
Abstract
Obesity is an ongoing global epidemic and has adverse consequences for cardiovascular health. Obesity is often associated with hypertension, which is, itself, a common condition and an important cause of morbidity and mortality worldwide. Although animal models of obesity have provided extensive data on the links between obesity and hypertension, a greater understanding of the pathways linking obesity and hypertension in humans is likely to assist translation of animal data, and may, itself, identify important treatment strategies. Ultimately, this could have a substantial impact on human health, both at an individual and population level. The current review will focus specifically on studies of experimental weight gain and weight loss in humans and the following key areas, which are strongly related to blood pressure: cardiovascular function, autonomic nervous system function, metabolic function and the impact of cardiorespiratory fitness.
Similar content being viewed by others

Introduction
Obesity is an ongoing epidemic worldwide. Globally, obesity rates have more than tripled in men and doubled in women, since 1975,1 with more than 1 in 3 US adults2 and 1 in 4 UK adults being obese.3 This is of concern because obesity has marked detrimental effects on cardiovascular (CV) health.4, 5 It is associated with an increased risk of developing type 2 diabetes, dyslipidaemia, stroke and heart disease.6, 7 A key factor underlying the adverse consequences of obesity on CV health is likely to be the presence of hypertension, which is, itself, a common condition. Indeed, hypertension is currently the leading risk factor for CV disease and an important cause of morbidity and mortality worldwide.8
There is extensive evidence from cross-sectional studies that obesity and hypertension co-exist. Epidemiological evidence demonstrates a positive association between body mass index (BMI) and blood pressure (BP).9, 10, 11 Hypertension is also more frequent in obese than lean individuals12 and, among subjects in the Framingham Heart study, the prevalence of obesity was more common in subjects with borderline or established hypertension than in normotensives.13 Moreover, calorie restriction leading to weight loss is commonly associated with a fall in BP,14 and weight loss is often encouraged as an effective, non-pharmacological strategy for preventing development of sustained hypertension.
Despite the extensive epidemiological evidence linking obesity and hypertension, the precise mechanisms of obesity-associated hypertension remain unclear. Although cross-sectional studies have contributed much to our understanding, longitudinal studies of weight gain and weight loss allow a much greater understanding of the relationship between obesity and BP, the direction of causality and the underlying pathophysiology. A wealth of overfeeding and calorie restriction studies have been undertaken in animals, which have contributed greatly to our understanding of some of the mechanisms involved in obesity-associated hypertension, particularly in regard to the hypothalamic-pituitary-adrenal axis (as reviewed extensively by Kotsis et al.15 and Esler et al.16). Moreover, animal models of obesity have been extremely useful in identifying and exploring novel targets for anti-obesity drugs.17 However, animals are not humans, and species differences do exist in CV structure and function, which may be differentially affected by weight gain and/or loss. Indeed, a number of mechanisms identified in animal models of obesity do not translate strongly to humans.16 Moreover, despite the success of anti-obesity therapies in preclinical models, translation into humans has been largely unsuccessful due to adverse safety profiles.18 As such, a greater understanding of the pathways linking obesity and hypertension in humans is likely to assist translation of animal data, and may, itself, identify important treatment strategies, which ultimately could have a substantial impact on health, both at an individual and population level.
A brief review of experimental weight gain studies previously examined the variation in susceptibility to gaining weight19 but did not examine the physiological consequences. Therefore, the current review will focus specifically on studies of experimental weight gain and weight loss in humans and the following key areas which are strongly related to BP: CV function, autonomic nervous system (ANS) function, metabolic function and the benefits of exercise. Weight gain studies are summarized in Table 1 and weight loss studies are summarized in Table 2.
CV function
Modest weight gain in humans is associated with significant changes in CV function. In a study of normal-weight males (n=14), weight gain of 5 kg was associated with increased systolic BP (5±1 mmHg, P<0.01) but not diastolic BP. In addition, beta stiffness index, a commonly used measure of arterial compliance, was increased by 13±6% and, interestingly, the degree of stiffness was associated with the level of abdominal visceral fat gain.20 This study confirmed the findings of earlier cross-sectional studies21, 22 that visceral adiposity was an important correlate of an elevated CV disease risk profile. In a further randomized-controlled trial (RCT) in 43 subjects, which consisted of weight gain or weight maintenance, an average 4.1 kg weight gain was associated with impaired flow-mediated dilatation (FMD), which was reversed with weight loss.23 The degree of FMD impairment was signficantly higher in subjects with predominantly visceral rather than subcutaneous fat accumulation. However, there were no significant changes in BP or heart rate, as a result of weight gain or loss, indicating that vascular responses are either more sensitive than, or may precede any effects on, BP. Indeed it is possible that the change in body weight, and in particular, amount of visceral fat may affect the material properties of blood vessels, independently of arterial distending pressure via mechanisms such as hyperinsulinemia,24, 25 hyperleptinaemia,26, 27, 28, 29 a greater sympathetic nervous system (SNS) activity,30, 31 and activation of the renin–angiotensin–aldosterone system.32, 33 Such mechanisms are thought to be typical of early vascular ageing (EVA); as reviewed by Nilsson et al.34
Many more studies have investigated the effects of experimental weight loss on blood pressure and CV function. In a meta-analysis, which included 25 RCTs, 4874 subjects were assessed to identify the effects of weight loss on BP.14 The mean age of the study populations ranged from 27–66 years and mean weight loss ranged from 0.6 to 11.9 kg. There was an average reduction in systolic BP of 4.44 mmHg and in diastolic BP of 3.57 mmHg, relating to a reduction in systolic BP/diastolic BP per kg of body mass of 1.05 and 0.92 mmHg, respectively. In a further study of 25 middle-aged and older individuals who were randomly assigned to losing weight (7.1 kg) via a hypocaloric diet vs. maintaining weight, brachial systolic BP and diastolic BP decreased only in the weight loss group, by 7 and 5 mmHg, respectively.35
A separate study36 compared the impact of two weight loss interventions on BP and other CV risk factors. The interventions were; (1) a goal of 10% body weight loss and (2) three meetings per year focusing on social support. Significant improvements were seen in CV risk factors such as BP, glycaemia, triglycerides and HDL cholesterol with modest weight losses of 5–10%, as well as increased odds of achieving a 5 mmHg decrease in systolic BP and diastolic BP, 0.5% reduction in HbA1c, a 40 mg dl−1 decrease in triglycerides and a 5 mg dl−1 increase in HDL cholesterol. The relationship between the degree of weight loss and improvement in the CV risk profile was almost exponential. In a further 344 overweight and obese males and females aged between 20 and 45 years, a mean weight loss of 6.7 kg over 12 months reduced aortic, but not brachial pulse wave velocity (PWV).37 In linear, mixed, log-transformed models which included age, sex, race and time as baseline, reductions in weight and BMI were signficantly associated with the reduction in aortic PWV. In further models, adjusting for variations in mean arterial pressure, the reduction in aortic PWV was positively associated with reduction in BMI and carotid artery diameter.
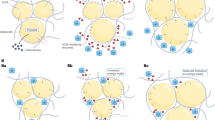
Existing intervention studies have examined the impact of weight gain or loss on novel aspects of CV function such as arterial stiffness and endothelial function. However, as the mean (arterial) BP is a function of cardiac output (CO) and peripheral vascular resistance (PVR), it is notable that the existing studies described above have not examined these key haemodynamic mechanisms. It is well-established that CO is higher in those with increased body size,38 and cross-sectional observations demonstrate a marked elevation of CO in overweight or obese subjects in the early stages of BP elevation.39, 40, 41 Moreover, longitudinal, observational data in 351 children (aged ~10 years) demonstrate a greater increase in SBP in those with the greatest increase in BMI over a two-year follow-up, and that this was related to increases in CO and stroke volume, rather than arterial stiffening.11 However, recent cross-sectional data in young adults demonstrate that not all overweight and obese subjects have elevated BP despite having an elevated CO, and it is the level of PVR, rather than CO, which distinguishes between different levels of BP in overweight and obese individuals42 (Figure 1). These data strongly suggest that the ability to modulate PVR in response to an increased CO accompanying overweight/obesity may be an important determinant of the overall level of BP. Clearly this hypothesis requires testing, and further studies in humans are required to explore potential modulatory influences on vascular structure and function during weight gain and whether these relate to measurable changes in BP.

Blood pressure (BP) is stratified by level of peripheral vascular resistance (PVR), not cardiac output (CO), in overweight/obese young adults (adapted from Middlemiss et al.42). Therefore, the ability to adapt to an elevated CO with weight gain may ultimately determine the level of blood pressure. A full color version of this figure is available at Hypertension Research online.
To summarize, the available evidence suggests that weight gain and weight loss induce changes in BP and other aspects of CV function. Interestingly, the magnitude of change is, in part, related to the extent of change in visceral fat levels. However, further longitudinal, interventional studies are warranted to explore, in greater detail, the association between weight gain and CV function. These studies should examine haemodynamic mechanisms directly related to BP, such as CO and PVR, as these may provide important early insights into how BP becomes elevated with weight gain in humans.
ANS function
Since Landsberg first proposed the involvement of the SNS as an adaptive, thermogenic response to overeating,43 the role of the SNS in obesity-associated hypertension has been intensively studied and reviewed.16, 44, 45 It has been speculated that the SNS is firstly downregulated, with reduced thermogenesis contributing to obesity, but eventually upregulated, contributing to hypertension.44 However, other studies have demonstrated that SNS activation is involved in the pathophysiology of hypertension in lean individuals.46, 47 Nevertheless, sympathetic overactivity has been widely implicated in obesity-associated hypertension, and data from animal models48, 49 (and reviewed by Vickers et al.17), cross-sectional studies in humans (as reviewed by Feldstein et al.44 and Kotsis et al.50) and interventional weight gain and loss studies largely support this view.
In an observational study of 1897 Japanese males, 353 individuals (18.6%) gained weight over the 12-month study period (defined as an increase in BMI of >10%).51 Interestingly, a rise in BP (increase in mean BP >10%) was detected only in ~60% of lean subjects, despite similar increases in BMI to those in whom pressure did not rise, indicating a variable response of BP to weight gain. Levels of plasma noradrenaline, a marker of SNS activity, insulin and leptin all rose with weight gain, irrespective of the change in BP. However, significant increases in heart rate and plasma noradrenaline were detected in those individuals with accompanying BP elevation, suggesting that SNS activation is likely to be a major mechanism of BP elevation with weight gain in humans.
In 12 healthy, nonobese males52 in whom 8 weeks of overfeeding by 4184 kJ day−1 (1000 kcal day−1) resulted in a mean 5 kg weight gain, increased muscle sympathetic nerve activity (MSNA) was observed, together with an increase in systolic BP from 114±2 to 119±2 mmHg. Interestingly, the reverse pattern was observed in another study involving an acute 3 day period of semi-starvation, followed by longer term (3–5 months) energy restriction in 30 moderately obese, borderline-hypertensive females. With longer-term energy restriction, there was a mean weight reduction of 5.8 kg. The acute and long-term energy restriction resulted in reduced body weight, diastolic BP and MSNA.53
In 41 adults with the metabolic syndrome, a very low-calorie diet (VLCD) of 800 cal day−1 for 9 weeks followed by maintenance of weight loss for 1 year,54 resulted in a mean weight loss of 14.6 kg. Night-time heart rate decreased and stayed reduced after 1 year. The high frequency spectrum of heart rate variability (HRV), which represents predominantly vagal activity,55 increased during the weight loss and maintenance periods. Both clinic and ambulatory BP decreased signficantly during the VLCD period, but only clinic systolic BP remained lower at 6-months. Furthermore, a study in 18 obese, hypertensive patients involved stress testing (cold pressor, deep breathing and hand-grip test) to assess the response of the autonomic nervous system. The effects of a short-term low-calorie diet (11 days) on HRV were examined before and during each stress test. The low frequency domain of HRV, a potential56, 57 but disputed58, 59 marker of SNS activity was significantly lower during the deep breathing and cold pressor tests on the low-calorie vs. regular-calorie diet.60 Adaptations in sympathetic and parasympathetic activity on weight loss may therefore be important in ‘setting’ the BP level.
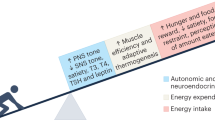
Taken together, the data from human weight gain and weight loss studies support the contention that SNS overactivity has a key role in driving increased BP with weight gain (Figure 2), although clearly, further studies in humans are required to clarify the relationship between the ANS and BP during weight gain/loss.

Pathophysiological mechanisms highlighted in interventional studies in humans by which weight gain may lead to high blood pressure (BP) and a detrimental impact on cardiometabolic health. *Metabolic syndrome includes high BP. SNS, sympathetic nervous system.
Metabolic function
It is likely that genetic,61 lifestyle and environmental factors15, 62 interact to modulate an individual’s BP and CV response to fat deposition, fat type and accumulation,63, 64 as has been demonstrated in animal models.65, 66, 67, 68, 69 Subjects with high levels of visceral adipose tissue (VAT) are at higher cardiometabolic risk, including higher incidence of hypertension.70, 71 In subjects in the Framingham Heart Study, Computed Tomography (CT) Substudy,70 ‘quality of fat’, thought to represent lipid density, macrophage accumulation and extent of arteriolar dysfunction was assessed in both visceral and subcutaneous regions, by CT attenuation. The odds ratio for hypertension, impaired fasting glucose, metabolic syndrome (a multiple risk factor syndrome including hypertension) and insulin resistance were all significantly greater with lower CT attenuation in visceral adipose tissue. Therefore, it may be that the quality, rather than quantity of adipose tissue is better related to metabolic health. In a study of 41 overfed 3180kJ (760 kcal day−1), nonobese males, lipid-storage-related gene expression that underlies visceral fat expansion was evaluated. Diacylgerol O-acyl-tranferase 2 (DGAT2) was positively associated with increased visceral fat.72 Metabolic characteristics of the subcutaneous fat were related to adipose tissue (AT) expansion and resultant increased storage of fat in the visceral depot. The results of this study support the ‘AT expansion theory,’ which hypothesizes that there is a limited capacity for the expansion of subcutaneous fat. This capacity differs from individual to individual, and once reached, will result in lipid deposition in ectopic sites, contributing to visceral fat deposition and development of the metabolic syndrome.73
Increased oxidative stress during fat accumulation has been established as an early initiator of metabolic syndrome in animal models,74 and cross-sectional studies in humans have demonstrated positive correlations between BMI and markers of systemic oxidative stress.74, 75, 76 However, factors that are known to cause oxidative stress, such as angiotensin II, which induce insulin resistance within adipose tissue77 do not necessarily cause weight gain.78 Consequently, the direction of causality still remains unclear and the role of oxidative stress in mediating vascular dysfunction in obesity requires clarification through longitudinal and interventional trials.
In another trial of overfeeding lasting 8 weeks, 28 lean, healthy adults (15 males) gained ~4 kg of body fat and an average 4.6±1.6 kg of body weight.79 Twenty-four hour insulin levels (measured as area-under-curve) positively correlated with body fat measured with dual energy X-ray absorptiometry, although low insulin sensitivity was not a precursor to upper body fat gain, despite previously reported cross-sectional associations between insulin resistance and upper body obesity.80
The evidence from interventional studies of weight loss in humans showing an improvement in metabolic function is strong. CV risk was assessed in 10 nondiabetic, morbidly obese women (age 38±13 years, pre-surgery weight 114±13 kg) before and following 36-months post bilio-pancreatic diversion (BPD).81 Insulin sensitivity more than doubled after surgery and leptin, IL-6, α-defensins, and C-reactive protein were all signficantly lowered with weight loss following surgery. These findings led the study authors to suggest that drastic weight loss via BPD demonstrates great potential in reducing the adverse inflammatory and metabolic effects of morbid obesity. These findings are supported by a meta-analysis of studies inducing weight loss via a variety of interventions,82 which demonstrated significant decreases in total cholesterol, low-density lipoprotein, very low-density lipoprotein and triglycerides. Furthermore, 845 bariatric surgery patients were analyzed 2 years post-surgery together with 845 weight-matched controls (BMI 41±4.6 kg m−2), with no intervention. Bariatric surgery patients and controls lost 28±15 and 0.5±8.9 kg, respectively. Systolic BP (7 mmHg), diastolic BP (5 mmHg), triglycerides (0.6 mmol), glucose (1 mmol l−1), insulin (10.7 mmol l−1), LDL cholesterol (0.19 mmol l−1), and HDL cholesterol (0.17 mmol l−1) were all signficantly lower in patients vs. controls.83 Ten years following the baseline visit, it was demonstrated that, compared with conventional therapy, bariatric surgery still appeared to be a viable option for the treatment of severe obesity and improvement in CV risk factors.84 However, it remains unclear whether a causal relationship exists between changes in metabolic parameters and changes in BP as a result of weight gain or loss. Indeed, risk factors tend to cluster and it may well be the case that metabolic and BP changes occur in parallel.
Impact of cardiorespiratory fitness
Regular aerobic exercise holds significant benefits for CV health. Indeed, several large outcome studies demonstrate that cardiorespiratory fitness (CRF) predicts mortality risk in the general population. The association between BMI, exercise capacity and mortality risk was assessed in 4183 hypertensive veterans (mean age 63.3±10.5 years; mean follow-up of 7.2 years) who undertook an incremental exercise test and were grouped according to body weight and level of fitness.85 There was a strong, inverse association between mortality risk and exercise capacity. Interestingly, mortality risks for the overweight-highly fit and obese-highly fit individuals were 60 and 78% lower than normal-weight, unfit individuals, highlighting that being overweight/obese but having high CRF confers a survival advantage over those are of normal weight but unfit, particularly in older individuals. In a further large outcome study involving 12 417 males (aged 40–70 years), CRF was assessed by a maximal exercise test. Compared with normal-weight, highly fit males, the underweight, unfit males had the highest mortality risk (4.5 (3.1–6.6)), whereas highly fit but overweight men had the lowest mortality risk (0.4 (0.3–0.6)). Altogether, these findings reflect the importance of obtaining and maintaining a high fitness level, regardless of weight status.86
Observational data from 25 639 individuals who participated in the EPIC-Norfolk Population and were followed up for 11.4 years87 demonstrate that physical inactivity and high abdominal adiposity independently correlate with high BP, which increased coronary heart disease (CHD) risk. A positive association existed between systolic and diastolic BP and waist circumference tertiles in both low and high fitness groups. Moreover, within each waist circumference tertile, active subjects had a lower systolic BP than inactive subjects. These data demonstrate the importance of cardiorespiratory fitness and an active lifestyle for reducing CV risk in the long-term and lend further support to the concept that CRF may counterbalance the effects of overweight and obesity.
The beneficial influence of CRF was prospectively explored in a study of experimental weight gain in 12 young males.88 As expected, at baseline, those with a signficantly higher fitness level had lower levels of body fat (−13±1.7 vs. 16.9±1.3 kg) and abdominal fat (49±6 vs. 80±14 cm2) than their less fit counterparts. However, despite similar weight gain, fitter study participants had smaller increases in systolic BP and diastolic BP compared with less fit participants (−1±3 vs. 5±1 mmHg). After weight gain, an inverse correlation was demonstrated between fitness and systolic BP (r=−0.64) and diastolic BP (r=−80) with these relationships remaining significant after adjusting for the amount of visceral fat. These findings are supported by cross-sectional data in 184 males and 223 females, where those with high levels of visceral fat had higher BP, which was independent of fitness level.89
Weight loss studies largely demonstrate the beneficial influence of exercise on CV health. A study of weight loss in 110 females was undertaken which included a dietary component combined with either aerobic exercise, resistance exercise or a combination of both.90 Regardless of the exercise type, in the diet plus exercise intervention, PWV, carotid intima-media-thickness, body weight, waist circumference, and total and low-density lipoprotein cholesterol levels were significantly decreased. High-density lipoprotein cholesterol levels and VO2max increased over the study period. However, the combination of aerobic and resistance exercise together with the dietary intervention was the most beneficial regime in overall weight management and in the reduction of subclinical cardiometabolic and atherosclerosis risk, particularly in females with abdominal obesity.
A calorie-restricted diet, exercise and a combination of diet and exercise (D+EX) were also assessed as potential weight loss strategies in 30 obese, hypertensive men over 24 weeks.91 The D+EX subgroup showed the most significant reductions in weight (21 kg), plasma noradrenaline and insulin concentrations, vs. the diet-only (16.2 kg) and exercise-only (16.6 kg) subgroups. In addition, after 4 weeks, subjects in the combined D+EX group had reductions in HOMA-IR (Homeostasis Model Assessment—Insulin Resistance) leptin, BMI, total body fat mass, waist-to-hip ratio and BP, which occurred earlier than the diet or exercise alone groups. After 24 weeks, BMI, total body fat mass and BP levels were significantly lower in the combined D+EX group than in the diet or exercise alone groups, with marked reductions in systolic BP (20 mmHg), diastolic BP (18 mmHg) and total fat mass (15.7 kg).
Not all trials involving lifestyle interventions have been completely successful in demonstrating benefits on CV outcome. The Look AHEAD trial92 examined 5145 type 2 diabetic patients to assess intentional weight loss on CV morbidity and mortality. Patients were randomly assigned to two groups; ‘intensive lifestyle intervention’ (ILI), which consisted of small group sessions, strict food prescriptions of 5021–7531 kJ day−1 (1200–1800 kcal day−1) via meal plans/replacements, ⩾175 min week−1 of moderate intensity physical activity and food/activity diary-recording; or ‘diabetes, support and education’ (DSE), as part of usual routine care. Patients were followed up after 13.5 years. The ILI and DSE group lost 4.7 and 2.1% of their initial weight, respectively. However, no beneficial effects on all-cause mortality, or other CV disease outcomes were seen as a result of the intensive intervention.
Nevertheless, the majority of studies strongly support the notion that physical inactivity may be as important a risk factor for CV disease as being overweight or obese. However, there is a shortage of prospective, interventional studies examining whether CRF can prevent or attenuate the adverse physiological consequences of gaining weight.
Implications for therapy
The mechanisms of obesity-associated hypertension in humans highlighted in this review hold a number of important implications for selecting the ideal non-pharmacological or pharmacological therapies. Weight loss and, perhaps more importantly, increased levels of physical activity have beneficial effects on BP, and may avoid the need for drug therapy and associated side-effects, which could become considerable over the life-course. However, in humans, weight loss programmes tend to be unsuccessful in the longer term93 and drug therapy is probably required in the majority of cases. Tailoring therapy towards the underlying pathophysiology would seem to be sensible and, in this regard, centrally-acting therapies targeted towards reducing SNS outflow or β-adrenoreceptor blocking drugs may be useful. They may also attenuate key haemodynamic abnormalities such as a high CO, which may be particularly beneficial in young individuals. However, there is evidence to suggest that β-adrenoreceptors are downregulated in patients with neurogenic hypertension, presumably due to prolonged SNS activation.44 Moreover, β-adrenoreceptors are associated with weight gain and increased insulin resistance94 and, therefore, may not be the ideal choice in the longer term. Sympathetic de-activation with catheter-based renal denervation is an emerging therapeutic area, although with an uncertain future.95 Irrespective of the nature of therapeutic interventions, further studies in humans, which are underpinned by sound scientific rationale, are clearly required.
Summary
There is still much that we do not understand in the complex, multifactorial phenomenon of obesity. Moreover, obesity and hypertension are becomingly increasingly prevalent, although the precise mechanisms underlying obesity-associated hypertension remain relatively poorly understood. Although a wealth of data from animal models exists, further well-controlled mechanistic studies in humans are necessary. Unfortunately, overfeeding studies in humans are uncommon due to the adverse psychological, physiological and aesthetic associations with gaining weight. Nevertheless, the studies examined in this review have demonstrated that weight gain and weight loss are associated with significant changes in BP and associated aspects of CV function, and that activation of the SNS appears to have a key role in these changes. In addition, the type and location of fat accumulation, the type of macronutrient consumed and method of weight reduction all have significant implications for CV and metabolic function. Indeed, visceral fat accumulation, in particular, has been highlighted as a predominant factor in the obesity milieu. Finally, it appears that physical inactivity, irrespective of body weight or BMI status is an important determinant of CV risk, an effect likely to be mediated via unfavorable changes in CV function leading to elevated BP. It is clear, however, that the physiological challenge of obesity can be ameliorated by weight loss be it via dietary, lifestyle or surgical methods, and gaining physical fitness.
References
NCD Risk Factor Collaboration (NCD-RisC). Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016; 387: 1377–1396.
Ogden CL, Carroll MD, Kit BK, Flegal KM . Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 2014; 311: 806–814.
Centre for Public Health Excellence at NICE (UK), National Collaborating Centre for Primary Care (UK). Obesity: The Prevention, Identification, Assessment and Management of Overweight and Obesity in Adults and Children. National Institute for Health and Clinical Excellence Guidance: London, UK, 2006.
Krzesinski P, Stanczyk A, Piotrowicz K, Gielerak G, Uzieblo-Zyczkowska B, Skrobowski A . Abdominal obesity and hypertension: a double burden to the heart. Hypertens Res 2016; 39: 349–355.
Dimitriadis K, Tsioufis C, Mazaraki A, Liatakis I, Koutra E, Kordalis A, Kasiakogias A, Flessas D, Tentolouris N, Tousoulis D . Waist circumference compared with other obesity parameters as determinants of coronary artery disease in essential hypertension: a 6-year follow-up study. Hypertens Res 2016; 39: 475–479.
Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, McQueen M, Budaj A, Pais P, Varigos J, Lisheng L INTERHEART Study Investigators.. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case–control study. Lancet 2004; 364: 937–952.
Ford ES, Ajani UA, Croft JB, Critchley JA, Labarthe DR, Kottke TE, Giles WH, Capewell S . Explaining the decrease in U.S. deaths from coronary disease, 1980–2000. N Engl J Med 2007; 356: 2388–2398.
Weidmann P, de Courten M, Bohlen L . Insulin resistance, hyperinsulinemia and hypertension. J Hypertens Suppl 1993; 11: S27–S38.
Landsberg L, Aronne LJ, Beilin LJ, Burke V, Igel LI, Lloyd-Jones D, Sowers J . Obesity-related hypertension: pathogenesis, cardiovascular risk, and treatment: a position paper of The Obesity Society and the American Society of Hypertension. J Clin Hypertens 2013; 15: 14–33.
Mancia G, Fagard R, Narkiewicz K, Redon J, Zanchetti A, Bohm M, Christiaens T, Cifkova R, De Backer G, Dominiczak A, Galderisi M, Grobbee DE, Jaarsma T, Kirchhof P, Kjeldsen SE, Laurent S, Manolis AJ, Nilsson PM, Ruilope LM, Schmieder RE, Sirnes PA, Sleight P, Viigimaa M, Waeber B, Zannad F,, Task Force Members. ESH/ESC Guidelines for the management of arterial hypertension: the Task Force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens 2013; 31: 1281–1357.
McGavock JM, Torrance B, McGuire KA, Wozny P, Lewanczuk RZ . The relationship between weight gain and blood pressure in children and adolescents. Am J Hypertens 2007; 20: 1038–1044.
Stamler R, Stamler J, Riedlinger WF, Algera G, Roberts RH . Weight and blood pressure. Findings in hypertension screening of 1 million Americans. JAMA 1978; 240: 1607–1610.
Kannel WB . Risk stratification in hypertension: new insights from the Framingham Study. Am J Hypertens 2000; 13: 3S–10S.
Neter JE, Stam BE, Kok FJ, Grobbee DE, Geleijnse JM . Influence of weight reduction on blood pressure: a meta-analysis of randomized controlled trials. Hypertension 2003; 42: 878–884.
Kotsis V, Nilsson P, Grassi G, Mancia G, Redon J, Luft F, Schmieder R, Engeli S, Stabouli S, Antza C, Pall D, Schlaich M, Jordan J,, WG on Obesity, Diabetes, the High Risk Patient, European Society of Hypertension.. New developments in the pathogenesis of obesity-induced hypertension. J Hypertens 2015; 33: 1499–1508.
Esler MD, Eikelis N, Lambert E, Straznicky N . Neural mechanisms and management of obesity-related hypertension. Curr Cardiol Rep 2008; 10: 456–463.
Vickers SP, Jackson HC, Cheetham SC . The utility of animal models to evaluate novel anti-obesity agents. Br J Pharmacol 2011; 164: 1248–1262.
Heal DJ, Gosden J, Smith SL . Regulatory challenges for new drugs to treat obesity and comorbid metabolic disorders. Br J Clin Pharmacol 2009; 68: 861–874.
Vanltallie TB . Resistance to weight gain during overfeeding: a NEAT explanation. Nutr Rev 2001; 59: 48–51.
Orr JS, Gentile CL, Davy BM, Davy KP . Large artery stiffening with weight gain in humans: role of visceral fat accumulation. Hypertension 2008; 51: 1519–1524.
Sironi AM, Petz R, De Marchi D, Buzzigoli E, Ciociaro D, Positano V, Lombardi M, Ferrannini E, Gastaldelli A . Impact of increased visceral and cardiac fat on cardiometabolic risk and disease. Diabet Med 2012; 29: 622–627.
Rothney MP, Catapano AL, Xia J, Wacker WK, Tidone C, Grigore L, Xia Y, Ergun DL . Abdominal visceral fat measurement using dual-energy X-ray: association with cardiometabolic risk factors. Obesity 2013; 21: 1798–1802.
Romero-Corral A, Sert-Kuniyoshi FH, Sierra-Johnson J, Orban M, Gami A, Davison D, Singh P, Pusalavidyasagar S, Huyber C, Votruba S, Lopez-Jimenez F, Jensen MD, Somers VK . Modest visceral fat gain causes endothelial dysfunction in healthy humans. J Am Coll Cardiol 2010; 56: 662–666.
Paolisso G, Manzella D, Rizzo MR, Barbieri M, Varricchio G, Gambardella A, Varricchio M . Effects of insulin on the cardiac autonomic nervous system in insulin-resistant states. Clin Sci 2000; 98: 129–136.
Emdin M, Gastaldelli A, Muscelli E, Macerata A, Natali A, Camastra S, Ferrannini E . Hyperinsulinemia and autonomic nervous system dysfunction in obesity: effects of weight loss. Circulation 2001; 103: 513–519.
Hall JE, Hildebrandt DA, Kuo J . Obesity hypertension: role of leptin and sympathetic nervous system. Am J Hypertens 2001; 14: 103S–115S.
Tallam LS, Stec DE, Willis MA, da Silva AA, Hall JE . Melanocortin-4 receptor-deficient mice are not hypertensive or salt-sensitive despite obesity, hyperinsulinemia, and hyperleptinemia. Hypertension 2005; 46: 326–332.
Eikelis N, Schlaich M, Aggarwal A, Kaye D, Esler M . Interactions between leptin and the human sympathetic nervous system. Hypertension 2003; 41: 1072–1079.
Haynes WG . Role of leptin in obesity-related hypertension. Exp Physiol 2005; 90: 683–688.
Lambert E, Straznicky N, Schlaich M, Esler M, Dawood T, Hotchkin E, Lambert G . Differing pattern of sympathoexcitation in normal-weight and obesity-related hypertension. Hypertension 2007; 50: 862–868.
Alvarez GE, Ballard TP, Beske SD, Davy KP . Subcutaneous obesity is not associated with sympathetic neural activation. Am J Physiol Heart Circ Physiol 2004; 287: H414–H418.
Engeli S, Negrel R, Sharma AM . Physiology and pathophysiology of the adipose tissue renin-angiotensin system. Hypertension 2000; 35: 1270–1277.
Grassi G, Seravalle G, Dell'Oro R, Trevano FQ, Bombelli M, Scopelliti F, Facchini A, Mancia G . Study C. Comparative effects of candesartan and hydrochlorothiazide on blood pressure, insulin sensitivity, and sympathetic drive in obese hypertensive individuals: results of the CROSS study. J Hypertens 2003; 21: 1761–1769.
Nilsson PM, Boutouyrie P, Cunha P, Kotsis V, Narkiewicz K, Parati G, Rietzschel E, Scuteri A, Laurent S . Early vascular ageing in translation: from laboratory investigations to clinical applications in cardiovascular prevention. J Hypertens 2013; 31: 1517–1526.
Dengo AL, Dennis EA, Orr JS, Marinik EL, Ehrlich E, Davy BM, Davy KP . Arterial destiffening with weight loss in overweight and obese middle-aged and older adults. Hypertension 2010; 55: 855–861.
Wing RR, Lang W, Wadden TA, Safford M, Knowler WC, Bertoni AG, Hill JO, Brancati FL, Peters A, Wagenknecht L,, Look AHEAD Research Group.. Benefits of modest weight loss in improving cardiovascular risk factors in overweight and obese individuals with type 2 diabetes. Diabetes Care 2011; 34: 1481–1486.
Cooper JN, Buchanich JM, Youk A, Brooks MM, Barinas-Mitchell E, Conroy MB, Sutton-Tyrrell K . Reductions in arterial stiffness with weight loss in overweight and obese young adults: potential mechanisms. Atherosclerosis 2012; 223: 485–490.
Collis T, Devereux RB, Roman MJ, de Simone G, Yeh J, Howard BV, Fabsitz RR, Welty TK . Relations of stroke volume and cardiac output to body composition: the strong heart study. Circulation 2001; 103: 820–825.
McEniery CM, Yasmin, Wallace S, Maki-Petaja K, McDonnell B, Sharman JE, Retallick C, Franklin SS, Brown MJ, Lloyd RC, Cockcroft JR, Wilkinson IB,, ENIGMA Study Investigators.. Increased stroke volume and aortic stiffness contribute to isolated systolic hypertension in young adults. Hypertension 2005; 46: 221–226.
Messerli FH . Cardiovascular effects of obesity and hypertension. Lancet 1982; 1: 1165–1168.
Messerli FH, Christie B, DeCarvalho JG, Aristimuno GG, Suarez DH, Dreslinski GR, Frohlich ED . Obesity and essential hypertension. Hemodynamics, intravascular volume, sodium excretion, and plasma renin activity. Arch Intern Med 1981; 141: 81–85.
Middlemiss JE, Miles KL, McDonnell BJ, Yasmin, Maki-Petaja KM, Cockcroft JR, Wilkinson IB, McEniery CM,, ENIGMA Study Investigators.. Mechanisms underlying elevated SBP differ with adiposity in young adults: the Enigma study. J Hypertens 2016; 34: 290–297.
Landsberg L . Diet, obesity and hypertension: an hypothesis involving insulin, the sympathetic nervous system, and adaptive thermogenesis. Q J Med 1986; 61: 1081–1090.
Feldstein C, Julius S . The complex interaction between overweight, hypertension, and sympathetic overactivity. J Am Soc Hypertens 2009; 3: 353–365.
Mancia G, Grassi G . The autonomic nervous system and hypertension. Circ Res 2014; 114: 1804–1814.
Weber MA, Neutel JM, Smith DH . Contrasting clinical properties and exercise responses in obese and lean hypertensive patients. J Am Coll Cardiol 2001; 37: 169–174.
Reims HM, Fossum E, Hoieggen A, Moan A, Eide I, Kjeldsen SE . Adrenal medullary overactivity in lean, borderline hypertensive young men. Am J Hypertens 2004; 17: 611–618.
Borne AT, Truett AA, Monteiro MP, Volaufova J, West DB . Changes in skeletal muscle vascular resistance with weight gain: associations with insulin and sympathetic activity. Obes Res 1999; 7: 68–75.
Hwang IS, Ho H, Hoffman BB, Reaven GM . Fructose-induced insulin resistance and hypertension in rats. Hypertension 1987; 10: 512–516.
Kotsis V, Stabouli S, Papakatsika S, Rizos Z, Parati G . Mechanisms of obesity-induced hypertension. Hypertens Res 2010; 33: 386–393.
Masuo K, Mikami H, Ogihara T, Tuck ML . Weight gain-induced blood pressure elevation. Hypertension 2000; 35: 1135–1140.
Gentile CL, Orr JS, Davy BM, Davy KP . Modest weight gain is associated with sympathetic neural activation in nonobese humans. Am J Physiol Regul Integr Comp Physiol 2007; 292: R1834–R1838.
Andersson B, Elam M, Wallin BG, Bjorntorp P, Andersson OK . Effect of energy-restricted diet on sympathetic muscle nerve activity in obese women. Hypertension 1991; 18: 783–789.
Laaksonen DE, Laitinen T, Schonberg J, Rissanen A, Niskanen LK . Weight loss and weight maintenance, ambulatory blood pressure and cardiac autonomic tone in obese persons with the metabolic syndrome. J Hypertens 2003; 21: 371–378.
Malliani A, Pagani M, Lombardi F, Cerutti S . Cardiovascular neural regulation explored in the frequency domain. Circulation 1991; 84: 482–492.
Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. Heart rate variability: standards of measurement, physiological interpretation and clinical use. Circulation 1996; 93: 1043–1065.
Parati G, Saul JP, Di Rienzo M, Mancia G . Spectral analysis of blood pressure and heart rate variability in evaluating cardiovascular regulation. A critical appraisal. Hypertension 1995; 25: 1276–1286.
Reyes del Paso GA, Langewitz W, Mulder LJ, van Roon A, Duschek S . The utility of low frequency heart rate variability as an index of sympathetic cardiac tone: a review with emphasis on a reanalysis of previous studies. Psychophysiology 2013; 50: 477–487.
Houle MS, Billman GE . Low-frequency component of the heart rate variability spectrum: a poor marker of sympathetic activity. Am J Physiol 1999; 276: H215–H223.
Ashida T, Ono C, Sugiyama T . Effects of short-term hypocaloric diet on sympatho-vagal interaction assessed by spectral analysis of heart rate and blood pressure variability during stress tests in obese hypertensive patients. Hypertens Res 2007; 30: 1199–1203.
Pausova Z, Syme C, Abrahamowicz M, Xiao Y, Leonard GT, Perron M, Richer L, Veillette S, Smith GD, Seda O, Tremblay J, Hamet P, Gaudet D, Paus T . A common variant of the FTO gene is associated with not only increased adiposity but also elevated blood pressure in French Canadians. Circ Cardiovasc Genet 2009; 2: 260–269.
Shimizu I, Yoshida Y, Minamino T . A role for circadian clock in metabolic disease. Hypertens Res 2016; 39: 483–491.
Patel P, Abate N . Body fat distribution and insulin resistance. Nutrients 2013; 5: 2019–2027.
Fujii M, Ohnishi H, Saitoh S, Akasaka H, Miura T, Mori M . The combination of abdominal obesity and high-sensitivity C-reactive protein predicts new-onset hypertension in the general Japanese population: the Tanno–Sobetsu study. Hypertens Res 2015; 38: 426–432.
Erlich Y, Rosenthal T . Effect of angiotensin-converting enzyme inhibitors on fructose induced hypertension and hyperinsulinaemia in rats. Clin Exp Pharmacol Physiol Suppl 1995; 22: S347–S349.
Suzuki M, Nomura C, Odaka H, Ikeda H . Effect of an insulin sensitizer, pioglitazone, on hypertension in fructose-drinking rats. Jpn J Pharmacol 1997; 74: 297–302.
Verma S, Bhanot S, McNeill JH . Antihypertensive effects of metformin in fructose-fed hyperinsulinemic, hypertensive rats. J Pharmacol Exp Ther 1994; 271: 1334–1337.
Martinez FJ, Rizza RA, Romero JC . High-fructose feeding elicits insulin resistance, hyperinsulinism, and hypertension in normal mongrel dogs. Hypertension 1994; 23: 456–463.
Dai S, McNeill JH . Fructose-induced hypertension in rats is concentration- and duration-dependent. J Pharmacol Toxicol Methods 1995; 33: 101–107.
Rosenquist KJ, Pedley A, Massaro JM, Therkelsen KE, Murabito JM, Hoffmann U, Fox CS . Visceral and subcutaneous fat quality and cardiometabolic risk. JACC Cardiovasc Imaging 2013; 6: 762–771.
Fox CS, Massaro JM, Hoffmann U, Pou KM, Maurovich-Horvat P, Liu CY, Vasan RS, Murabito JM, Meigs JB, Cupples LA, D'Agostino RB Sr., O'Donnell CJ . Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation 2007; 116: 39–48.
Alligier M, Gabert L, Meugnier E, Lambert-Porcheron S, Chanseaume E, Pilleul F, Debard C, Sauvinet V, Morio B, Vidal-Puig A, Vidal H, Laville M . Visceral fat accumulation during lipid overfeeding is related to subcutaneous adipose tissue characteristics in healthy men. J Clin Endocrinol Metab 2013; 98: 802–810.
Slawik M, Vidal-Puig AJ . Adipose tissue expandability and the metabolic syndrome. Genes Nutr 2007; 2: 41–45.
Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I . Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 2004; 114: 1752–1761.
Keaney JF Jr., Larson MG, Vasan RS, Wilson PW, Lipinska I, Corey D, Massaro JM, Sutherland P, Vita JA, Benjamin EJ, Framingham S . Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol 2003; 23: 434–439.
Olusi SO . Obesity is an independent risk factor for plasma lipid peroxidation and depletion of erythrocyte cytoprotectic enzymes in humans. Int J Obes Relat Metab Disord 2002; 26: 1159–1164.
Sowers JR, Whaley-Connell A, Hayden MR . The role of overweight and obesity in the cardiorenal syndrome. Cardiorenal Med 2011; 1: 5–12.
Aroor AR, DeMarco VG . Oxidative stress and obesity: the chicken or the egg? Diabetes 2014; 63: 2216–2218.
Votruba SB, Jensen MD . Insulin sensitivity and regional fat gain in response to overfeeding. Obesity 2011; 19: 269–275.
Gastaldelli A, Miyazaki Y, Pettiti M, Matsuda M, Mahankali S, Santini E, DeFronzo RA, Ferrannini E . Metabolic effects of visceral fat accumulation in type 2 diabetes. J Clin Endocrinol Metab 2002; 87: 5098–5103.
Manco M, Fernandez-Real JM, Equitani F, Vendrell J, Valera Mora ME, Nanni G, Tondolo V, Calvani M, Ricart W, Castagneto M, Mingrone G . Effect of massive weight loss on inflammatory adipocytokines and the innate immune system in morbidly obese women. J Clin Endocrinol Metab 2007; 92: 483–490.
Dattilo AM, Kris-Etherton PM . Effects of weight reduction on blood lipids and lipoproteins: a meta-analysis. Am J Clin Nutr 1992; 56: 320–328.
Sjostrom CD, Lissner L, Wedel H, Sjostrom L . Reduction in incidence of diabetes, hypertension and lipid disturbances after intentional weight loss induced by bariatric surgery: the SOS Intervention Study. Obes Res 1999; 7: 477–484.
Sjostrom L, Lindroos AK, Peltonen M, Torgerson J, Bouchard C, Carlsson B, Dahlgren S, Larsson B, Narbro K, Sjostrom CD, Sullivan M, Wedel H Swedish Obese Subjects Study Scientific Group.. Swedish Obese Subjects Study Scientific G. Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N Engl J Med 2004; 351: 2683–2693.
Faselis C, Doumas M, Kokkinos JP, Panagiotakos D, Kheirbek R, Sheriff HM, Hare K, Papademetriou V, Fletcher R, Kokkinos P . Exercise capacity and progression from prehypertension to hypertension. Hypertension 2012; 60: 333–338.
McAuley PA, Kokkinos PF, Oliveira RB, Emerson BT, Myers JN . Obesity paradox and cardiorespiratory fitness in 12,417 male veterans aged 40 to 70 years. Mayo Clin Proc 2010; 85: 115–121.
Rheaume C, Arsenault BJ, Despres JP, Faha, Boekholdt SM, Wareham NJ, Khaw KT, Chir M . Impact of abdominal obesity and systemic hypertension on risk of coronary heart disease in men and women: the EPIC-Norfolk Population Study. J Hypertens 2014; 32: 2224–2230.
Gentile CL, Orr JS, Davy BM, Davy KP . Cardiorespiratory fitness influences the blood pressure response to experimental weight gain. Obesity 2007; 15: 3005–3012.
Rheaume C, Arsenault BJ, Belanger S, Perusse L, Tremblay A, Bouchard C, Poirier P, Despres JP . Low cardiorespiratory fitness levels and elevated blood pressure: what is the contribution of visceral adiposity? Hypertension 2009; 54: 91–97.
Choo J, Lee J, Cho JH, Burke LE, Sekikawa A, Jae SY . Effects of weight management by exercise modes on markers of subclinical atherosclerosis and cardiometabolic profile among women with abdominal obesity: a randomized controlled trial. BMC Cardiovasc Disord 2014; 14: 82.
Masuo K, Rakugi H, Ogihara T, Lambert GW . Different mechanisms in weight loss-induced blood pressure reduction between a calorie-restricted diet and exercise. Hypertens Res 2012; 35: 41–47.
Look ARG, Wing RR, Bolin P, Brancati FL, Bray GA, Clark JM, Coday M, Crow RS, Curtis JM, Egan CM, Espeland MA, Evans M, Foreyt JP, Ghazarian S, Gregg EW, Harrison B, Hazuda HP, Hill JO, Horton ES, Hubbard VS, Jakicic JM, Jeffery RW, Johnson KC, Kahn SE, Kitabchi AE, Knowler WC, Lewis CE, Maschak-Carey BJ, Montez MG, Murillo A, Nathan DM, Patricio J, Peters A, Pi-Sunyer X, Pownall H, Reboussin D, Regensteiner JG, Rickman AD, Ryan DH, Safford M, Wadden TA, Wagenknecht LE, West DS, Williamson DF, Yanovski SZ . Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N Engl J Med 2013; 369: 145–154.
Elfhag K, Rossner S . Who succeeds in maintaining weight loss? A conceptual review of factors associated with weight loss maintenance and weight regain. Obes Rev 2005; 6: 67–85.
Elliott WJ, Meyer PM . Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet 2007; 369: 201–207.
Grassi G, Mark A, Esler M . The sympathetic nervous system alterations in human hypertension. Circ Res 2015; 116: 976–990.
Douketis JD, Macie C, Thabane L, Williamson DF . Systematic review of long-term weight loss studies in obese adults: clinical significance and applicability to clinical practice. Int J Obes (Lond) 2005; 29: 1153–1167.
Acknowledgements
This work was funded by the NIHR Cambridge Biomedical Research Centre.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Middlemiss, J., McEniery, C. Feeling the pressure: (patho) physiological mechanisms of weight gain and weight loss in humans. Hypertens Res 40, 226–236 (2017). https://doi.org/10.1038/hr.2016.142
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2016.142
Keywords
This article is cited by
-
Long-term effects of photobiomodulation therapy on blood pressure in obese rats induced by a high-fat diet
Lasers in Medical Science (2024)
-
A history of obesity leaves an inflammatory fingerprint in liver and adipose tissue
International Journal of Obesity (2018)
-
Wrist circumference is associated with increased systolic blood pressure in children with overweight/obesity
Hypertension Research (2018)
-
Obesity-Associated Hypertension: the Upcoming Phenotype in African-American Women
Current Hypertension Reports (2017)
