
Abstract
Evidence has consistently supported the association of obstructive sleep apnea syndrome (OSAS) with an increased prevalence of hypertension. It has also been shown that the severity of OSAS is directly correlated with the degree of blood pressure (BP) elevation and that hypertension occurring in subjects with OSAS is more likely to be severe, resistant to antihypertensive treatment and associated with alterations in day-to-night BP changes. Proposed mechanisms for the pathogenesis of OSAS-related hypertension include the activation of the sympathetic nervous system, alterations in autonomic cardiovascular (CV) modulation, the activation of the renin–angiotensin–aldosterone system, endothelial dysfunction, systemic and vascular inflammation, oxidative stress, metabolic abnormalities, arterial stiffness and alterations in cardiac function and structure. Given the adverse prognostic implications of OSAS-related hypertension for CV morbidity and mortality, the confirmation of resistant hypertension by using ambulatory BP monitoring (ABPM) and the identification of alterations in day-to-night BP changes is of the utmost importance to implement more aggressive strategies for achieving BP control. In turn, the proper identification and implementation of specific treatment strategies for OSAS (that is, continuous positive airway pressure) in subjects with resistant hypertension may promote BP control and optimize CV protection. The present paper will review the evidence supporting the association of OSAS with resistant hypertension and the proposed mechanisms for this association. It will also address the role of ABPM in the confirmation of resistant hypertension in subjects with OSAS and whether the proper identification and management of OSAS in subjects with resistant hypertension will improve BP control.
Similar content being viewed by others

Introduction
Obstructive sleep apnea syndrome (OSAS), which combines intermittent obstruction of the upper airways at night with somnolence during the day, is one of the suggested causes of secondary hypertension1 and has been associated with a severe and resistant hypertension.2, 3, 4 Evidence from longitudinal studies has indicated that OSAS may also predict the development of future hypertension.3, 5, 6, 7 However, despite the wealth of epidemiological evidence supporting the association of OSAS and hypertension, there is only partial understanding of the pathophysiological mechanisms underlying this relationship. In contrast, although OSAS and resistant hypertension have been shown to be independent predictors of cardiovascular (CV) prognosis, evidence is still needed to determine the actual prognostic relevance of their interaction independently of the other CV risk factors that are often clustered in the context of OSAS. Because of the frequent association of OSAS with resistant hypertension and in recognition of their probable synergistic effects on CV risk, current guidelines for the management of arterial hypertension have included OSAS among the modifiable causes to be ruled out during the diagnostic approach to resistant hypertension.1, 8 However, whether specific treatment strategies for OSAS (that is, the implementation of continuous positive airway pressure (CPAP)) will be effective in achieving BP control in combination with antihypertensive treatment has not been consistently shown. The present paper is aimed at critically reviewing the evidence supporting OSAS as a cause of resistant hypertension as well as the proposed mechanisms of this association. It also addresses the importance of implementing ambulatory blood pressure (BP) monitoring (ABPM) and home BP monitoring to determine whether the failure to achieve OBP control in a subject with OSAS actually corresponds to true resistant hypertension. Finally, it addresses whether and to what extent the proper management of OSAS either by pharmacological or nonpharmacological strategies is likely to improve BP control in subjects with resistant hypertension.
OSAS and BP levels
OSAS is defined as the presence of recurrent obstructive breathing events generated by complete upper airway obstruction during sleep and is accompanied by daytime symptoms and sleep hypoventilation syndrome.9 The alterations in breathing patterns in individuals with OSAS may influence many regulatory mechanisms involved in BP regulation. OSA events occurring at night (that is, alternating obstructive apnea and hyperventilation episodes during sleep) have been shown to be accompanied by acute changes in autonomic and hemodynamic parameters, which in turn induce marked increases in nighttime BP levels.10 Indeed, hypertension related to OSAS is predominantly nocturnal and frequently accompanied by a nondipper profile of BP (that is, the nocturnal BP falls <10% compared with daytime BP levels).11, 12 Nonetheless, the increase in BP levels in OSA subjects is not limited to the nighttime hours when the OSA episodes occur but is also sustained during the day. Case–control studies using 24-h ABPM have provided evidence that, compared with matched control subjects, OSAS patients show significantly higher ABP levels not only during nighttime sleep but also during daytime wakefulness.13, 14, 15
Epidemiological evidence for the association between OSAS and hypertension
Both in the general population and in cohorts of OSAS patients, a number of studies of different natures have confirmed the association between OSAS and hypertension.2, 4, 5, 16, 17, 18 Overall, these studies have indicated a variable frequency of hypertension in subjects with OSAS that can range from 35 to 80%.11, 19 Conversely, when properly investigated, OSAS has been shown to be present in up to 40% of hypertensive subjects.20 Whether the association between OSAS and hypertension is explained by other CV risk factors in addition to OSAS is still a matter of debate. For instance, OSAS is often associated with obesity, which in turn can explain ∼65–75% of cases of essential hypertension.21, 22 Indeed, some studies have indicated that much of the relationship between OSAS and hypertension can be explained by the associated obesity.6 Conversely, other reports have indicated that the association between obesity and arterial hypertension can be explained by the presence of OSAS in a substantial number of subjects. In addition to body mass index (BMI), other factors such as sex and age have been shown to significantly influence the relationship between OSAS and elevated BP levels.23 Evidence for this influence has been provided from cross-sectional16 and longitudinal studies6, 24 indicating that OSAS is more strongly associated with hypertension and resistant hypertension in young- to middle-aged adults (<50 years of age).23 This association is more frequently observed in men than in women.25 It is thus clear that disentangling the independent contributions to BP elevation of OSAS, obesity and other CV risk factors is rather difficult. Despite this difficulty, several longitudinal studies have supported the association between OSAS and hypertension independently of other potential contributing factors, such as BMI, also indicating that OSAS is not only associated with an increased risk of prevalent hypertension but may also be an independent and significant predictor of the future development of hypertension, particularly if not properly treated3, 5, 6, 7 (Figure 1).

Predicted increases in systolic blood pressure (SBP) and diastolic blood pressure (DBP) associated with sleep-disordered breathing for three body mass index (BMI) categories in the Wisconsin Sleep Cohort Study. Modified from Young et al.5 by permission.
In particular, in the Wisconsin Sleep Cohort Study, a dose–response relationship between sleep-disordered breathing at baseline and the development of hypertension after 4 years of follow-up was reported independently of baseline hypertension status, BMI, neck and waist circumference, age, sex or other potential confounders, suggesting that sleep-disordered breathing is likely to be a risk factor for hypertension and resultant CV morbidity in the general population.3
OSAS as a cause of resistant hypertension: proposed mechanisms
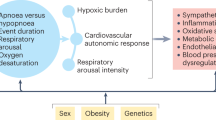
Several studies have identified OSAS as an important risk factor for resistant hypertension and have shown a dose–response relationship between OSAS severity and the degree of BP elevation.15 It has also been shown that hypertension occurring in individuals with OSAS is more likely to be severe, resistant to treatment and associated with alterations in day-to-night BP changes (that is, nocturnal hypertension and nondipping profile of BP on 24 h ABPM).15, 26, 27 Conversely, an extremely high level of OSA (∼80%) has been reported among adult patients with drug-resistant hypertension.25 It has also been shown that the rates of BP control decrease as the severity of sleep-related breathing disorders increases.2 Although all the above evidence supports a potential role of OSAS in the pathogenesis of hypertension and drug-resistant hypertension, the mechanisms by which OSAS promotes arterial hypertension are not completely understood. Evidence provided by experimental and clinical studies has indicated that the pathogenesis of OSAS-related hypertension is likely to be multifactorial, involving alterations in several regulatory systems: the activation of the sympathetic nervous system, alterations in autonomic CV modulation, the activation of the renin–angiotensin–aldosterone system, endothelial dysfunction, systemic and vascular inflammation, oxidative stress, metabolic abnormalities, arterial stiffness and alterations in cardiac function and structure. Figure 2 outlines the main mechanisms by which OSAS contributes to resistant hypertension.

Mechanisms by which obstructive sleep apnea syndrome (OSAS) contributes to resistant hypertension. CV, cardiovascular; LV, left ventricular; RAAS, renin–angiotensin–aldosterone system; ROS, reactive oxygen species; ET-1, endothelin-1; NOS, nitric oxide synthase; NO, nitric oxide; LVH, left ventricular hypertrophy; BP, blood pressure.
Sympathetic nervous system activation
The activation of the sympathetic nervous system is considered a major pathophysiological mechanism underlying the alterations in BP regulation reported in OSAS (Figure 2). This has been consistently demonstrated by several studies implementing direct techniques for the assessment of the sympathetic nervous system (that is, the recording of efferent postganglionic muscle sympathetic nerve activity via microneurography and norepinephrine plasma levels) in which an increase in central sympathetic drive was positively correlated with increases in BP levels independently of other contributing factors. The sympathetic activation in OSAS is largely explained by the stimulation of the peripheral and central chemoreflexes, triggered by the reductions in arterial oxygen content and hypercapnia, respectively. Moreover, sleep fragmentation, related to repeated arousals after each apnea/hypopnea event, might have an additional role in this context. The resultant increases in sympathetic drive to the heart and peripheral vasculature lead to significant increases in heart rate and vascular tone, which in turn are responsible for the marked increases in BP levels during the resumption of ventilation after each apneic episode28 (Figure 3a).

(a) Recordings of sympathetic nerve activity (SNA), respiration (RESP) and blood pressure (BP) during 3 min of stage II sleep, showing continuous oscillations in BP and SNA in response to the repetitive obstructive sleep apneas (OSAs). These oscillations occurred continuously during sleep, throughout all sleep stages. (b) Recordings of SNA during wakefulness in patients with OSAS and matched controls showing high levels of SNA in patients with OSA. Taken from Somers et al.28 by permission.
This increase in central sympathetic drive has also been shown to be associated with alterations in circadian BP variation (that is, the absence of nocturnal BP fall or an increase in BP at night), and nocturnal hypertension is frequently observed in OSAS patients.29 In addition, several studies using microneurography recordings have indicated that the sympathetic activation in OSAS subjects is not only limited to nighttime but may persist even after resuming a normal breathing pattern during daytime wakefulness, despite normal arterial oxygen saturation and carbon dioxide levels28, 30 (Figure 3b). Remarkably, in several studies, the long-term implementation of CPAP resulted in marked reductions in sympathetic nerve traffic28 and BP levels31 during nighttime and daytime,32 further supporting the pathogenic role of sympathetic activation in BP elevation associated with OSAS.
Alterations in autonomic CV modulation
In normal physiological conditions, the control of BP levels is achieved through a complex interplay between the central and reflex neural influences, leading to a continuous modulation of efferent sympathetic and parasympathetic nerve activity and the associated activity of neurohormonal systems primarily regulated by the hypothalamus. In OSAS, the sustained chemoreflex activation, the related adrenergic overactivity and the resulting hypertension may blunt and/or reset arterial and cardiopulmonary reflexes, which in turn may lead to chemoreflex potentiation.33, 34 In addition, the dysfunction of neural reflexogenic areas (that is, baroreflex impairment) may lead to reduced sympathoinhibition,35, 36 further contributing to adrenergic overdrive and BP elevation (Figure 2). In particular, the observation of a reduced cardiac baroreflex sensitivity (BRS; as assessed by the sequence method) and the absence of 24-h baroreflex modulation (that is, blunted increase in BRS during sleep compared with its values during wakefulness) in OSAS patients35 has provided indirect support for the concept that baroreflex dysfunction in addition to chemoreceptor stimulation by hypoxia may contribute to the acute and long-term sympathetic activation in OSAS patients (Figure 4). The depressed BRS during sleep may in turn contribute to the pathophysiology of hypertension in OSAS patients.

Relationship between spontaneous baroreflex sensitivity (BRS) and the severity of obstructive sleep apnea syndrome (OSAS), as quantified by the apnea-hypopnea index (AHI). Data are shown as individual values from 11 patients during periods of wakefulness (W), nonrapid eye movement (REM) sleep (NREM) and REM sleep (REM). Taken from Parati et al.35 by permission.
This concept has been further supported by the results of interventional studies in OSAS patients showing a significant improvement in BRS after the long-term implementation of CPAP treatment.37, 38
Other independent studies applying the spectral analysis of the variability of microneurography, BP and heart rate have provided additional evidence that the autonomic CV modulation is impaired in OSA based on the demonstration of significant increases in heart rate and sympathetic drive, reduced HRV and a marked increase in BP variability (more than double the variance in healthy controls)39 (Figure 5).

RR interval, systolic blood pressure (SBP) and their variances, and normalized low-frequency (LF) and high-frequency (HF) components of RR interval in control subjects, patients with mild OSA and patients with moderate-to-severe OSA. *P<0.05 versus control subjects. †P<0.05 versus mild OSA. Data are the means±s.e.m. Modified from Narkiewicz et al.39 by permission.
Further evidence that sleep-related breathing disorders may induce alterations in autonomic CV modulation has been provided by a recent study in untreated subjects with sleep-related breathing disorders of differing severity, which indicated that excessive daytime sleepiness is accompanied by lower BRS and a significantly higher low-to-high-frequency power ratio of heart rate variability (which is believed to be a marker of sympathetic activity) during the different stages of nocturnal sleep40 (Figure 6).

Trends of baroreflex sensitivity (BRS) and of the ratio between low- and high-frequency powers of RRI (LF/HF) in healthy controls without sleep-related breathing disorders (square symbols), in patients with excessive daytime sleepiness (EDS, open circles) and in subjects not affected by EDS (nEDS, solid circles). Taken from Lombardi et al.40 by permission.
Activation of renin–angiotensin–aldosterone system (increase in aldosterone levels)
The frequent association of OSAS with hyperaldosteronism reported in patients with resistant hypertension has led to the suggestion that both factors may interact to contribute to BP elevation.41, 42, 43 Although evidence is still needed to determine the cause of this association, it has been hypothesized that OSAS may contribute to the pathogenesis of resistant hypertension by stimulating aldosterone excretion44 (Figure 2). Evidence supporting this concept has been provided by several studies showing positive and significant correlations between plasma aldosterone concentrations and OSAS severity in patients with resistant hypertension but no correlations in normotensive subjects or in hypertensive patients treated with BP controlled.45 It is likely that aldosterone excess promotes fluid accumulation in the neck and thus increases upper airway resistance, which may increase the severity of OSA and the related increase in BP levels.20, 46 Indirect evidence favoring this concept has been provided by interventional studies in subjects with OSAS and resistant hypertension in which the addition of spironolactone to antihypertensive treatment resulted in significant reductions in the severity of OSA (that is, reductions in apnea-hypopnea index and the number of central and obstructive events) in addition to the BP-lowering effects.47 Despite this finding, evidence is still needed to convincingly demonstrate a causal relationship between excess aldosterone in OSAS and resistant hypertension.
Endothelial dysfunction
Intermittent hypoxia and the associated neural and humoral alterations and recurring BP surges during OSA episodes may contribute to endothelial function impairment. In turn, the inhibition of nitric oxide (NO) production, decreased vasodilatation and increased vasoconstriction associated with endothelial dysfunction may substantially contribute to BP elevation (Figure 2). Several studies assessing brachial artery endothelium-dependent flow-mediated dilation (FMD, an indirect marker of endothelial NO-mediated reactivity) and forearm blood flow responses to different stimuli (that is, the infusion of acetylcholine, sodium nitroprusside or nitroglycerin) have shown that compared with healthy controls, patients with OSAS often exhibit an impairment of resistance-vessel endothelium-dependent vasodilation.48, 49 Even when accounting for important confounding factors, such as body weight, brachial artery flow-mediated dilation has been shown to be significantly lower in normal-weight OSAS patients than in OSAS-free controls.50 Additional evidence that OSAS may significantly influence both indices of macrovascular and microvascular endothelial function was provided by a recent study showing abnormal myocardial perfusion, attenuated brachial artery reactivity and reduced cutaneous perfusion response in OSAS patients compared with healthy controls.51 Remarkably, several interventional studies have shown substantial improvements in different indices of endothelial function following the implementation of regular CPAP use in subjects with hypertension and OSAS,49, 50, 51 which indirectly supports a role for endothelial dysfunction in the pathogenesis of arterial hypertension in OSAS.
Vascular inflammation and oxidative stress
Recurring episodes of hypoxia/reoxygenation during the transient cessation of breathing in OSA may also reduce NO availability, promoting vascular endothelial inflammation and elevated oxidative stress48, 49, 52, 53, 54 (Figure 2). When compared with OSAS-free controls and regardless of the presence of obesity, OSAS patients have been shown to have reduced expression of endothelial NO synthase and phosphorylated endothelial NO synthase (proteins that regulate basal NO production and activity) as well as increased levels of nitrotyrosine (a marker of oxidative stress) and nuclear factor κ-light-chain enhancer of activated B cells (a marker of inflammation).50 Most importantly, after 1 month of regular treatment with CPAP, flow-mediated dilation, the expression of endothelial NO synthase and phosphorylated endothelial NO synthase were significantly increased, whereas the levels of nitrotyrosine and nuclear factor κ-light-chain enhancer of activated B cells were decreased.50 It has also been proposed that intermittent hypoxia/hypercapnia associated with OSAS may contribute to the pathogenesis of hypertension by increasing endothelin-1 production. This hypothesis has been supported by experimental studies in rats showing significant increases in the plasma levels of endothelin-1 (a potent vasoconstrictor) and higher BP levels in rats exposed to intermittent hypoxia (that is, cycles of hypoxia/hypercapnia for 8 h a day over 11 days) compared with those breathing normoxic air.55
Data from several studies have indicated that the selective activation of inflammatory pathways may be an additional important molecular mechanism for the pathogenesis of arterial hypertension in OSAS. This concept has been supported by translational studies showing a selective activation of the proinflammatory transcription nuclear factor κ-light-chain enhancer of activated B cells in HeLa cells exposed to intermittent hypoxia/reoxygenation cycles.56 In addition, compared with healthy controls, subjects with OSAS showed significantly higher levels of circulating proinflammatory cytokines (that is, tumor necrosis factor-α and the adaptive factor erythropoietin) and higher levels of circulating neutrophils. Interestingly, the levels of tumor necrosis factor-α normalized after 6 weeks of continuous treatment with CPAP.56 Other studies have shown that compared with healthy controls, the serum levels of inflammatory markers (that is, C-reactive protein) are significantly higher in OSAS patients and independently associated with OSAS severity.57 Interestingly, interventional studies have shown significant reductions in serum levels of C-reactive protein and interleukin-6 following the implementation of regular CPAP treatment.58 Finally, evidence has also been provided that OSAS may induce the activation of adhesion molecules participating in inflammation. These data have been supported by case–control studies showing significantly higher levels of intercellular adhesion molecule-1, vascular cell adhesion molecule-1 and L-selectin in OSA patients compared with healthy controls.59
Arterial stiffness
Increased arterial stiffness is a recognized risk factor contributing to the pathogenesis of arterial hypertension.60, 61, 62 A recent systematic review of relevant studies indicated an independent effect of OSAS on arterial stiffness, which in turn may contribute to BP elevation and resistant hypertension63 (Figure 2). A number of studies have consistently reported significantly higher values of carotid-femoral pulse wave velocity, which is considered the ‘gold-standard’ measure of aortic stiffness, in patients with OSAS compared with healthy controls.63, 64 Of note, the increase in carotid-femoral pulse wave velocity has been shown to be directly related to the severity of the disease and to be even higher in subjects with OSAS and associated hypertension or in the presence of other CV risk factors.65 In the Asian population, several studies implementing brachial-ankle PWV have also reported significant associations between OSAS and increased arterial stiffness.66 Even when comparisons have been performed between individuals with or without OSAS entirely free from other CV risk factors, an independent effect of OSAS on arterial stiffening has been reported.67 Remarkably, in randomized interventional studies, the effective treatment of OSAS with CPAP has been associated with significant decreases in arterial stiffness.68, 69 In one of these studies, CPAP was also associated with significant reductions in sympathetic nerve activity and ambulatory BP and with significant improvements in arterial BRS.68
Metabolic factors
In addition to the hemodynamic changes, OSAS has been frequently associated with metabolic alterations (that is, alterations in glucose metabolism, insulin resistance and leptin resistance), which in turn may contribute to the pathogenesis of arterial hypertension (Figure 2). Although altered glucose metabolism is thought to be a consequence of other conditions associated with OSAS (that is, an increased BMI, metabolic syndrome and/or type 2 diabetes) rather than a consequence of OSAS, evidence has shown that OSAS, independently of the presence of confounding factors, is associated with alterations in glucose metabolism that may promote the development of type 2 diabetes.70 In addition, interventional studies have shown the efficacy of regular CPAP treatment in improving glucose metabolism in OSAS patients.70 Compared with healthy controls, OSAS patients have also been shown to have greater insulin and leptin resistance71, 72, 73 even after accounting for body fat content.74 Although the aforementioned metabolic alterations should theoretically contribute to the pathogenesis of hypertension in OSAS, their relative contribution to BP elevation still needs to be further explored independently of other concomitant factors.
Contribution of genetic variation to OSAS-related hypertension
Hypertension and OSAS represent complex polygenic disorders. The pathophysiology for both implies joint actions of different genes acting at different levels and their interactions with intrinsic and extrinsic environmental factors. Although genetic variation may explain ∼30–40% of BP variation, identifying a genetic etiology for OSAS-related hypertension is difficult.75 Genome-wide association studies have yielded inconsistent results, and identified associations with some candidate genes have not always been replicated. The limited amount of data, the small sample size of the studies and the failure to properly phenotype the clinical characteristics of OSAS patients have prevented the identification of a genetic etiology of OSAS-related hypertension.
Prognostic relevance of the combination of OSAS and hypertension
Evidence from several studies has supported an independent association between OSAS and CV disease.76 OSAS, particularly if severe, has been linked to fatal and nonfatal CV events,77, 78, 79, 80, 81 the development and progression of congestive heart failure79 and with all-cause mortality.82, 83 However, because the link between OSAS and CV disease may be related to age, obesity and visceral adiposity, in some of these studies the associations have loose strength when adjusting for these factors. With regard to subclinical organ damage, evidence has also been provided that OSAS is independently associated with cardiac (that is, left ventricular hypertrophy and dysfunction),69, 84, 85 vascular (that is, increased carotid intima-media thickness, increased arterial stiffness),63 renal organ damage (that is, increased urinary albumin excretion)86, 87 and with endothelial dysfunction (that is, blunted endothelium-dependent dilatation).63 Evidence has also shown that patients with drug-resistant hypertension are at a considerably greater risk of CV complications, including myocardial infarction, stroke, congestive heart failure and chronic kidney disease than patients whose BP is pharmacologically well controlled.88, 89, 90 In consideration of the increased CV risk associated with OSAS and resistant hypertension, current guidelines for the management of arterial hypertension include OSAS among the modifiable causes to be addressed in subjects with resistant hypertension to properly manage both conditions.1, 8 It should be mentioned, however, that no studies have specifically addressed how and to what extent the coincidence of hypertension with OSAS increases the risk of CV disease independently of other CV risk factors that are often clustered in the context of OSAS. Evidence from longitudinal interventional studies in OSAS controlling for potential confounders (that is, visceral obesity, increased BMI) are thus needed not only to determine the prognostic relevance of the interaction between OSAS and hypertension but also to determine whether treating OSAS in resistant hypertension confers significant benefits in terms of CV protection.
Prognostic relevance of alterations in day-to-night BP profiles and nocturnal hypertension in OSAS
As mentioned above, nocturnal sympathetic activation during OSAS episodes significantly contributes to increases in BP during sleep, thus attenuating the physiologic nocturnal dipping of BP (average 10–20% decrease from daytime BP values) or even increasing nocturnal BP levels. It is thus not surprising that a nondipping BP profile (that is, nocturnal BP falls <10% compared with daytime BP levels) is often seen in OSA patients independently of the presence of hypertension.91 Remarkably, the degree of impairment in the nocturnal BP decrease has been found to be related to the severity of OSAS.92 In contrast, an increased incidence of alterations in day-to-night BP profiles and nocturnal hypertension has been reported in subjects with resistant hypertension regardless of the presence of OSAS.90, 93, 94 It is thus expected that alterations in day-to-night BP changes might be even more pronounced in subjects with OSAS and resistant hypertension. From a prognostic point of view, the identification of nocturnal hypertension and alterations in day-to-night BP changes in subjects with OSAS-related hypertension is of utmost relevance, considering the evidence showing the superior prognostic value of nocturnal BP compared with awake or 24 h average BP in predicting CV morbidity and mortality,95, 96, 97, 98, 99, 100 the development of CV events95, 96, 101, 102, 103 and overall mortality.95, 96, 97, 102, 104, 105 The identification of a ‘nondipping’ BP pattern in OSAS patients is also important if we consider that subjects whose nocturnal BP decrease is blunted have been reported to have a higher prevalence of subclinical organ damage,106, 107 and an increased risk of CV events108 and mortality,100 which is even higher in patients whose BP increases rather than decreases at night (often referred to as risers or ‘inverted dippers’). Despite the very high prevalence of nocturnal hypertension and alterations in day-to-night BP changes in OSAS patients, these morbidities are often undiagnosed (that is, masked resistant hypertension) because BP measurements are usually measured during the day at a clinical visit. Given their relevant prognostic value, alterations in circadian BP should be properly investigated in patients with OSAS-resistant hypertension through the use of 24-h ABPM to guide antihypertensive treatment toward the normalization and optimization of CV protection.
Diagnostic approach to OSAS-related resistant hypertension
Confirming the diagnosis of OSAS in subjects with resistant hypertension is relevant to implement specific treatment strategies (that is, CPAP, weight reduction). This confirmation might allow the achievement of BP control and reduce the CV risk of these subjects. Polysomnography is currently considered the standard technique for the diagnosis of OSAS and requires the simultaneous monitoring of several CV and respiratory variables during nocturnal sleep (that is, sleep, air flow, respiratory effort, oxygen saturation and brain activity through electroencephalogram). On the basis of the number of apneas and hypopneas lasting >10 s during each hour of recording, the severity of the disease is graded using the apnea-hypopnea index.109 Whether polysomnography should be employed systematically in individuals with resistant hypertension is still a matter of debate in the absence of cost-effectiveness studies supporting this suggestion. According to a recent position paper of the European Respiratory Society/European Society of Hypertension,110 polysomnography should be performed in all subjects with a high pretest probability of OSA based on structured questionnaires (for example, Epworth and Berlin questionnaires).
Considering the extremely high frequency of alterations in ambulatory BP profiles during the nighttime in subjects with resistant hypertension and OSAS, the task force of the European Respiratory Society/European Society of Hypertension also recommends performing ABPM to identify alterations in day-to-night BP changes in subjects with resistant hypertension to guide the decision to perform polysomnography in subjects with a low probability of OSA based on questionnaires. Indeed, in subjects with a low pretest probability of OSAS, polysomnography is only recommended for those who present alterations in day-to-night BP changes (that is, nondipping pattern of BP; Figure 7).

Proposed algorithm for the diagnostic management of patients with hypertension associated with obstructive sleep apnea (OSA). BP, blood pressure; SBP, systolic BP; DBP, diastolic BP; ABPM, ambulatory BP monitoring; PSG, polysomnography. # denotes according to clinical evaluation and questionnaires, for example, Epworth and Berlin; ¶ indicates hypertension guidelines recommend the use of home BP monitoring in most hypertensive patients. Reproduced by permission from Parati et al.110
It is worth mentioning that before starting the instrumental tests to rule out OSAS, a first step in the diagnostic approach of the patient with resistant hypertension consists of confirming whether resistance to antihypertensive treatment is real or false. Current guidelines for the management of arterial hypertension define resistant hypertension as the persistence of elevated BP (that is, ⩾140/90 mm Hg for diastolic/systolic BP) despite the concomitant use of three optimally dosed antihypertensive medications from different classes, one of which should ideally be a diuretic, at near-maximal doses.1, 8 However, this definition is based on office (clinically obtained) BP measurements, which have acknowledged limitations, including the inherent inaccuracy of the technique, the observer’s bias and digit preference, variable interference as a result of the ‘white-coat effect’ and the inability of this approach to collect information on BP during subjects’ usual activities or over an extended time.111 Thus, for the confirmation of truly resistant hypertension, out-of-office BP measuring techniques, such as ABPM and/or home BP monitoring (which are not affected by the limitations of OBP), should be performed in addition to OBP. On the basis of measurements obtained with these methods, a substantial and sometimes larger than expected number of subjects initially diagnosed with resistant hypertension or with BP control based on OBP may actually have false resistant hypertension or white-coat resistant hypertension (that is, elevated OBP but normal out-of-office BP values) or masked resistant hypertension (that is, normal OBP but elevated out-of-office BP values).90, 112, 113
From a prognostic point of view, the identification of OSAS patients with true resistant hypertension in addition to those with masked resistant hypertension (normal OBP and elevated ABP or HBP)114, 115 is of the greatest relevance, considering the evidence showing these conditions to be associated with a higher incidence of target organ damage,116, 117 a higher risk of future CV and renal events,103, 118, 119 and ultimately higher health-care costs.27, 120, 121
Effects of different therapeutic strategies on OSAS-related resistant hypertension
Effects of lifestyle changes and weight loss on OSAS-related hypertension
Obesity is the single most important cause of OSAS and BP elevation. It is thus expected that weight loss will reduce the severity of OSAS and reduce BP levels. Indeed, in subjects who achieve significant reductions in body weight either through dietary,122 pharmacological123 or surgical124 measures, considerable reductions have been reported for various indices of OSA severity (that is, apnea-hypopnea index) and for BP levels. In particular, bariatric surgery has been shown to be a highly effective measure to achieve OSAS improvement and BP control, as supported by a large meta-analysis of 136 randomized controlled trials.125 Of note, reductions in BP levels were mainly related to weight loss (that is, each 1% reduction in body weight decreased the SBP by 1 mm Hg and the DBP by 2 mm Hg).125 However, despite its efficacy, bariatric surgery is reserved for selected patient groups (that is, patients with type 2 diabetes mellitus and severe obesity (BMI >35 kg/m2) and moderately obese patients (BMI: 30–35 kg/m2) who are inadequately controlling their body weights by conventional medical and behavioral therapies).
Effects of CPAP treatment on OSAS-related hypertension
Nasal CPAP is currently considered the optimal treatment for OSA.126 When properly implemented, CPAP not only provides relatively instant relief of clinical symptoms127 and reduction in the severity of OSA but also improves many of the acute and chronic pathophysiological alterations induced by OSAS. Of note, CPAP use has been shown to induce marked and acute reductions in microneurography not only during nighttime sleep but also during daytime wakefulness if maintained over a long term28 (Figure 8). Several studies have also shown the effectiveness of CPAP in improving baroreflex impairment,68 systemic inflammation,50, 56, 58 endothelial dysfunction,49, 50, 51 RAAS activation,128 arterial stiffness68, 69 and metabolic alterations.70

Elimination of apneas by continuous positive airway pressure (CPAP) reduces muscle sympathetic nerve activity (SNA) and prevents blood pressure (BP) surge during rapid eye movement (REM) sleep. Taken from Somers et al.28 by permission.
Although improvements in these pathophysiological alterations should theoretically translate into substantial BP reductions, most interventional trials in OSAS and subsequent meta-analyses have indicated that, although CPAP has a significant effect on BP levels, the overall effect on 24 h, daytime and nighttime systolic and diastolic ambulatory BP levels is rather small (of the order of 1–2 mm Hg).129, 130, 131 Notably, one of these meta-analyses showed that CPAP therapy may have substantial effects in selected groups of OSAS patients.130 Effective CPAP treatment in patients with moderate-to-severe OSAS has been shown to induce important reductions in both daytime and nighttime BP levels.132 This has also been the case for subjects with resistant hypertension for whom regular CPAP implementation has resulted in marked reductions in ambulatory BP levels not only during the night but also during the day.133 In a recent study addressing the effects of a 1-year treatment with CPAP, no effects on BP levels were observed in patients with BP controlled at baseline, but marked and significant reductions in BP levels were observed in subjects with resistant hypertension.134
A critical aspect when assessing the clinical effects of CPAP is guaranteeing patients adhere to therapy. Given the mechanical nature of CPAP (that is, facial interface mask and the pressure required to prevent airway collapse), it is not always well accepted by patients, especially those free of OSA-related symptoms. Indeed, CPAP compliance has been shown to be directly related to the severity of OSAS.135 However, several studies have indicated that to observe an effect of CPAP on BP, this treatment should be for months and for a sufficient number of hours per night, and the BP levels should be ideally assessed by ABPM. Proof of this has been provided by several studies of OSAS in which the benefits of CPAP are evident only for subjects who have confirmed resistant hypertension (that is, persistent elevation in both the office and the out-of-office BP levels) and have used CPAP for at least 3 months and for more than 5.8 h per night.136 For nonsleepy hypertensive patients with OSA, the most significant reductions in BP have been observed when using CPAP for more than 5.6 h per night.135 Further studies are needed to better determine whether CPAP implementation in OSAS patients with hypertension is associated with better BP control or a reduction in the number of antihypertensive medications needed to achieve BP control.
Finally, oral appliance therapy has been proposed as an alternative to CPAP in patients with mild-to-moderate OSA. Although a recent meta-analysis showed a favorable effect of oral appliances on BP levels,137 most of the studies reporting the BP-lowering effects of this technique have been limited by small sample sizes, observational nature and short follow-up periods. Thus, evidence from randomized controlled trials is still needed to confirm the effects of oral appliances on BP levels (especially ambulatory BP) in OSAS patients.
Effects of renal denervation in OSAS-related resistant hypertension
Sympathetic activation in OSAS causes an increase in sympathetic drive to the heart, the peripheral vasculature and the kidneys. With respect to kidneys, the sympathetic nerves arriving in the renal area have been identified as a major contributing factor to the pathophysiology of hypertension both in experimental models and in human studies.138 This has been the basis for the development of interventional strategies aimed at modulating renal sympathetic nerve activity through radiofrequency catheter-based renal sympathetic denervation (RDN).139 In subjects with uncontrolled hypertension, RDN has been shown to induce significant reductions in renal sympathetic efferent-nerve activity, whole-body sympathetic-nerve activity, norepinephrine spillover and substantial and sustained reductions in BP levels.140 A small interventional study in OSAS patients who were refractory to lifestyle modifications, weight loss, pharmacological treatment and CPAP has also suggested that RDN may represent an effective strategy for the management of resistant hypertension, as it induced significant and sustained changes in BP levels at 3 and 6 months of follow-up.141 Remarkably, the changes in BP levels reported in this study have also been accompanied by improvements in OSAS severity as indicated by the significant reductions in apnea-hypopnea index 3 and 6 months after denervation.141 Potential mechanisms for improving OSAS severity following RDN might include reducing salt retention and total body fluid (which in turn might reduce peripharyngeal fluid accumulation and upper airway obstruction), as well as reducing insulin resistance and improving the metabolic profile. RDN may thus represent a potentially useful option for the management of resistant hypertension in OSAS patients who are refractory to lifestyle modifications, weight loss, pharmacological treatment and CPAP. Nonetheless, given the very small sample size in this report, adequately powered longitudinal studies are needed to confirm the long-term impact of RDN on hypertension control and its benefits in terms of organ damage and the incidence of CV morbid-mortality in subjects with OSAS.
Do different antihypertensive drug classes have different effects on OSAS-related hypertension?
Different antihypertensive drug classes may have different effects on the pathophysiological mechanisms involved in the pathogenesis of OSAS-related hypertension. However, the few studies that have comparatively assessed the BP-lowering effects of different drug classes in OSAS have had small sample sizes, and their statistical power has limited consistent conclusions. In a randomized study assessing the effects of different classes of antihypertensive drugs (that is, β-blockers, calcium antagonists, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers and thiazidic diuretics) on office and ambulatory BP levels in patients with hypertension and OSAS, no significant differences between drug classes were observed in their ability to reduce office and daytime ambulatory BP levels. However, treatment with β-blockers was more effective at reducing nighttime ambulatory BP than the administration of other compounds, most likely because of their effects on sympathetic activation. In general, however, no consistent evidence has shown superior antihypertensive efficacy for any antihypertensive drug in OSA patients.142 The long-term effects of treatment with different antihypertensive agents on hypertension severity in OSAS have not been systematically addressed in clinical trials. Evidence is therefore still needed to identify preferred compounds for an adequate BP control in this group of high-risk patients.
Recent studies in resistant hypertension have suggested that spironolactone should be considered in all patients with uncontrolled hypertension on three or more antihypertensive agents.143 In some studies, the addition of spironolactone at doses of 25–50 mg per day to the current antihypertensive treatment in resistant hypertensive patients was shown to reduce the severity of OSAS in addition to lowering the BP.47 This finding is in line with the concept that aldosterone-mediated chronic fluid retention may influence the severity of OSA.
Conclusions
Evidence has consistently supported the association between OSAS and hypertension;2, 4, 5, 16, 17, 18 showing the occurrence of a dose–response relationship between OSAS severity and the degree of BP elevation.2, 26, 27 It has also been shown that hypertension in individuals with OSAS is more likely to be severe, resistant to treatment and associated with alterations in day-night BP changes.2, 26, 27 The pathogenesis of OSAS-related hypertension is likely to be multifactorial, involving alterations in several regulatory systems. However, the mechanisms by which OSAS promotes arterial hypertension still need to be better understood. Although OSAS and drug-resistant hypertension are independent predictors of CV morbidity and mortality, evidence from longitudinal studies is needed to determine the actual prognostic relevance of OSAS-related hypertension. In a subject with resistant hypertension and suspected OSAS, ABPM should be performed whenever possible for the confirmation of resistant hypertension, to identify alterations in day-to-night BP changes, and to determine the need for additional diagnostic procedures (that is, polysomnography) and/or the implementation of more aggressive pharmacological or interventional strategies for the management of resistant hypertension. In turn, the identification of OSAS and the proper implementation of specific treatment strategies (that is, CPAP) in subjects with resistant hypertension may promote the achievement of BP control and improve CV protection.
References
Mancia G, De Backer G, Dominiczak A, Cifkova R, Fagard R, Germano G, Grassi G, Heagerty AM, Kjeldsen SE, Laurent S, Narkiewicz K, Ruilope L, Rynkiewicz A, Schmieder RE, Boudier HA, Zanchetti A, Vahanian A, Camm J, De Caterina R, Dean V, Dickstein K, Filippatos G, Funck-Brentano C, Hellemans I, Kristensen SD, McGregor K, Sechtem U, Silber S, Tendera M, Widimsky P, Zamorano JL, Erdine S, Kiowski W, Agabiti-Rosei E, Ambrosioni E, Lindholm LH, Viigimaa M, Adamopoulos S, Bertomeu V, Clement D, Farsang C, Gaita D, Lip G, Mallion JM, Manolis AJ, Nilsson PM, O'Brien E, Ponikowski P, Redon J, Ruschitzka F, Tamargo J, van Zwieten P, Waeber B, Williams B . 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J. Hypertens 2007; 25: 1105–1187.
Grote L, Hedner J, Peter JH . Sleep-related breathing disorder is an independent risk factor for uncontrolled hypertension. J Hypertens 2000; 18: 679–685.
Peppard PE, Young T, Palta M, Skatrud J . Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med 2000; 342: 1378–1384.
Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S, D'Agostino RB, Newman AB, Lebowitz MD, Pickering TG . Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. JAMA 2000; 283: 1829–1836.
Young T, Peppard P, Palta M, Hla KM, Finn L, Morgan B, Skatrud J . Population-based study of sleep-disordered breathing as a risk factor for hypertension. Arch Int Med 1997; 157: 1746–1752.
O'Connor GT, Caffo B, Newman AB, Quan SF, Rapoport DM, Redline S, Resnick HE, Samet J, Shahar E . Prospective study of sleep-disordered breathing and hypertension: the Sleep Heart Health Study. Am J Respir Crit Care Med 2009; 179: 1159–1164.
Cano-Pumarega I, Duran-Cantolla J, Aizpuru F, Miranda-Serrano E, Rubio R, Martinez-Null C, de Miguel J, Egea C, Cancelo L, Alvarez A, Fernandez-Bolanos M, Barbe F . Obstructive sleep apnea and systemic hypertension: longitudinal study in the general population: the Vitoria Sleep Cohort. Am J Respir Crit Care Med 2011; 184: 1299–1304.
Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr, Jones DW, Materson BJ, Oparil S, Wright JT Jr, Roccella EJ . Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42: 1206–1252.
American Academy of Sleep Medicine. International Classification of Sleep Disorders: Diagnostic and Coding Manual 2nd edn Westchester: IL, USA. 2005.
Coccagna G, Mantovani M, Brignani F, Parchi C, Lugaresi E . Continuous recording of the pulmonary and systemic arterial pressure during sleep in syndromes of hypersomnia with periodic breathing. Bull Physiopathol Respir 1972; 8: 1159–1172.
Baguet JP, Hammer L, Levy P, Pierre H, Rossini E, Mouret S, Ormezzano O, Mallion JM, Pepin JL . Night-time and diastolic hypertension are common and underestimated conditions in newly diagnosed apnoeic patients. J Hypertens 2005; 23: 521–527.
Hla KM, Young T, Finn L, Peppard PE, Szklo-Coxe M, Stubbs M . Longitudinal association of sleep-disordered breathing and nondipping of nocturnal blood pressure in the Wisconsin Sleep Cohort Study. Sleep 2008; 31: 795–800.
Lavie P, Herer P, Hoffstein V . Obstructive sleep apnoea syndrome as a risk factor for hypertension: population study. BMJ 2000; 320: 479–482.
Davies CW, Crosby JH, Mullins RL, Barbour C, Davies RJ, Stradling JR . Case-control study of 24 hour ambulatory blood pressure in patients with obstructive sleep apnoea and normal matched control subjects. Thorax 2000; 55: 736–740.
Pankow W, Nabe B, Lies A, Becker H, Kohler U, Kohl FV, Lohmann FW . Influence of sleep apnea on 24-hour blood pressure. Chest 1997; 112: 1253–1258.
Bixler EO, Vgontzas AN, Lin HM, Ten Have T, Leiby BE, Vela-Bueno A, Kales A . Association of hypertension and sleep-disordered breathing. Arch Int Med 2000; 160: 2289–2295.
Duran J, Esnaola S, Rubio R, Iztueta A . Obstructive sleep apnea-hypopnea and related clinical features in a population-based sample of subjects aged 30 to 70 yr. Am J Respir Crit Care Med 2001; 163 (3 Pt 1): 685–689.
Tanigawa T, Tachibana N, Yamagishi K, Muraki I, Kudo M, Ohira T, Kitamura A, Sato S, Shimamoto T, Iso H . Relationship between sleep-disordered breathing and blood pressure levels in community-based samples of Japanese men. Hypertens Res 2004; 27: 479–484.
Kiely JL, McNicholas WT . Cardiovascular risk factors in patients with obstructive sleep apnoea syndrome. Eur Respir J 2000; 16: 128–133.
Calhoun DA . Obstructive sleep apnea and hypertension. Curr Hypertens Rep 2010; 12: 189–195.
Garrison RJ, Kannel WB, Stokes J 3rd, Castelli WP . Incidence and precursors of hypertension in young adults: the Framingham Offspring Study. Prev Med 1987; 16: 235–251.
Bramlage P, Pittrow D, Wittchen HU, Kirch W, Boehler S, Lehnert H, Hoefler M, Unger T, Sharma AM . Hypertension in overweight and obese primary care patients is highly prevalent and poorly controlled. Am J Hypertens 2004; 17: 904–910.
Kapa S, Sert Kuniyoshi FH, Somers VK . Sleep apnea and hypertension: interactions and implications for management. Hypertension 2008; 51: 605–608.
Haas DC, Foster GL, Nieto FJ, Redline S, Resnick HE, Robbins JA, Young T, Pickering TG . Age-dependent associations between sleep-disordered breathing and hypertension: importance of discriminating between systolic/diastolic hypertension and isolated systolic hypertension in the Sleep Heart Health Study. Circulation 2005; 111: 614–621.
Logan AG, Perlikowski SM, Mente A, Tisler A, Tkacova R, Niroumand M, Leung RS, Bradley TD . High prevalence of unrecognized sleep apnoea in drug-resistant hypertension. J Hypertens 2001; 19: 2271–2277.
Somers VK, White DP, Amin R, Abraham WT, Costa F, Culebras A, Daniels S, Floras JS, Hunt CE, Olson LJ, Pickering TG, Russell R, Woo M, Young T . Sleep apnea and cardiovascular disease: an American Heart Association/american College Of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council On Cardiovascular Nursing. In collaboration with the National Heart, Lung, and Blood Institute National Center on Sleep Disorders Research (National Institutes of Health). Circulation 2008; 118: 1080–1111.
Calhoun DA, Jones D, Textor S, Goff DC, Murphy TP, Toto RD, White A, Cushman WC, White W, Sica D, Ferdinand K, Giles TD, Falkner B, Carey RM . Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 2008; 51: 1403–1419.
Somers VK, Dyken ME, Clary MP, Abboud FM . Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest 1995; 96: 1897–1904.
Portaluppi F, Provini F, Cortelli P, Plazzi G, Bertozzi N, Manfredini R, Fersini C, Lugaresi E . Undiagnosed sleep-disordered breathing among male nondippers with essential hypertension. J Hypertens 1997; 15: 1227–1233.
Narkiewicz K, van de Borne PJ, Cooley RL, Dyken ME, Somers VK . Sympathetic activity in obese subjects with and without obstructive sleep apnea. Circulation 1998; 98: 772–776.
Ali NJ, Davies RJ, Fleetham JA, Stradling JR . The acute effects of continuous positive airway pressure and oxygen administration on blood pressure during obstructive sleep apnea. Chest 1992; 101: 1526–1532.
Narkiewicz K, Kato M, Phillips BG, Pesek CA, Davison DE, Somers VK . Nocturnal continuous positive airway pressure decreases daytime sympathetic traffic in obstructive sleep apnea. Circulation 1999; 100: 2332–2335.
Hedner JA, Wilcox I, Laks L, Grunstein RR, Sullivan CE . A specific and potent pressor effect of hypoxia in patients with sleep apnea. Am Rev Respir Dis 1992; 146 (5 Pt 1): 1240–1245.
Narkiewicz K, van de Borne PJ, Pesek CA, Dyken ME, Montano N, Somers VK . Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation 1999; 99: 1183–1189.
Parati G, Di Rienzo M, Bonsignore MR, Insalaco G, Marrone O, Castiglioni P, Bonsignore G, Mancia G . Autonomic cardiac regulation in obstructive sleep apnea syndrome: evidence from spontaneous baroreflex analysis during sleep. J Hypertens 1997; 15 (12 Pt 2): 1621–1626.
Narkiewicz K, Pesek CA, Kato M, Phillips BG, Davison DE, Somers VK . Baroreflex control of sympathetic nerve activity and heart rate in obstructive sleep apnea. Hypertension 1998; 32: 1039–1043.
Noda A, Nakata S, Koike Y, Miyata S, Kitaichi K, Nishizawa T, Nagata K, Yasuma F, Murohara T, Yokota M . Continuous positive airway pressure improves daytime baroreflex sensitivity and nitric oxide production in patients with moderate to severe obstructive sleep apnea syndrome. Hypertens Res 2007; 30: 669–676.
Ryan S, Ward S, Heneghan C, McNicholas WT . Predictors of decreased spontaneous baroreflex sensitivity in obstructive sleep apnea syndrome. Chest 2007; 131: 1100–1107.
Narkiewicz K, Montano N, Cogliati C, van de Borne PJ, Dyken ME, Somers VK . Altered cardiovascular variability in obstructive sleep apnea. Circulation 1998; 98: 1071–1077.
Lombardi C, Parati G, Cortelli P, Provini F, Vetrugno R, Plazzi G, Vignatelli L, Di Rienzo M, Lugaresi E, Mancia G, Montagna P, Castiglioni P . Daytime sleepiness and neural cardiac modulation in sleep-related breathing disorders. J Sleep Res 2008; 17: 263–270.
Gonzaga CC, Gaddam KK, Ahmed MI, Pimenta E, Thomas SJ, Harding SM, Oparil S, Cofield SS, Calhoun DA . Severity of obstructive sleep apnea is related to aldosterone status in subjects with resistant hypertension. J Clin Sleep Med 2010; 6: 363–368.
Pimenta E, Calhoun DA, Oparil S . Sleep apnea, aldosterone, and resistant hypertension. Prog Cardiovasc Dis 2009; 51: 371–380.
Goodfriend TL, Calhoun DA . Resistant hypertension, obesity, sleep apnea, and aldosterone: theory and therapy. Hypertension 2004; 43: 518–524.
Calhoun DA, Nishizaka MK, Zaman MA, Harding SM . Aldosterone excretion among subjects with resistant hypertension and symptoms of sleep apnea. Chest 2004; 125: 112–117.
Pratt-Ubunama MN, Nishizaka MK, Boedefeld RL, Cofield SS, Harding SM, Calhoun DA . Plasma aldosterone is related to severity of obstructive sleep apnea in subjects with resistant hypertension. Chest 2007; 131: 453–459.
Dudenbostel T, Calhoun DA . Resistant hypertension, obstructive sleep apnoea and aldosterone. J Hum Hypertens 2012; 26: 281–287.
Gaddam K, Pimenta E, Thomas SJ, Cofield SS, Oparil S, Harding SM, Calhoun DA . Spironolactone reduces severity of obstructive sleep apnoea in patients with resistant hypertension: a preliminary report. J Hum Hypertens 2010; 24: 532–537.
Kato M, Roberts-Thomson P, Phillips BG, Haynes WG, Winnicki M, Accurso V, Somers VK . Impairment of endothelium-dependent vasodilation of resistance vessels in patients with obstructive sleep apnea. Circulation 2000; 102: 2607–2610.
Ip MS, Tse HF, Lam B, Tsang KW, Lam WK . Endothelial function in obstructive sleep apnea and response to treatment. Am J Respir Crit Care Med 2004; 169: 348–353.
Jelic S, Lederer DJ, Adams T, Padeletti M, Colombo PC, Factor PH, Le Jemtel TH . Vascular inflammation in obesity and sleep apnea. Circulation 2010; 121: 1014–1021.
Butt M, Khair OA, Dwivedi G, Shantsila A, Shantsila E, Lip GY . Myocardial perfusion by myocardial contrast echocardiography and endothelial dysfunction in obstructive sleep apnea. Hypertension 2011; 58: 417–424.
Jelic S, Padeletti M, Kawut SM, Higgins C, Canfield SM, Onat D, Colombo PC, Basner RC, Factor P, LeJemtel TH . Inflammation, oxidative stress, and repair capacity of the vascular endothelium in obstructive sleep apnea. Circulation 2008; 117: 2270–2278.
Dyugovskaya L, Lavie P, Lavie L . Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. Am J Respir Crit Care Med 2002; 165: 934–939.
Minoguchi K, Yokoe T, Tazaki T, Minoguchi H, Tanaka A, Oda N, Okada S, Ohta S, Naito H, Adachi M . Increased carotid intima-media thickness and serum inflammatory markers in obstructive sleep apnea. Am J Respir Crit Care Med 2005; 172: 625–630.
Kanagy NL, Walker BR, Nelin LD . Role of endothelin in intermittent hypoxia-induced hypertension. Hypertension 2001; 37 (2 Part 2): 511–515.
Ryan S, Taylor CT, McNicholas WT . Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation 2005; 112: 2660–2667.
Shamsuzzaman AS, Winnicki M, Lanfranchi P, Wolk R, Kara T, Accurso V, Somers VK . Elevated C-reactive protein in patients with obstructive sleep apnea. Circulation 2002; 105: 2462–2464.
Yokoe T, Minoguchi K, Matsuo H, Oda N, Minoguchi H, Yoshino G, Hirano T, Adachi M . Elevated levels of C-reactive protein and interleukin-6 in patients with obstructive sleep apnea syndrome are decreased by nasal continuous positive airway pressure. Circulation 2003; 107: 1129–1134.
Ohga E, Nagase T, Tomita T, Teramoto S, Matsuse T, Katayama H, Ouchi Y . Increased levels of circulating ICAM-1, VCAM-1, and L-selectin in obstructive sleep apnea syndrome. J Appl Physiol 1999; 87: 10–14.
O'Rourke MF, Nichols WW . Aortic diameter, aortic stiffness, and wave reflection increase with age and isolated systolic hypertension. Hypertension 2005; 45: 652–658.
Mitchell GF, Lacourciere Y, Ouellet JP, Izzo JL Jr., Neutel J, Kerwin LJ, Block AJ, Pfeffer MA . Determinants of elevated pulse pressure in middle-aged and older subjects with uncomplicated systolic hypertension: the role of proximal aortic diameter and the aortic pressure-flow relationship. Circulation 2003; 108: 1592–1598.
Schillaci G, Bilo G, Pucci G, Laurent S, Macquin-Mavier I, Boutouyrie P, Battista F, Settimi L, Desamericq G, Dolbeau G, Faini A, Salvi P, Mannarino E, Parati G . Relationship between short-term blood pressure variability and large-artery stiffness in human hypertension: findings from 2 large databases. Hypertension 2012; 60: 369–377.
Doonan RJ, Scheffler P, Lalli M, Kimoff RJ, Petridou ET, Daskalopoulos ME, Daskalopoulou SS . Increased arterial stiffness in obstructive sleep apnea: a systematic review. Hypertens Res 2011; 34: 23–32.
Tsioufis C, Thomopoulos K, Dimitriadis K, Amfilochiou A, Tousoulis D, Alchanatis M, Stefanadis C, Kallikazaros I . The incremental effect of obstructive sleep apnoea syndrome on arterial stiffness in newly diagnosed essential hypertensive subjects. J Hypertens 2007; 25: 141–146.
Drager LF, Bortolotto LA, Figueiredo AC, Silva BC, Krieger EM, Lorenzi-Filho G . Obstructive sleep apnea, hypertension, and their interaction on arterial stiffness and heart remodeling. Chest 2007; 131: 1379–1386.
Shiina K, Tomiyama H, Takata Y, Usui Y, Asano K, Hirayama Y, Nakamura T, Yamashina A . Concurrent presence of metabolic syndrome in obstructive sleep apnea syndrome exacerbates the cardiovascular risk: a sleep clinic cohort study. Hypertens Res 2006; 29: 433–441.
Nagahama H, Soejima M, Uenomachi H, Higashi Y, Yotsumoto K, Samukawa T, Arima T . Pulse wave velocity as an indicator of atherosclerosis in obstructive sleep apnea syndrome patients. Intern Med 2004; 43: 184–188.
Kohler M, Pepperell JC, Casadei B, Craig S, Crosthwaite N, Stradling JR, Davies RJ . CPAP and measures of cardiovascular risk in males with OSAS. Eur Respir J 2008; 32: 1488–1496.
Drager LF, Bortolotto LA, Figueiredo AC, Krieger EM, Lorenzi GF . Effects of continuous positive airway pressure on early signs of atherosclerosis in obstructive sleep apnea. Am J Respir Crit Care Med 2007; 176: 706–712.
Rasche K, Keller T, Tautz B, Hader C, Hergenc G, Antosiewicz J, Di Giulio C, Pokorski M . Obstructive sleep apnea and type 2 diabetes. Eur J Med Res 2010; 15 (Suppl 2): 152–156.
Basoglu OK, Sarac F, Sarac S, Uluer H, Yilmaz C . Metabolic syndrome, insulin resistance, fibrinogen, homocysteine, leptin, and C-reactive protein in obese patients with obstructive sleep apnea syndrome. Ann Thorac Med 2011; 6: 120–125.
Zirlik S, Hauck T, Fuchs FS, Neurath MF, Konturek PC, Harsch IA . Leptin, obestatin and apelin levels in patients with obstructive sleep apnoea syndrome. Med Sci Monit 2011; 17: CR159–CR164.
Bonsignore MR, Esquinas C, Barcelo A, Sanchez-de-la-Torre M, Paterno A, Duran-Cantolla J, Marin JM, Barbe F . Metabolic syndrome, insulin resistance and sleepiness in real-life obstructive sleep apnoea. Eur Respir J 2012; 39: 1136–1143.
Phillips BG, Kato M, Narkiewicz K, Choe I, Somers VK . Increases in leptin levels, sympathetic drive, and weight gain in obstructive sleep apnea. Am J Physiol Heart Circ Physiol 2000; 279: H234–H237.
Riha RL, Diefenbach K, Jennum P, McNicholas WT . Genetic aspects of hypertension and metabolic disease in the obstructive sleep apnoea-hypopnoea syndrome. Sleep Med Rev 2008; 12: 49–63.
McNicholas WT, Bonsigore MR . Sleep apnoea as an independent risk factor for cardiovascular disease: current evidence, basic mechanisms and research priorities. Eur Respir J 2007; 29: 156–178.
Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V . Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005; 353: 2034–2041.
Selim B, Won C, Yaggi HK . Cardiovascular consequences of sleep apnea. Clin Chest Med 2010; 31: 203–220.
Gottlieb DJ, Yenokyan G, Newman AB, O'Connor GT, Punjabi NM, Quan SF, Redline S, Resnick HE, Tong EK, Diener-West M, Shahar E . Prospective study of obstructive sleep apnea and incident coronary heart disease and heart failure: the sleep heart health study. Circulation 2010; 122: 352–360.
Redline S, Yenokyan G, Gottlieb DJ, Shahar E, O'Connor GT, Resnick HE, Diener-West M, Sanders MH, Wolf PA, Geraghty EM, Ali T, Lebowitz M, Punjabi NM . Obstructive sleep apnea-hypopnea and incident stroke: the sleep heart health study. Am J Respir Crit Care Med 2010; 182: 269–277.
Capampangan DJ, Wellik KE, Parish JM, Aguilar MI, Snyder CR, Wingerchuk D, Demaerschalk BM . Is obstructive sleep apnea an independent risk factor for stroke? A critically appraised topic. Neurologist 2010; 16: 269–273.
Marshall NS, Wong KK, Liu PY, Cullen SR, Knuiman MW, Grunstein RR . Sleep apnea as an independent risk factor for all-cause mortality: the Busselton Health Study. Sleep 2008; 31: 1079–1085.
Young T, Finn L, Peppard PE, Szklo-Coxe M, Austin D, Nieto FJ, Stubbs R, Hla KM . Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin sleep cohort. Sleep 2008; 31: 1071–1078.
Otto ME, Belohlavek M, Romero-Corral A, Gami AS, Gilman G, Svatikova A, Amin RS, Lopez-Jimenez F, Khandheria BK, Somers VK . Comparison of cardiac structural and functional changes in obese otherwise healthy adults with versus without obstructive sleep apnea. Am J Cardiol 2007; 99: 1298–1302.
Tavil Y, Kanbay A, Sen N, Ciftci TU, Abaci A, Yalcin MR, Kokturk O, Cengel A . Comparison of right ventricular functions by tissue Doppler imaging in patients with obstructive sleep apnea syndrome with or without hypertension. Int J Cardiovasc Imaging 2007; 23: 469–477.
Agrawal V, Vanhecke TE, Rai B, Franklin BA, Sangal RB, McCullough PA . Albuminuria and renal function in obese adults evaluated for obstructive sleep apnea. Nephron Clin Pract 2009; 113: c140–c147.
Sim JJ, Rasgon SA, Derose SF . Managing sleep apnoea in kidney diseases. Nephrology 2010; 15: 146–152.
Persell SD . Prevalence of resistant hypertension in the United States, 2003-2008. Hypertension 2011; 57: 1076–1080.
Egan BM, Zhao Y, Axon RN, Brzezinski WA, Ferdinand KC . Uncontrolled and apparent treatment resistant hypertension in the United States, 1988 to 2008. Circulation 2011; 124: 1046–1058.
de la Sierra A, Segura J, Banegas JR, Gorostidi M, de la Cruz JJ, Armario P, Oliveras A, Ruilope LM . Clinical features of 8295 patients with resistant hypertension classified on the basis of ambulatory blood pressure monitoring. Hypertension 2011; 57: 898–902.
Wolf J, Hering D, Narkiewicz K . Non-dipping pattern of hypertension and obstructive sleep apnea syndrome. Hypertens Res 2010; 33: 867–871.
Lavie-Nevo K, Pillar G . Evening-morning differences in blood pressure in sleep apnea syndrome: effect of gender. Am J Hypertens 2006; 19: 1064–1069.
Muxfeldt ES, Cardoso CR, Salles GF . Prognostic value of nocturnal blood pressure reduction in resistant hypertension. Arch Int Med 2009; 169: 874–880.
Muxfeldt ES, Bloch KV, Nogueira AR, Salles GF . Twenty-four hour ambulatory blood pressure monitoring pattern of resistant hypertension. Blood Press Monit 2003; 8: 181–185.
Staessen JA, Thijs L, Fagard R, O'Brien ET, Clement D, de Leeuw PW, Mancia G, Nachev C, Palatini P, Parati G, Tuomilehto J, Webster J . Predicting cardiovascular risk using conventional vs ambulatory blood pressure in older patients with systolic hypertension. Systolic Hypertension in Europe Trial Investigators. JAMA 1999; 282: 539–546.
Sega R, Facchetti R, Bombelli M, Cesana G, Corrao G, Grassi G, Mancia G . Prognostic value of ambulatory and home blood pressures compared with office blood pressure in the general population: follow-up results from the Pressioni Arteriose Monitorate e Loro Associazioni (PAMELA) study. Circulation 2005; 111: 1777–1783.
Kikuya M, Ohkubo T, Asayama K, Metoki H, Obara T, Saito S, Hashimoto J, Totsune K, Hoshi H, Satoh H, Imai Y . Ambulatory blood pressure and 10-year risk of cardiovascular and noncardiovascular mortality: the Ohasama study. Hypertension 2005; 45: 240–245.
Fagard RH, Celis H, Thijs L, Staessen JA, Clement DL, De Buyzere ML, De Bacquer DA . Daytime and nighttime blood pressure as predictors of death and cause-specific cardiovascular events in hypertension. Hypertension 2008; 51: 55–61.
Boggia J, Li Y, Thijs L, Hansen TW, Kikuya M, Bjorklund-Bodegard K, Richart T, Ohkubo T, Kuznetsova T, Torp-Pedersen C, Lind L, Ibsen H, Imai Y, Wang J, Sandoya E, O'Brien E, Staessen JA . Prognostic accuracy of day versus night ambulatory blood pressure: a cohort study. Lancet 2007; 370: 1219–1229.
Hansen TW, Li Y, Boggia J, Thijs L, Richart T, Staessen JA . Predictive role of the nighttime blood pressure. Hypertension 2011; 57: 3–10.
Clement DL, De Buyzere ML, De Bacquer DA, de Leeuw PW, Duprez DA, Fagard RH, Gheeraert PJ, Missault LH, Braun JJ, Six RO, Van Der Niepen P, O'Brien E . Prognostic value of ambulatory blood-pressure recordings in patients with treated hypertension. N Engl J Med 2003; 348: 2407–2415.
Fagard RH, Van Den Broeke C, De Cort P . Prognostic significance of blood pressure measured in the office, at home and during ambulatory monitoring in older patients in general practice. J Hum Hypertens 2005; 19: 801–807.
Redon J, Campos C, Narciso ML, Rodicio JL, Pascual JM, Ruilope LM . Prognostic value of ambulatory blood pressure monitoring in refractory hypertension: a prospective study. Hypertension 1998; 31: 712–718.
Dolan E, Stanton A, Thijs L, Hinedi K, Atkins N, McClory S, Den Hond E, McCormack P, Staessen JA, O'Brien E . Superiority of ambulatory over clinic blood pressure measurement in predicting mortality: the Dublin outcome study. Hypertension 2005; 46: 156–161.
Hansen TW, Jeppesen J, Rasmussen S, Ibsen H, Torp-Pedersen C . Ambulatory blood pressure and mortality: a population-based study. Hypertension 2005; 45: 499–504.
Sander D, Kukla C, Klingelhofer J, Winbeck K, Conrad B . Relationship between circadian blood pressure patterns and progression of early carotid atherosclerosis: A 3-year follow-up study. Circulation 2000; 102: 1536–1541.
Lurbe E, Redon J, Kesani A, Pascual JM, Tacons J, Alvarez V, Batlle D . Increase in nocturnal blood pressure and progression to microalbuminuria in type 1 diabetes. N Engl J Med 2002; 347: 797–805.
Metoki H, Ohkubo T, Kikuya M, Asayama K, Obara T, Hashimoto J, Totsune K, Hoshi H, Satoh H, Imai Y . Prognostic significance for stroke of a morning pressor surge and a nocturnal blood pressure decline: the Ohasama study. Hypertension 2006; 47: 149–154.
Parati G, Lombardi C, Hedner J, Bonsignore MR, Grote L, Tkacova R, Levy P, Riha R, Bassetti C, Narkiewicz K, Mancia G, McNicholas WT . Position paper on the management of patients with obstructive sleep apnea and hypertension: joint recommendations by the European Society of Hypertension, by the European Respiratory Society and by the members of European COST (COoperation in Scientific and Technological research) ACTION B26 on obstructive sleep apnea. J Hypertens 2012; 30: 633–646.
Parati G, Lombardi C, Hedner J, Bonsignore M, Grote L, Tkacova R, Levy P, Riha R, Bassetti C, Narkiewicz K, Mancia G, McNicholas WT, on behalf of the EU COST Action B26 members11. Recommendations for the management of patients with obstructive sleep apnoea and hypertension. Eur Respir J 2013; 41: 523–538.
O'Brien E, Asmar R, Beilin L, Imai Y, Mallion JM, Mancia G, Mengden T, Myers M, Padfield P, Palatini P, Parati G, Pickering T, Redon J, Staessen J, Stergiou G, Verdecchia P, European Society of Hypertension Working Group on Blood Pressure Monitoring.. European Society of Hypertension recommendations for conventional, ambulatory and home blood pressure measurement. J Hypertens 2003; 21: 821–848.
Oikawa T, Obara T, Ohkubo T, Kikuya M, Asayama K, Metoki H, Komai R, Murai K, Hashimoto J, Totsune K, Imai YJ . Characteristics of resistant hypertension determined by self-measured blood pressure at home and office blood pressure measurements: the J-HOME study. J Hypertens 2006; 24: 1737–1743.
de la Sierra A, Banegas JR, Oliveras A, Gorostidi M, Segura J, de la Cruz JJ, Armario P, Ruilope LM . Clinical differences between resistant hypertensives and patients treated and controlled with three or less drugs. J Hypertens 2012; 30: 1211–1216.
Pickering TG, Davidson K, Gerin W, Schwartz JE . Masked hypertension. Hypertension 2002; 40: 795–796.
Parati G, Ulian L, Santucciu C, Omboni S, Mancia G . Difference between clinic and daytime blood pressure is not a measure of the white coat effect. Hypertension 1998; 31: 1185–1189.
Muxfeldt ES, Bloch KV, Nogueira Ada R, Salles GF . True resistant hypertension: is it possible to be recognized in the office? Am J Hypertens 2005; 18 (12 Pt 1): 1534–1540.
Oliveras A, Armario P, Hernandez-Del Rey R, Arroyo JA, Poch E, Larrousse M, Roca-Cusachs A, de la Sierra A . Urinary albumin excretion is associated with true resistant hypertension. J Hum Hypertens 2010; 24: 27–33.
Pierdomenico SD, Lapenna D, Bucci A, Di Tommaso R, Di Mascio R, Manente BM, Caldarella MP, Neri M, Cuccurullo F, Mezzetti A . Cardiovascular outcome in treated hypertensive patients with responder, masked, false resistant, and true resistant hypertension. Am J Hypertens 2005; 18: 1422–1428.
Salles GF, Cardoso CR, Muxfeldt ES . Prognostic influence of office and ambulatory blood pressures in resistant hypertension. Arch Int Med 2008; 168: 2340–2346.
Cuspidi C, Macca G, Sampieri L, Michev I, Salerno M, Fusi V, Severgnini B, Meani S, Magrini F, Zanchetti A . High prevalence of cardiac and extracardiac target organ damage in refractory hypertension. J Hypertens 2001; 19: 2063–2070.
Moser M, Setaro JF . Clinical practice. Resistant or difficult-to-control hypertension. N Engl J Med 2006; 355: 385–392.
Johansson K, Hemmingsson E, Harlid R, Trolle Lagerros Y, Granath F, Rossner S, Neovius M . Longer term effects of very low energy diet on obstructive sleep apnoea in cohort derived from randomised controlled trial: prospective observational follow-up study. BMJ 2011; 342: d3017.
Yee BJ, Phillips CL, Banerjee D, Caterson I, Hedner JA, Grunstein RR . The effect of sibutramine-assisted weight loss in men with obstructive sleep apnoea. Int J Obes 2007; 31: 161–168.
Pannain S, Mokhlesi B . Bariatric surgery and its impact on sleep architecture, sleep-disordered breathing, and metabolism. Best Pract Res Clin Endocrinol Metab 2010; 24: 745–761.
Buchwald H, Avidor Y, Braunwald E, Jensen MD, Pories W, Fahrbach K, Schoelles K . Bariatric surgery: a systematic review and meta-analysis. JAMA 2004; 292: 1724–1737.
Leech JA, Onal E, Lopata M . Nasal CPAP continues to improve sleep-disordered breathing and daytime oxygenation over long-term follow-up of occlusive sleep apnea syndrome. Chest 1992; 102: 1651–1655.
Kribbs NB, Pack AI, Kline LR, Getsy JE, Schuett JS, Henry JN, Maislin G, Dinges DF . Effects of one night without nasal CPAP treatment on sleep and sleepiness in patients with obstructive sleep apnea. Am Rev Respir Dis 1993; 147: 1162–1168.
Bradley TD, Floras JS . Obstructive sleep apnoea and its cardiovascular consequences. Lancet 2009; 373: 82–93.
Alajmi M, Mulgrew AT, Fox J, Davidson W, Schulzer M, Mak E, Ryan CF, Fleetham J, Choi P, Ayas NT . Impact of continuous positive airway pressure therapy on blood pressure in patients with obstructive sleep apnea hypopnea: a meta-analysis of randomized controlled trials. Lung 2007; 185: 67–72.
Haentjens P, Van Meerhaeghe A, Moscariello A, De Weerdt S, Poppe K, Dupont A, Velkeniers B . The impact of continuous positive airway pressure on blood pressure in patients with obstructive sleep apnea syndrome: evidence from a meta-analysis of placebo-controlled randomized trials. Arch Int Med 2007; 167: 757–764.
Bazzano LA, Khan Z, Reynolds K, He J . Effect of nocturnal nasal continuous positive airway pressure on blood pressure in obstructive sleep apnea. Hypertension 2007; 50: 417–423.
Becker HF, Jerrentrup A, Ploch T, Grote L, Penzel T, Sullivan CE, Peter JH . Effect of nasal continuous positive airway pressure treatment on blood pressure in patients with obstructive sleep apnea. Circulation 2003; 107: 68–73.
Logan AG, Tkacova R, Perlikowski SM, Leung RS, Tisler A, Floras JS, Bradley TD . Refractory hypertension and sleep apnoea: effect of CPAP on blood pressure and baroreflex. Eur Respir J 2003; 21: 241–247.
Dernaika TA, Kinasewitz GT, Tawk MM . Effects of nocturnal continuous positive airway pressure therapy in patients with resistant hypertension and obstructive sleep apnea. J Clin Sleep Med 2009; 5: 103–107.
Barbe F, Duran-Cantolla J, Capote F, de la Pena M, Chiner E, Masa JF, Gonzalez M, Marin JM, Garcia-Rio F, de Atauri JD, Teran J, Mayos M, Monasterio C, del Campo F, Gomez S, de la Torre MS, Martinez M, Montserrat JM . Long-term effect of continuous positive airway pressure in hypertensive patients with sleep apnea. Am J Respir Crit Care Med 2010; 181: 718–726.
Lozano L, Tovar JL, Sampol G, Romero O, Jurado MJ, Segarra A, Espinel E, Rios J, Untoria MD, Lloberes P . Continuous positive airway pressure treatment in sleep apnea patients with resistant hypertension: a randomized, controlled trial. J Hypertens 2010; 28: 2161–2168.
Iftikhar IH, Hays ER, Iverson MA, Magalang UJ, Maas AK . Effect of oral appliances on blood pressure in obstructive sleep apnea: a systematic review and meta-analysis. J Clin Sleep Med 2013; 9: 165–174.
DiBona GF, Kopp UC . Neural control of renal function. Physiol Rev 1997; 77: 75–197.
Doumas M, Faselis C, Papademetriou V . Renal sympathetic denervation in hypertension. Curr Opin Nephrol Hypertens 2011; 20: 647–653.
Schlaich MP, Sobotka PA, Krum H, Lambert E, Esler MD . Renal sympathetic-nerve ablation for uncontrolled hypertension. N Engl J Med 2009; 361: 932–934.
Witkowski A, Prejbisz A, Florczak E, Kadziela J, Sliwinski P, Bielen P, Michalowska I, Kabat M, Warchol E, Januszewicz M, Narkiewicz K, Somers VK, Sobotka PA, Januszewicz A . Effects of renal sympathetic denervation on blood pressure, sleep apnea course, and glycemic control in patients with resistant hypertension and sleep apnea. Hypertension 2011; 58: 559–565.
Ziegler MG, Milic M, Sun P . Antihypertensive therapy for patients with obstructive sleep apnea. Curr Opin Nephrol Hypertens 2011; 20: 50–55.
Pimenta E, Calhoun DA . Treatment of resistant hypertension. J Hypertens 2010; 28: 2194–2195.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Parati, G., Ochoa, J., Bilo, G. et al. Obstructive sleep apnea syndrome as a cause of resistant hypertension. Hypertens Res 37, 601–613 (2014). https://doi.org/10.1038/hr.2014.80
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2014.80
Keywords
This article is cited by
-
Resistant hypertension: consensus document from the Korean society of hypertension
Clinical Hypertension (2023)
-
miR-485-5p alleviates obstructive sleep apnea syndrome with hypertension by inhibiting PI3K/AKT signaling pathway via downregulating HIF3A expression
Sleep and Breathing (2023)
-
Association between arterial stiffness and sleep apnoea in patients with resistant hypertension
Journal of Human Hypertension (2022)
-
Nocturnal nasal congestion is associated with uncontrolled blood pressure in patients with hypertension comorbid obstructive sleep apnea
European Archives of Oto-Rhino-Laryngology (2022)
-
Obstructive sleep apnea and associated factors among hypertensive patients attending a tertiary cardiac center in Tanzania: a comparative cross-sectional study
Sleep Science and Practice (2021)
