 1). Of all proteins in the NCBI database, Synechococcus PCC 7335 homologs had the highest similarity to those of Acaryochloris sp. HICR111A in all cases (Table 2). Phylogenetic analysis of Acaryochloris sp. HICR111A Nif proteins confirmed a very high sequence similarity, locating the respective orthologs from both strains in the same clade. For these analysis, we concatenated the NifHDK sequences from 19 different cyanobacteria and used the homologs from Rhodospirillum rubrum as outgroup (Figure 3b). Remarkable is not only the tight clustering of Synechococcus PCC 7335 and Acaryochloris sp. HICR111A Nif proteins in a single clade, but also the totally unexpected closer relatedness to the respective orthologs in Trichodesmium erythraeum (Figure 3b). Furthermore, a discrepancy to the general phylogeny of these species is evident when comparing NifHDK phylogeny to the 16 S rRNA-based phylogeny (Figure 3a). This pattern of presence/absence of nif genes throughout different clades can only be explained by events of selective gene gains or losses and horizontal gene transfer must have played an important role in these processes. Of special interest was also the fact that Acaryochloris sp. HICR111A expresses a fused version of the genes nifE and nifN (Figure 1). Of all fully sequenced diazotrophic cyanobacteria, only four strains have such a fused version of nifEN (Figure 3c). Remarkably, besides Acaryochloris sp. HICR111A, these are Synechococcus PCC 7335 (GenBank accession: EDX83984), Trichodesmium erythraeum (NCBI reference sequence: YP_723620.1), and Anabaena variabilis (nifEN2, GenBank accession: AAA93024) which has one fused and one separate version in the nif2 and nif1 clusters, respectively. The expression of nifEN was verified experimentally for Trichodesmium (Dominic et al., 2000) and is evident at the DNA sequence level in the other three organisms. The phylogenetic analysis (Figure 3c) of the NifEN protein shows that the occurrence of fused nifEN coincides with higher sequence similarity, clearly separating the Acaryochloris nif genes from those of typical unicellular diazotrophic cyanobacteria. This strengthens the hypothesis that the Acaryochloris nif cluster had originally been acquired by horizontal gene transfer.

Bayesian trees showing the phylogeny of 16S ribosomal RNA sequences and of concatenated amino acid sequences of nitrogenase structural proteins NifHDK and NifEN. (a) Phylogenetic analysis of 40 sequences of 16S ribosomal RNA from N2 fixing and other cyanobacteria. Rhodospirillum rubrum ATCC 11170 served as outgroup; one subtree showing 11 species of marine picocyanobacteria (Prochlorococcus and Synechococcus) was collapsed for better overview. (b) Phylogenetic analysis of 21 amino acid sequences of concatenated NifHDK proteins from 19 different cyanobacteria and Rhodospirillum rubrum ATCC 11170 (outgroup). For Anabaena variabilis, NifHDK protein sequences from nif clusters 1 (expressed in the heterocysts) and 2 (expressed in the vegetative cells under anaerobic conditions) were included. Variable C- and N-termini were cut out of the alignment (with a maximum of 55 and 33 amino acids deleted at the NifH-NifD and NifD-NifK junctions, respectively). (c) Phylogenetic analysis of the fused NifEN protein in N2 fixing cyanobacteria and Rhodospirillum rubrum (outgroup). For all strains without the fused version of these genes, amino-acid sequences were concatenated for this analysis. Strains that express the fused nifEN are indicated by a gray arrow. In all trees, Acaryochloris strains and members of the Nostocales are highlighted. Synechococcus sp. 7335 is framed for comparison with Acaryochloris. All strains from (b) and (c) are included in (a).
Expression of Acaryochloris sp. HICR111A nif genes is controlled by the light/dark cycle
Horizontally transferred genes are not necessarily actively expressed genes. They might just have been acquired and may now be in the process of decline. The incompatibility between photosynthetic oxygen production and the well-known oxygen sensitivity of the nitrogenase enzyme should also be considered. Therefore, we set out to measure the expression of these genes in two different ways.
First, we measured expression of nifD in N-deplete (−N) versus N-replete (+N) conditions in cultures growing under natural light/dark cycles (German summer, varying light intensities and length of day/night). Using quantitative RT-PCR, we observed a several 1000-fold induction of gene expression in the –N cultures during the dark, starting already toward the end of the light phase (Figure 4a) and detected the appearance of a strongly accumulating transcript of about 2.3 kb in northern blot hybridization in −N cultures only (Figure 4b). The large size of the nifD transcript characterizes it as a likely polycistronic mRNA. Similar results were obtained for other probed nif genes (not shown). The fact that nif genes were already induced before the onset of the dark phase indicates that the circadian clock probably played a role in the timing of the expression, the key components of which are encoded by the kai genes present in this genome. Follow-up experiments, including hoaxing experiments with shifted light and dark phases, should give further insight.

Transcript levels of nifD by quantitative real-time reverse transcription PCR (RT-qPCR) and Northern blot analysis in two different experiments. (a) Relative transcript levels of nifD obtained by qPCR in Acaryochloris HICR111A grown with (+N) and without (−N) combined nitrogen (nitrate) in summer under natural light conditions. The shaded gray area shows the measured light intensity during the sampling period. The data were normalized to 16S rRNA and calibrated to the 12:00 o′clock nitrate sample. (b) Northern blot analysis of nifD transcripts during the dark phase in Acaryochloris HICR111A grown under controlled conditions with a defined 12-h/12-h light/dark cycle. –N: grown without combined nitrogen; NO: grown with nitrate (NO3−). 5S rRNA was hybridized as internal standard.
To prove that the induction of gene expression detected at the mRNA level actually results in enzymatically active nitrogenase, we measured nitrogenase activity during six 2-h periods and, in parallel, analyzed the expression of nif genes in each sample. This experiment was done twice with two different cultures. Figure 5a shows that both cultures, despite different periods of adaptation to the final light and temperature conditions (9 and 22 days, respectively) and differing time points of their shift to N-deplete medium (9 and 7 days, respectively), were performing dinitrogen fixation in the second half of the 12-h dark period. In contrast, the expression of three different genes, nifB, nifD and fdxH, measured at the RNA level, started to increase with the beginning of the dark period, whereas the gene for a typical photosynthetic protein, psbA, chosen for control, showed an inverse behavior with high expression during the light period and a minimum at night (Figures 5b and c). Thus, compared to the induction of nifB, nifD, and fdxH expression at the RNA level, the peaks of dinitrogen fixation are offset by about 6 hours. The maximum observed nitrogenase activity rate was 485 nmol C2H4/mg Chl d /2h−1. This relatively low N2 fixation rate corresponds to the slow growth of Acaryochloris sp. HICR111A observed in culture. Our measurements were done under microaerobic conditions. In an aerobic environment the small diameter of the cells would make it impossible for Acaryochloris sp. HICR111A to maintain an anaerobic intracellular environment for nitrogenase, as the diffusion of oxygen would be higher than the scavenging of O2 by respiration. However, Acaryochloris sp. HICR111A forms aggregates in culture (Mohr et al., 2010). Such aggregates may enhance anoxic conditions in the dark and hence support the formation of active nitrogenase. The energy required for N2 fixation in Acaryochloris sp. HICR111A is most likely obtained from the aerobic respiration of the intracellular glycogen reserve, possibly involving the enzymatic activity of the putative glycogen phosphorylase encoded by the glgP gene next to the nif gene cluster. However, we can also not exclude the possibility of anaerobic fermentation when aggregates of biofilms of this organism turn anoxic at night as observed in other cases (Stal and Moezelaar, 1997).

Nitrogenase activity and real-time reverse transcription PCR (RT-qPCR) analysis of relative transcript levels from genes involved in dinitrogen fixation and photosynthesis. Shown are two different experiments with Acaryochloris HICR111A cultures grown under a 12-h/12-h light-dark cycle. Nitrogenase activity (acetylene reduction) and transcript levels were measured at six points in time during the 12-h/12-h light–dark cycle. (a) Nitrogenase activity. The culture used for measurements on 2 June 2011 had adapted to the final temperature and light conditions for 9 days and was shifted to nitrogen-deplete medium 9 days before the assay, the culture used for measurements on 3 June 2011 had adapted to the final temperature and light conditions for 22 days and was shifted to nitrogen-deplete medium 7 days before the assay. Data points are set in the middle of the acetylene incubation time (total incubation 2 h). (b and c) Relative transcript levels of nifB, nifD, and fdxH (nif cluster genes) as well as psbA in nitrogen-deplete culture samples. All transcript data were normalized to 16 S rRNA and calibrated to their respective mRNA levels in the one sample of nitrate-supplemented culture harvested at 12:00 and 22:00 o′clock for the first (a) and second (b) experiment, respectively (relative expression for these samples is 1). The black and white bars above each graph indicate light and dark periods.
Conclusions
Acaryochloris sp. HICR111A differs from other chlorophyll d-containing Acaryochloris strains by the presence of a complete and fully functional cluster of nitrogenase genes that might have been acquired through horizontal gene transfer from other unicellular marine cyanobacteria. Nitrogenase genes were expressed and nitrogenase activity was detected in the second half of the dark phase. Nitrogenase activity was comparatively low but in line with the slow growth rate of this organism. We conclude that Acaryochloris sp. HICR111A may thrive diazotrophically in its natural habitat by forming aggregates or biofilms at or close to the intertidal zone of tropical reefs that are characterized by low oxygen levels during the night.
Accession codes
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ . (1990). Basic local alignment search tool. J Mol Biol 215: 403–410.
Bergman B, Gallon J, Rai A, Stal LJ . (1997). N2 Fixation by non-heterocystous cyanobacteria. FEMS Microbiol Rev 19: 139–185.
Berman-Frank I, Lundgren P, Chen Y-B, Küpper H, Kolber Z, Bergman B et al. (2001). Segregation of nitrogen fixation and oxygenic photosynthesis in the marine cyanobacterium Trichodesmium. Science 294: 1534–1537.
Berman-Frank I, Lundgren P, Falkowski P. . (2003). Nitrogen fixation and photosynthetic oxygen evolution in cyanobacteria. Res Microbiol 154: 157–164.
Dominic B, Zani S, Chen Y-B, Mellon MT, Zehr JP . (2000). Organization of the nif genes of the nonheterocystous cyanobacterium Trichodesmium sp. IMS101. J Phycol 36: 693–701.
Drummond A, Ashton B, Buxton S, Cheung M, Cooper A, Duran C et al. (2011). Geneious v5.4 http://www.geneious.com.
Hall TA . (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41: 95–98.
Hess WR . (2011). Cyanobacterial genomics for ecology and biotechnology. Curr Opin Microbiol 14: 608–614.
Huelsenbeck JP, Ronquist F . (2001). : Bayesian inference of phylogenetic trees. Bioinformatics 17: 754–755.
Kalendar R, Lee D, Schulman AH . (2011). Java web tools for PCR, in silico PCR, and oligonucleotide assembly and analysis. Genomics 98: 137–144.
Karl D, Michaels A, Bergman B, Capone D, Carpenter E, Letelier R et al. (2002). Dinitrogen fixation in the world's oceans. Biogeochemistry 57–58: 47–98.
Kashiyama Y, Miyashita H, Ohkubo S, Ogawa NO, Chikaraishi Y, Takano Y et al. (2008). Evidence of global chlorophyll d. Science 321: 658.
Livak KJ, Schmittgen TD . (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408.
Miller SR, Augustine S, Olson TL, Blankenship RE, Selker J, Wood AM . (2005). Discovery of a free-living chlorophyll d-producing cyanobacterium with a hybrid proteobacterial/cyanobacterial small-subunit rRNAgene. Proc Natl Acad Sci USA 102: 850.
Miller SR, Wood AM, Blankenship RE, Kim M, Ferriera S . (2011). Dynamics of gene duplication in the genomes of chlorophyll d-producing cyanobacteria: implications for the ecological niche. Genome Biol Evol 3: 601–613.
Miyashita H, Ikemoto H, Kurano N, Adachi K, Chihara M, Miyachi S . (1996). Chlorophyll d as a major pigment. Nature 383: 402.
Mohr R, Voß B, Schliep M, Kurz T, Maldener I, Adams DG et al. (2010). A new chlorophyll d-containing cyanobacterium: evidence for niche adaptation in the genus Acaryochloris. ISME J 4: 1456–1469.
Pinto F, Thapper A, Sontheim W, Lindblad P . (2009). Analysis of current and alternative phenol based RNA extraction methodologies for cyanobacteria. BMC Mol Biol 10: 79.
Ramaswamy KS, Carrasco CD, Fatma T, Golden JW . (1997). Cell-type specificity of the Anabaena fdxN-element rearrangement requires xisH and xisI. Mol Microbiol 23: 1241–1249.
Riemann L, Farnelid H, Steward GF . (2010). Nitrogenase genes in non-cyanobacterial plankton: prevalence, diversity and regulation in marine waters. Aquat Microb Ecol 61: 235–247.
Rippka R, Deruelles J, Waterbury JB, Herdman M, Stanier RY . (1979). Generic assignments, strain histories and properties of pure cultures of cyanobacteria. Microbiology 111: 1–61.
Ritchie RJ . (2006). Consistent sets of spectrophotometric chlorophyll equations for acetone, methanol and ethanol solvents. Photosynth Res 89: 27–41.
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA et al. (2000). Artemis: sequence visualization and annotation. Bioinformatics 16: 944–945.
Staal M, Lintel-Hekkert ST, Harren F, Stal LJ . (2001). Nitrogenase activity in cyanobacteria measured by the acetylene reduction assay: a comparison between batch incubation and on-line monitoring. Environ Microbiol 3: 343–351.
Stal LJ . (1988). Nitrogen fixation in cyanobacterial mats. Meth Enzymol 167: 474–484.
Stal LJ, Moezelaar R . (1997). Fermentation in cyanobacteria. FEMS Microbiol Rev 21: 179–211.
Steward WDP, Fitzgerald GP, Burris RH . (1967). In situ studies on N2 fixation using the acetylene reduction technique. PNAS 58: 2071–2078.
Swingley WD, Chen M, Cheung PC, Conrad AL, Dejesa LC, Hao J et al. (2008). Niche adaptation and genome expansion in the chlorophyll d-producing cyanobacterium Acaryochloris marina. Proc Natl Acad Sci USA 105: 2005.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S . (2011). 5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739.
Tripp HJ, Bench SR, Turk KA, Foster RA, Desany BA, Niazi F et al. (2010). Metabolic streamlining in an open-ocean nitrogen-fixing cyanobacterium. Nature 464: 90–94.
Zehr JP . (2011). Nitrogen fixation by marine cyanobacteria. Trends Microbiol 19: 162–173.
Zehr JP, Kudela RM . (2011). Nitrogen cycle of the open ocean: from genes to ecosystems. Annu Rev Marine Sci 3: 197–225.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pfreundt, U., Stal, L., Voß, B. et al. Dinitrogen fixation in a unicellular chlorophyll d-containing cyanobacterium. ISME J 6, 1367–1377 (2012). https://doi.org/10.1038/ismej.2011.199
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2011.199
Keywords
This article is cited by
-
Photosynthesis at the far-red region of the spectrum in Acaryochloris marina
Biological Research (2017)
-
Sequential splicing of a group II twintron in the marine cyanobacterium Trichodesmium
Scientific Reports (2015)
-
The primary transcriptome of the marine diazotroph Trichodesmium erythraeum IMS101
Scientific Reports (2014)
-
Subsurface Associations of Acaryochloris-Related Picocyanobacteria with Oil-Utilizing Bacteria in the Arabian Gulf Water Body: Promising Consortia in Oil Sediment Bioremediation
Microbial Ecology (2013)
-
Chlorophyll d and Acaryochloris marina: current status
Photosynthesis Research (2013)
