
Abstract
Angiotensin II (AngII) stimulates vascular smooth muscle cell (VSMC) proliferation; however, the effect of AngII on cell proliferation in the presence of mechanical force is not clear. We investigated the mechanism of AngII-induced cell proliferation mediated by mechanical stretch in VSMCs of both normotensive and hypertensive rats. VSMCs obtained from the thoracic aortas of 8-week-old Wistar–Kyoto (WKY) rats and spontaneously hypertensive rats (SHR) were stretched by a Flex culture system. Mechanical stretch significantly upregulated protein expression of AngII type 1 (AT1) receptor, epidermal growth factor (EGF) receptor and mitogen-activated protein kinase phosphatase-1 in both SHR and WKY VSMCs; however, there was no significant difference in these changes between the cells from SHR and WKY. Mechanical stretch attenuated AngII-induced phosphorylation of extracellular signal-regulated kinase (ERK) 1/2, ERK kinase (MEK) and EGF receptor; it also attenuated [3H] thymidine incorporation and cell proliferation in VSMC of WKY. In contrast, the effects of AngII were augmented by mechanical stretch in VSMC of SHR. AngII-induced ERK 1/2 phosphorylation and cell proliferation in SHR were inhibited by pretreatment with an AT1 receptor blocker, candesartan and an inhibitor of MEK, PD98059. Moreover, pretreatment with an EGF receptor tyrosine kinase inhibitor, AG1478, also blocked upregulation of AngII-induced ERK 1/2 phosphorylation induced by stretch in SHR VSMCs. This study demonstrates that mechanical stretch augments SHR VSMC proliferation through an AT1/EGF receptor/ERK-dependent pathway. These findings may provide new insights into the signaling mechanisms whereby AngII exerts its growth-promoting effects on vasculature in a hypertensive state.
Similar content being viewed by others


Introduction
Angiotensin II (AngII) has an important role in the pathogenesis of a variety of cardiovascular diseases. Treatment with AngII type 1 (AT1) receptor blockers elicits therapeutic benefits in the treatment of hypertension and cardiovascular disorders.1, 2 In spontaneously hypertensive rats (SHR), vascular reactivity to AngII has been found to be increased, although responses to endothelin-1, vasopressin and norepinephrine are decreased or unchanged.3, 4 The AngII-augmented vascular response is not specific to SHR, in either the prehypertensive or the established hypertensive phases, and it has been demonstrated in other models of hypertension, including stroke-prone SHR, renal hypertensive rats, deoxycorticosterone acetate/salt-hypertensive rats and AngII-infused hypertensive rats.5, 6, 7, 8 These findings suggest that the vascular response to AngII is activated in a hypertensive state.
AngII acts not only as a vasoconstrictive peptide but also as a vascular growth factor.9, 10 In particular, AngII stimulates vascular smooth muscle cell (VSMC) proliferation, which is an important process in vascular remodeling in hypertension.11 In this regard, many studies have emphasized the roles of Ras/Raf-1/mitogen-activated protein kinase cascades, especially that of extracellular signal-regulated protein kinase (ERK) 1/2, in mediating VSMC proliferation induced by AngII.12 ERK1/2 phosphorylation is increased in the vasculature of various animal models of hypertension and in cultured VSMCs from SHR.13, 14 Furthermore, enhanced AngII-induced contraction and growth is associated with increased ERK1/2 activation.15
VSMCs are major cellular components of blood vessels and they are subjected to a dynamic mechanical environment modulated by pulsatile pressure. The accompanying stress regulates normal vascular tone;16 it also contributes to atherogenesis,17 vascular remodeling associated with hypertension18 and acute rupture of atherosclerotic lesions.19 Despite the fact that VSMCs reside in a dynamic state and are constantly subjected to pulsatile pressures in vivo, the changes of VSMC signaling pathways and phenotypes have not been well investigated under conditions of mechanical force. We previously demonstrated that AngII and mechanical stretch synergistically increase oxidative stress in VSMCs by upregulation of the AT1 receptor.20 However, the effect of AngII on VSMC proliferation in the presence of mechanical stretch is still unclear.
In this study, we hypothesized that mechanical stretch at the vessel wall affects AngII-induced VSMC proliferation differentially in normotensive and hypertensive animals. To test this hypothesis, we investigated the cellular and molecular mechanisms underlying the cell proliferation induced by the contribution of mechanical stretch and AngII using VSMCs from normotensive Wistar–Kyoto (WKY) rats and SHR.
Methods
Cell culture and mechanical stretch
VSMCs were isolated from the thoracic aortas of 8-week-old male SHR and WKY rats by enzymatic digestion as previously described.20 Blood pressure was not significantly different between SHR and WKY rats at this age (data not shown). Cells were grown in Dulbecco's modified Eagle's medium (Sigma-Aldrich, St Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT, USA), penicillin (100 U/ml; Invitrogen, Carlsbad, CA, USA) and streptomycin (100 μg/ml, Invitrogen) at 37 °C under 5% CO2 /95% air in a humidified incubator. Only VSMCs from the third to sixth passages were used. The cells were then cultured in six-well collagen I-coated BioFlex plates containing a flexible silicone elastomer substratum (Flexcell International, Hillsborough, NC, USA). Cells that achieved 70% confluence were subjected to cyclic stretch (60 cycles per min, 5 or 15% elongation) for 2 h using a computer-controlled mechanical strain unit (Flexcell International). After a cyclic stretch, the growth medium was replaced with medium containing 0.1% fetal bovine serum for 24 h. In some experiments, VSMCs were preincubated with various inhibitors, as indicated, before stimulation with AngII.
Western blot analysis
After stimulation with AngII, the proteins were extracted as previously described.21 The protein content of the supernatant was determined with the Bradford assay, using bovine serum albumin as the standard. The proteins were subjected to SDS–PAGE and detected by an immunoblot assay with the following antibodies: anti-phospho-ERK1/2, anti-ERK1/2, anti-phospho-epidermal growth factor (EGF)-receptor (Tyr-992), anti-EGF-receptor, anti-phospho-ERK kinase (MEK), anti-MEK (Cell Signaling Technology, Danvers, MA, USA), anti-AT1 receptor (Millipore, Billerica, MA, USA) and anti-mitogen-activated protein kinase phosphatase-1 (MKP)-1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). To confirm equal loading, membranes were reprobed with an antibody against β-actin (Sigma-Aldrich). The protein bands were visualized using an ECL plus system (GE Healthcare, Buckinghamshire, UK). Band intensities were quantified by densitometry of the immunoblots using NIH ImageJ software.
[3H] Thymidine incorporation and cell counts
DNA synthesis was determined by measuring [3H] thymidine incorporation as previously described.21 In brief, quiescent VSMCs cultured in six-well plates were treated with AngII (100 nmol l−1) containing 1 μCi ml−1 [3H] thymidine for 24 h. The radioactivity of the cell lysates was determined using a liquid scintillation counter.
Cells were counted with a Coulter Counter Multisizer 3 (Beckman-Coulter, Miami, FL, USA). The cells were washed in phosphate-buffered saline and lifted from the culture dish after 3 min of incubation with 0.5% trypsin and 0.53-mmol l−1 EDTA (Invitrogen). The trypsin was inactivated by the addition of an equal volume of medium, and cells were briefly centrifuged to form a pellet. Cells were resuspended and counted using a Coulter Counter.
AngII content in the medium of VSMCs
The culture medium (3 ml per dish) was collected from dishes before and after stretching the cells. We measured AngII concentration by radioimmunoassay using a specific antibody for AngII—monoiodinated 125I-labeled AngII—and AngII standard as previously described.22 In brief, each lyophilized antiserum was diluted sufficiently to yield a specific binding of 28–35% after incubation with a 2500 cpm label for 48 h at 4 °C. Bound and free AngII were separated with dextran-coated charcoal (10 mg of charcoal and 1 mg of dextran in 1 ml of assay buffer/tube). After centrifugation, the supernatants were decanted and counted for 5 min in an automated gamma counter.
Statistical analysis
Values are presented as means±s.e. Multiple group comparison was done by a one-way analysis of variance. The two-tailed Student’s t-test was used to compare drug-treated and vehicle-treated specimens under identical conditions. Values of P<0.05 were considered statistically significant.
Results
Effect of mechanical stretch on AngII-induced VSMC proliferation and ERK 1/2 phosphorylation
First, we measured [3H] thymidine incorporation as a marker of DNA synthesis in VSMCs. As shown in Figure 1a, AngII (100 nmol l−1, 24 h) significantly increased DNA synthesis in unstretched WKY and SHR VSMCs (n=6, P<0.05). In addition, treatment with 5 or 15% elongation stretch augmented AngII-induced [3H] thymidine incorporation in SHR VSMCs, whereas diminishing it in WKY VSMCs (n=6, P<0.05). However, treatment with stretch alone did not increase [3H] thymidine incorporation in cells from both WKYs and SHR. We also determined the cell numbers using a Coulter Counter. As shown in Figure 1b, AngII (100 nmol l−1, 24 h) significantly increased cell number in unstretched WKY VSMCs (n=6, P<0.05). Although mechanical stretch attenuated AngII-induced cell proliferation in WKY VSMCs, AngII increased cell numbers in SHR VSMCs.

Effect of mechanical stretch on AngII-induced (100 nmol l−1, 24 h) [3H] thymidine incorporation (a) and cell numbers (b) in VSMCs from SHR and WKY rats. VSMCs were subjected to mechanical stretch (5 or 15% elongation). The measurements of [3H] thymidine incorporation and cell numbers are described in the Methods section. In all panels, the bar graphs represent means±s.e. (n=6), expressed as the fold increase in incorporation over that in unstimulated cells. *P<0.05 vs. control in unstretched VSMCs. #P<0.05 vs. unstretched VSMCs with AngII. **P<0.05 vs. VSMCs without AngII.
Next, we investigated ERK 1/2 activation, which was determined by phosphorylation. AngII (100 nmol/l) enhanced ERK 1/2 phosphorylation, with a peak at 10 min, in both unstretched WKY and SHR VSMCs. Mechanical stretch (5 or 15% elongation, 2 h) significantly increased AngII-induced ERK 1/2 phosphorylation in SHR VSMCs (Figure 2a; n=6, P<0.01). However, mechanical stretch significantly attenuated AngII-induced ERK 1/2 phosphorylation in WKY VSMCs (Figure 2b; n=6, P<0.01).

Effect of mechanical stretch on AngII (100 nmol l−1)-induced ERK 1/2 phosphorylation in VSMCs from SHR (a) and WKY rats (b). VSMCs were subjected to mechanical stretch (5 or 15% elongation) for 2 h. Western blotting with phospho-ERK 1/2 (top panel) or total ERK 1/2 (lower panel) antibodies. In all panels, the bar graphs represent means±s.e. (n=6), expressed as the fold increase in phosphorylation over that in unstimulated cells. (c) The culture medium from SHR VSMCs after stretch (2 h) was measured for AngII content by radioimmunoassay. Values are expressed as means±s.e. (n=6). *P<0.05 vs. unstretched VSMCs.
We measured AngII content in the culture medium of stretched VSMCs and there was no significant increase in AngII after stretching (Figure 2c). Moreover, 0.01 nmol l−1 AngII (10 min) did not significantly increase ERK 1/2 phosphorylation in unstretched SHR and WKY VSMCs (data not shown).
Involvement of AT1 receptor/ERK 1/2-dependent pathway in AngII-induced DNA synthesis in stretched VSMCs
To investigate the role of the AT1 receptor in mechanical stretch-mediated regulation of AngII-induced WKY and SHR VSMC proliferation, we examined the effect of mechanical stretch on AT1 receptor protein expression. Treatment with 5 or 15% elongative mechanical stretch increased AT1 receptor protein expression in both WKY and SHR VSMCs (n=6, P<0.01), although the increases between cells from WKYs and SHR were not significant (Figure 3a). Moreover, an ARB, candesartan (100 nmol l−1 for 3 h) and a specific inhibitor of MEK, PD98059 (10 μmol l−1 for 30 min), abolished both AngII-induced ERK 1/2 phosphorylation and [3H] thymidine incorporation in stretched SHR VSMCs (Figures 3b and c). Inhibitors alone did not affect phosphorylation or thymidine incorporation (data not shown). These results suggest that the upregulation of AngII-induced cell proliferation in stretched SHR VSMCs is mediated through AT1 receptor-mediated ERK 1/2 phosphorylation.

The involvement of AT1 receptor/ERK 1/2-dependent pathway in AngII-induced DNA synthesis in stretched VSMCs. VSMCs were subjected to mechanical stretch (5 or 15% elongation) for 2 h. (a) Western blotting with AT1 receptor (top panel) or β-Actin (lower panel) antibodies. (b) VSMCs were treated with 100 nmol l−1 AngII for 10 min. Western blotting with phospho-ERK 1/2 (top panel) or total ERK 1/2 (lower panel) antibodies. (a, b) In all panels, bar graphs represent means±s.e. (n=6), expressed as the fold increase in expression over that in unstimulated cells. (c) Effect of candesartan and PD98059 on [3H] thymidine incorporation. The bar graphs represent means±s.e. (n=6), expressed as the fold increase in incorporation over that in unstimulated cells. *P<0.05 vs. control in unstretched VSMCs. #P<0.05 vs. stretched VSMCs with AngII.
Effect of mechanical stretch on VSMC MKP-1 expression and AngII-induced MEK phosphorylation
We further investigated signal transduction pathways of mechanical stretch regulation on AngII-induced ERK 1/2 activation. First, we examined the effect of mechanical stretch on AngII-induced phosphorylation of MEK, which is an upstream activator of ERK 1/2 in WKY and SHR VSMCs. As shown in Figure 4a, mechanical stretch upregulated AngII-induced MEK phosphorylation in SHR VSMCs, but decreased phosphorylation in WKY VSMCs. These observations are in line with the changes in AngII-induced ERK 1/2 phosphorylation. In addition, we examined the effect of mechanical stretch on the expression of MKP-1, which is a dual-specific protein phosphatase and downregulates ERK 1/2 activation.23 Treatment with mechanical stretch for 2 h upregulated MKP-1 protein expression in both WKY and SHR VSMCs (n=6, P<0.01), but there was no significant difference in protein expression between cells from WKY and SHR (Figure 4b). In contrast, mechanical stretch had no effect on protein expression levels of Ras or Raf-1, two upstream activators of ERK/MEK (data not shown). These results suggest that the upstream signaling of ERK/MEK contributes to the mechanical stretch-mediated regulation of AngII-induced VSMC proliferation.

Effect of mechanical stretch on MKP-1 expression and AngII (100 nmol l−1, 10 min)-induced MEK phosphorylation. (a) Western blotting with phospho-MEK (top panel) or total MEK (lower panel) antibodies. (b) Western blotting with MKP-1 (top panel) or β-Actin (lower panel) antibodies. (a, b): In all panels, bar graphs represent means±s.e. (n=6), expressed as the fold increase in expression over that in unstimulated cells. *P<0.05 vs. control in unstretched VSMCs. #P<0.05 vs. stretched VSMCs with AngII.
Involvement of EGF receptor transactivation in mechanical stretch regulation of AngII-induced VSMC ERK 1/2 phosphorylation
To examine the upstream signaling of ERK/MEK regulated by AngII, we focused on the EGF receptor. It is generally accepted that Gq-coupled receptors activate ERK 1/2 in VSMCs through transactivation of the EGF receptor.24, 25, 26 Therefore, we investigated the effect of mechanical stretch on AngII-induced EGF receptor phosphorylation. Mechanical stretch (5 or 15% elongation for 2 h) upregulated EGF receptor protein expression in both WKY and SHR VSMCs (n=6, P<0.01; Figure 5a). However, mechanical stretch increased AngII-induced EGF receptor phosphorylation in SHR VSMCs, but decreased it in WKY VSMCs (n=6, P<0.01). In addition, pretreatment with an EGF receptor tyrosine kinase inhibitor, AG1478 (25 nmol l−1), markedly inhibited AngII-induced ERK 1/2 phosphorylation in SHR VSMCs, with or without stretch (Figure 5b). AG1478 alone did not affect ERK 1/2 phosphorylation (data not shown). These results suggest that EGF receptor transactivation is required for mechanical stretch-mediated regulation of AngII-induced proliferation of VSMCs.

The involvement of EGF receptor transactivation in mechanical stretch regulation of AngII (100 nmol l−1, 10 min)-induced ERK 1/2 phosphorylation. (a) Western blotting with phospho-EGF receptor (top panel), total EGF receptor (middle panel) or β-Actin (lower panel) antibodies. (b) Western blotting with phospho-ERK 1/2 (top panel) or total ERK 1/2 (lower panel) antibodies. (a, b): In all panels, bar graphs represent means±s.e. (n=6), expressed as the fold increase in expression over that in unstimulated cells. *P<0.05 vs. control in unstretched VSMCs. #P<0.05 vs. stretched VSMCs with AngII.
Discussion
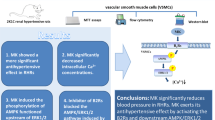
VSMCs in the arterial wall are constantly exposed to a variety of flow-related mechanical stimuli27 that can induce significant changes in cell growth and alter cell phenotype.20, 28, 29 In this study, we demonstrated several significant findings regarding the response of VSMCs to mechanical force (Figure 6). First, mechanical stretch upregulates expression of AT1 receptor, EGF receptor and MKP-1; however, these changes are not significantly different between WKY and SHR VSMCs. Second, mechanical stretch augments AngII-induced cell proliferation in SHR VSMCs, but suppresses proliferation in WKY VSMCs. Third, mechanical stretch upregulates AngII-induced VSMC proliferation in SHR through an AT1/EGF-receptor/ERK-dependent pathway. These findings provide novel information about different VSMC signal transduction pathways in both normotensive and hypertensive subjects. They also suggest a possible mechanism for the progression of atherosclerosis in a hypertensive state.

Proposed cell signaling pathways in VSMCs from SHR and WKY rats. See Discussion for details.
AngII is crucial in maintaining the structural and functional integrity of the vessel wall and has important roles in VSMC contraction and proliferation. Clinical evidence has revealed that treatment with AT1 receptor blockers reduces morbidity and mortality in patients with hypertension, congestive cardiac failure and myocardial infarction.30, 31 In this study, we found that mechanical stretch augments AngII-induced VSMC proliferation in SHR. It has been reported that AngII stimulation augments DNA synthesis without affecting cell division in VSMCs.32, 33 Because this effect is enhanced in VSMCs from SHR, we confirmed AngII-induced VSMC proliferation by measuring DNA synthesis and performing cell counts. In addition, the augmentative effect of mechanical stretch is blocked by treatment with candesartan and PD98059, suggesting that the AT1 receptor and ERK 1/2-dependent pathways essentially contribute to AngII-induced VSMC proliferation induced by mechanical stretch. Moreover, as shown in Figures 1, 2a and b, mechanical stretch augments both cell proliferation and ERK 1/2 phosphorylation in an elongation-dependent manner. This suggests that ARB lowers blood pressure by attenuating mechanical force and that it could have a protective role in vascular proliferation.
In agreement with previous studies,20 we found that mechanical stretch upregulated AT1 receptor expression to similar levels in both SHR and WKY VSMCs. Schiffrin et al.34 reported that vascular tissue AT1 receptor density and affinity are not significantly different between adult SHR and WKY rats, suggesting that upregulation of AngII-mediated vascular proliferation in SHR occurs primarily at the post-receptor level, during signal transduction. Gq-coupled receptors, including the AT1 receptor, activate ERK 1/2 in VSMCs through transactivation of the EGF receptor.25, 35 Kagiyama et al.36 demonstrated that the EGF receptor-activated ERK pathway has a critical role in the development of hypertension and left ventricular hypertrophy, but not in established left ventricular hypertrophy in adult SHR (13–20 weeks old). The authors also showed that blockade of the EGF receptor significantly attenuated left ventricular hypertrophy in young SHR (5–12 weeks old). In this study, we found that mechanical stretch upregulated EGF receptor protein expression in VSMCs from both WKY and SHR rats. Interestingly, AngII-induced EGF receptor phosphorylation was augmented in SHR VSMCs but was rather diminished in WKY VSMCs. Moreover, the changes in downstream signaling of EGF receptor transactivation and [3H] thymidine incorporation were associated with the changes in AngII-induced EGF receptor transactivation. These data suggest that EGF receptor transactivation is a key factor responsible for AngII-induced VSMC proliferation mediated by mechanical stretch. Previous reports indicated that several candidates are involved in EGF receptor transactivation, such as Src activation or reactive oxygen species.24, 25 Therefore, it is possible that different responses to mechanical stretch by these candidates regulate EGF receptor transactivation and cell proliferation in AngII-treated VSMCs.
In conclusion, this study shows that mechanical stretch directly regulates AngII-induced VSMC proliferation and that the AT1/EGF receptor/ERK-dependent signaling pathway is involved in proliferation in VSMCs of SHR. This investigation provides new insights into the potential mechanisms responsible for the development of hypertension, atherosclerosis and the progression of proliferative cardiovascular diseases. The present results also suggest that AngII has a pivotal role in the development of hypertension and the progression of atherosclerosis under the conditions in which VSMCs are mechanically stretched. These data support the clinical evidence demonstrating the therapeutic importance for sufficiently lowering blood pressure with AT1 receptor blockers in hypertensive patients.
References
de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T . International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev 2000; 52: 415–472.
Kobori H, Nangaku M, Navar LG, Nishiyama A . The intrarenal renin–angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 2007; 59: 251–287.
Touyz RM, Deng LY, Li JS, Schiffrin EL . Differential effects of vasopressin and endothelin-1 on vascular contractile and calcium responses in pressurized small arteries from spontaneously hypertensive rats. J Hypertens 1996; 14: 983–991.
Resink TJ, Scott-Burden T, Baur U, Burgin M, Buhler FR . Enhanced responsiveness to angiotensin II in vascular smooth muscle cells from spontaneously hypertensive rats is not associated with alterations in protein kinase C. Hypertension 1989; 14: 293–303.
Berecek KH, Schwertschlag U, Gross F . Alterations in renal vascular resistance and reactivity in spontaneous hypertension of rats. Am J Physiol 1980; 238: H287–H293.
Couture R, Regoli D . Vascular reactivity to angiotensin and noradrenaline in spontaneously and renal hypertensive rats. Clin Exp Hypertens 1980; 2: 45–63.
Couture R, Regoli D . Vascular reactivity to angiotensin and noradrenaline in rats maintained on a sodium free diet or made hypertensive with desoxycorticosterone acetate and salt (DOCA/salt). Clin Exp Hypertens 1980; 2: 25–43.
Ichihara A, Inscho EW, Imig JD, Michel RE, Navar LG . Role of renal nerves in afferent arteriolar reactivity in angiotensin-induced hypertension. Hypertension 1997; 29: 442–449.
Okamoto K, Kato S, Arima N, Fujii T, Morimatsu M, Imaizumi T . Cyclin-dependent kinase inhibitor, p21Waf1, regulates vascular smooth muscle cell hypertrophy. Hypertens Res 2004; 27: 283–291.
Daemen MJ, Lombardi DM, Bosman FT, Schwartz SM . Angiotensin II induces smooth muscle cell proliferation in the normal and injured rat arterial wall. Circ Res 1991; 68: 450–456.
Touyz RM, Deng LY, He G, Wu XH, Schiffrin EL . Angiotensin II stimulates DNA and protein synthesis in vascular smooth muscle cells from human arteries: role of extracellular signal-regulated kinases. J Hypertens 1999; 17: 907–916.
Duff JL, Marrero MB, Paxton WG, Schieffer B, Bernstein KE, Berk BC . Angiotensin II signal transduction and the mitogen-activated protein kinase pathway. Cardiovasc Res 1995; 30: 511–517.
Xu Q, Liu Y, Gorospe M, Udelsman R, Holbrook NJ . Acute hypertension activates mitogen-activated protein kinases in arterial wall. J Clin Invest 1996; 97: 508–514.
Hamaguchi A, Kim S, Yano M, Yamanaka S, Iwao H . Activation of glomerular mitogen-activated protein kinases in angiotensin II-mediated hypertension. J Am Soc Nephrol 1998; 9: 372–380.
Touyz RM, El Mabrouk M, He G, Wu XH, Schiffrin EL . Mitogen-activated protein/extracellular signal-regulated kinase inhibition attenuates angiotensin II-mediated signaling and contraction in spontaneously hypertensive rat vascular smooth muscle cells. Circ Res 1999; 84: 505–515.
D’Angelo G, Meininger GA . Transduction mechanisms involved in the regulation of myogenic activity. Hypertension 1994; 23: 1096–1105.
Zarins CK, Zatina MA, Giddens DP, Ku DN, Glagov S . Shear stress regulation of artery lumen diameter in experimental atherogenesis. J Vasc Surg 1987; 5: 413–420.
Dzau VJ, Gibbons GH, Morishita R, Pratt RE . New perspectives in hypertension research. Potentials of vascular biology. Hypertension 1994; 23: 1132–1140.
Richardson PD, Davies MJ, Born GV . Influence of plaque configuration and stress distribution on fissuring of coronary atherosclerotic plaques. Lancet 1989; 2: 941–944.
Hitomi H, Fukui T, Moriwaki K, Matsubara K, Sun GP, Rahman M, Nishiyama A, Kiyomoto H, Kimura S, Ohmori K, Abe Y, Kohno M . Synergistic effect of mechanical stretch and angiotensin II on superoxide production via NADPH oxidase in vascular smooth muscle cells. J Hypertens 2006; 24: 1089–1095.
Nishiyama A, Yao L, Fan Y, Kyaw M, Kataoka N, Hashimoto K, Nagai Y, Nakamura E, Yoshizumi M, Shokoji T, Kimura S, Kiyomoto H, Tsujioka K, Kohno M, Tamaki T, Kajiya F, Abe Y . Involvement of aldosterone and mineralocorticoid receptors in rat mesangial cell proliferation and deformability. Hypertension 2005; 45: 710–716.
Nishiyama A, Seth DM, Navar LG . Renal interstitial fluid concentrations of angiotensins I and II in anesthetized rats. Hypertension 2002; 39: 129–134.
Duff JL, Monia BP, Berk BC . Mitogen-activated protein (MAP) kinase is regulated by the MAP kinase phosphatase (MKP-1) in vascular smooth muscle cells. Effect of actinomycin D and antisense oligonucleotides. J Biol Chem 1995; 270: 7161–7166.
Eguchi S, Iwasaki H, Inagami T, Numaguchi K, Yamakawa T, Motley ED, Owada KM, Marumo F, Hirata Y . Involvement of PYK2 in angiotensin II signaling of vascular smooth muscle cells. Hypertension 1999; 33: 201–206.
Ushio-Fukai M, Griendling KK, Becker PL, Hilenski L, Halleran S, Alexander RW . Epidermal growth factor receptor transactivation by angiotensin II requires reactive oxygen species in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2001; 21: 489–495.
Voisin L, Foisy S, Giasson E, Lambert C, Moreau P, Meloche S . EGF receptor transactivation is obligatory for protein synthesis stimulation by G protein-coupled receptors. Am J Physiol Cell Physiol 2002; 283: C446–C455.
Learoyd BM, Taylor MG . Alterations with age in the viscoelastic properties of human arterial walls. Circ Res 1966; 18: 278–292.
Yasuda N, Miura S, Akazawa H, Tanaka T, Qin Y, Kiya Y, Imaizumi S, Fujino M, Ito K, Zou Y, Fukuhara S, Kunimoto S, Fukuzaki K, Sato T, Ge J, Mochizuki N, Nakaya H, Saku K, Komuro I . Conformational switch of angiotensin II type 1 receptor underlying mechanical stress-induced activation. EMBO Rep 2008; 9: 179–186.
Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I . Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol 2004; 6: 499–506.
Cohn JN, Tognoni G . A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med 2001; 345: 1667–1675.
Mochizuki S, Dahlof B, Shimizu M, Ikewaki K, Yoshikawa M, Taniguchi I, Ohta M, Yamada T, Ogawa K, Kanae K, Kawai M, Seki S, Okazaki F, Taniguchi M, Yoshida S, Tajima N . Valsartan in a Japanese population with hypertension and other cardiovascular disease (Jikei Heart Study): a randomised, open-label, blinded endpoint morbidity-mortality study. Lancet 2007; 369: 1431–1439.
Hixon ML, Muro-Cacho C, Wagner MW, Obejero-Paz C, Millie E, Fujio Y, Kureishi Y, Hassold T, Walsh K, Gualberto A . Akt1/PKB upregulation leads to vascular smooth muscle cell hypertrophy and polyploidization. J Clin Invest 2000; 106: 1011–1020.
Hixon ML, Gualberto A . Vascular smooth muscle polyploidization–from mitotic checkpoints to hypertension. Cell Cycle 2003; 2: 105–110.
Schiffrin EL, Thome FS, Genest J . Vascular angiotensin II receptors in SHR. Hypertension 1984; 6: 682–688.
Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK . Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res 2002; 91: 406–413.
Kagiyama S, Qian K, Kagiyama T, Phillips MI . Antisense to epidermal growth factor receptor prevents the development of left ventricular hypertrophy. Hypertension 2003; 41: 824–829.
Acknowledgements
We are grateful to Takeda Pharmaceutical for supplying us with candesartan. We thank Izumi Shibata and Yuki Shibayama for their excellent technical assistance. This work was supported in part by a grant-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (20590253) and by grants from the Japan Research Foundation for Clinical Pharmacology, the Kagawa University Project Research Fund 2009 (to Akira Nishiyama) and the Sanju alumni Research Grant (to Hirofumi Hitomi).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Liu, G., Hitomi, H., Hosomi, N. et al. Mechanical stretch potentiates angiotensin II-induced proliferation in spontaneously hypertensive rat vascular smooth muscle cells. Hypertens Res 33, 1250–1257 (2010). https://doi.org/10.1038/hr.2010.187
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2010.187
Keywords
This article is cited by
-
Biomechanical signal communication in vascular smooth muscle cells
Journal of Cell Communication and Signaling (2020)
-
Activation of the D4 dopamine receptor attenuates proliferation and migration of vascular smooth muscle cells through downregulation of AT1a receptor expression
Hypertension Research (2015)
