
Abstract
We describe an 8-year-old Japanese boy with a de novo recurrent missense mutation in CSNK2A1, c.593A>G, that is causative of Okur–Chung neurodevelopmental syndrome. He exhibited distinctive facial features, severe growth retardation with relative macrocephaly, and friendly, hyperactive behavior. His dysmorphic features might suggest a congenital histone modification defect syndrome, such as Kleefstra, Coffin–Siris, or Rubinstein–Taybi syndromes, which are indicative of functional interactions between the casein kinase II, alpha 1 gene and histone modification factors.
Similar content being viewed by others


Okur–Chung neurodevelopmental syndrome (OCNDS, OMIM #617062) is an autosomal-dominant disorder caused by heterozygous mutations in the casein kinase II, alpha-1 gene (CSNK2A1) located on chromosome 20p13. The clinical findings include delayed psychomotor development, intellectual disability (ID), speech delay, behavioral abnormalities, cortical malformations, and variable dysmorphic facial features. Thirteen cases of OCNDS have been reported in two large cohorts with undiagnosed developmental delay.1,2 A total of 12 de novo CSNK2A1 variants have been identified, including 11 missense and 1 splice site mutation. Nine missense variants reside in the glycine-rich ATP-binding loop or activation site of CSNK2A1. These regions are highly conserved among species and involved in the regulation and activation of casein kinase II. An additional novel de novo mutation in CSNK2A1 was reported in the active site of the protein, which is highly conserved.3
Herein, we describe a Japanese male with ID, motor and speech delay; severe growth retardation; behavioral problems; distinctive facial features; abnormal magnetic resonance imaging findings; and a CSNK2A1 mutation. The patient is a boy of 8 years and 4 months born at 40 weeks and 6 days of gestation. He was born by normal spontaneous vaginal delivery that was uneventful. He was conceived by in vitro fertilization to healthy, non-consanguineous Japanese parents. His birth weight, length, and head circumference were 2740 g, 47.5 cm, and 33.0 cm, respectively. All measurements were within normal limits. His developmental milestones were delayed. He gained head control at 4 months, reached for a toy at 9–10 months, and sat by himself at 12 months. He was referred to a pediatric neurologist at 1 year and 4 months due to considerable developmental delay. At this time, his height was 71.5 cm (–2.7 s.d.), his weight was 8.07 kg (–2.1 s.d.), and his head circumference was 45.5 cm (–1.0 s.d.). He also had hypotonia and decreased muscle bulk (Figure 1a, b). Magnetic resonance imaging revealed a reduced anterior pituitary gland and delayed myelination (Figure 1c, d). He was nonverbal but interacted with his family and friends.

The proband and cranial magnetic resonance imaging (MRI). The patient at 3 years and 7 months (a) and 7 years and 10 months of age (b), revealing synophrys, hypertrichosis, down-slanting palpebral fissures, and a bulbous nose. Axial T2-weighted view (c) and saggital T1-weighted view (d) of cranial MRI revealing a reduced anterior pituitary gland and delayed myelination.
He was referred to our clinic at 2 years 10 months for genetic disorder evaluation. His height was 81.8 cm (–3.0 s.d.), his weight was 10.3 kg (–2.1 s.d.), and his head circumference was relatively large at 48.0 cm (–0.8 s.d.). He was nonverbal and walked only one or two steps with support. He had distinct facial features, including synophrys, hypertrichosis, down-slanting palpebral fissures, and a bulbous nose. His behavior was very friendly. These findings were suggestive of Kleefstra syndrome, Coffin–Siris syndrome, or Rubinstein–Taybi syndrome. He walked alone at 3 years and started to speak meaningful words and two-word sentences at the age of 6 years and 1 month.
At 7 years and 4 months, he was socialized into school. Here his behavior was hyperactive. He spoke two-word phrases and could count from 1 to 20. He was evaluated as having a severe ID (Intelligent Quotient 21–35) on the Tanaka–Binet scale. At 8 years and 4 months of age, his height and weight were 109.5 cm (<–2 s.d.) and 17.0 kg (–2 s.d.), respectively. His speech improved significantly after he attended school. He could answer simple questions but was unable to express complex phrases appropriately. His chromosomal analysis was 46,XY, and cytogenetic microarray analysis using the SurePrint G3 8x60k Microarray Kit (Agilent Technologies, Santa Clara, CA, USA) revealed no pathogenic copy number variations. A skeletal survey also revealed no abnormal findings.
Peripheral blood samples were collected from the patient and his parents after obtaining written informed consent. The institutional review board of Kanagawa Children’s Medical Center approved this study. Genomic DNA was extracted from peripheral blood for trio-based whole-exome sequencing using standard protocols. Genomic DNA was captured using a SureSelect Human All Exon V5+UTRs Kit (Agilent Technologies), and exon-enriched genomic DNA libraries were sequenced on a HiSeq2500 system (Illumina, San Diego, CA, USA) using 101-bp paired-end reads. Mapping to the human reference genome (UCSC hg19, NCBI build 37.1) was performed using the Burrows–Wheeler Aligner (version 0.7.10) (http://bio-bwa.sourceforge.net/index.shtml). PCR duplications were removed using Picard (version 1.118) (http://broadinstitute.github.io/picard/). All variants were called using the Genome Analysis Toolkit (version 3.2-2) with the UnifiedGenotyper and HaplotypeCaller algorithms (https://software.broadinstitute.org/gatk/) and were annotated using ANNOVAR (22 March 2015) (http://annovar.openbioinformatics.org/en/latest/). From all variants within exons and ±10 bp of intronic regions from exon–intron boundaries, those registered in the NHLBI GO Exome Sequencing Project (ESP) 6500, the 1000 Genomes Project, dbSNP138, the Human Genetic Variation Database (HGVD), and our in-house Japanese exome data (78 individuals) and call variants from parents’ samples were removed. The candidate variant was confirmed by Sanger sequencing on an Applied Biosystems 3730xl DNA Analyzer (Life Technologies, Carlsbad, CA, USA). Sequence data were analyzed using the Applied Biosystem’s Variant Reporter software (Gene Codes Corporation, Ann Arbor, MI, USA).
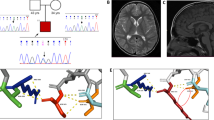
The mean read depth against RefSeq protein-coding regions was 53.31–75.44 reads with 72.8–98.1% being covered by ⩾20 reads. We referred to an online human genome mutation database (https://portal.biobase-international.com/cgi-bin/portal/login.cgi) as a reference for disease-causing mutations in all filtered variants and identified a putative CSNK2A1 mutation (NM_001895.3: c.593 A>G, p.K198R). De novo occurrences of this missense mutation were confirmed by Sanger sequencing (Figure 2a). This mutation has been previously reported as causative for OCNDS in two different cohort studies.1,2 The mutation was not registered in the NHLBI-ESP 6500, the 1000 Genomes Project, dbSNP138, the HGVD, or our in-house Japanese exome data. This mutation occurred within amino acids that are evolutionarily conserved among 10 different species (Figure 2b) and causes a substitution (p.K198R) that is localized in the activation segment of the protein (Figure 2c). Therefore, we confirmed the mutation to be pathogenic in this case.

CSNK2A1 mutation. (a) Electropherogram of the patient and his parents. (b) The mutation occurred at an amino acid residue that is evolutionarily conserved in nine different species. The altered nucleotide is highlighted in the gray box. (c) De novo mutation (c.593A>G, p.K198R) identified in the patient. The CSNK2A1 protein contains four domains: the ATP/GTP binding loop, basic cluster, active site, and activation segment.
Previous studies have demonstrated a wide range of dysmorphic features and behavioral problems, including microcephaly in some patients (Supplementary Table S1). We recognized three novel findings in our patient. First, he exhibited distinctive facial features, such as synophrys, hypertrichosis, down-slanting palpebral fissures, and a bulbous nose, which were suggestive of Kleefstra syndrome, Coffin–Siris syndrome, and Rubinstein–Taybi syndrome. Interestingly, one previously reported case of OCNDS with the same variant (Patient 2) exhibited similar features, especially mild synophrys.1 Second, our patient has behaved in a friendly and interactive manner since a young age. In this respect, his personality resembles that of Coffin–Siris syndrome and Rubinstein–Taybi syndrome patients. His hyperactive behavior has also become more noticeable over time, especially since starting school. Third, he presented with severe growth retardation with relative macrocephaly (Supplementary Figure S1), which was unique to our patient (Supplementary Table S1).
CSNK2A1 mutations were initially recognized in somatic cancers.4 CSNK2A1 is expressed in the brain and encodes the catalytic subunit of protein kinase casein kinase II, which plays an important role in cell proliferation and apoptosis.5 Although the biological mechanisms associated with ID are not fully understood, our patient’s phenotype, especially ID and synophrys, is similar to that of Kleefstra syndrome. Kleefstra syndrome can be caused by a haploinsufficiency of EHMT1, which encodes a histone methyltransferase that is capable of histone 3 lysine 9 dimethylation.6,7 Moreover, several studies have demonstrated that histone lysine methylation genes are associated with ID.8,9 These results indicate that facial dysmorphism is common in patients with the same variant and in congenital histone modification defect syndromes. This finding might suggest the possibility of a functional interaction between the casein kinase II, alpha 1 gene and histone modification factors.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Okur V, Cho MT, Henderson L, Retterer K, Schneider M, Sattler S et al. De novo mutations in CSNK2A1 are associated with neurodevelopmental abnormalities and dysmorphic features. Hum Genet 2016; 135: 699–705.
Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017; 542: 433–438.
Trinh J, Hüning I, Budler N, Hingst V, Lohmann K, Gillessen-Kaesbach G . A novel de novo mutation in CSNK2A1: reinforcing the link to neurodevelopmental abnormalities and dysmorphic features. J Hum Genet 2017; 62: 1005–1006.
Benveniste EN, Gray GK, McFarland BC. Protein kinase CK2 and dysregulated oncogenic inflammatory signaling pathways. In: Ahmed K, Issinger OG, Szyszka R (eds). Protein Kinase CK2 Cellular Function in Normal and Disease States, vol 12. Springer: Switzerland, 2015, e-book.
Ceglia I, Flajolet M, Rebholz H . Predominance of CK2alpha over CK2alpha′ in the mammalian brain. Mol Cell Biochem 2011; 356: 169–175.
Kleefstra T, Kramer JM, Neveling K, Willemsen MH, Koemans TS, Vissers LE et al. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am J Hum Genet 2012; 13: 73–82.
Stewart DR, Kleefstra T . The chromosome 9q subtelomere deletion syndrome. Am J Med Genet C Semin Med Genet 2007; 15: 383–392.
Kim JH, Lee JH, Lee IS, Lee SB, Cho KS . Histone lysine methylation and neurodevelopmental disorders. Int J Mol Sci 2017; 18: 1404.
Vallianatos CN, Iwase S . Disrupted intricacy of histone H3K4 methylation in neurodevelopmental disorders. Epigenomics 2015; 7: 503–519.
Data Citations
Kurosawa, Kenji HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.1917 (2018)
Acknowledgements
We thank the patient and his family for participating in this work. We also thank Dr. S Tsuji (Department of Neurology, Graduate School of Medicine, The University of Tokyo), Dr. S Morishita and Dr. J Yoshimura (Department of Computational Biology, Graduate School of Frontier Sciences, The University of Tokyo) for their advice; Dr. N Aida (Division of Radiology, Kanagawa Children’s Medical Center, Yokohama, Japan) for radiology evaluation; and Ms. K Ida and Ms. M Umegae for technical assistance. This work was supported by MEXT KAKENHI (No. 221S0002) (to KK), CREST, JST (to KK), the Naito Foundation (to YT), and the Yokohama Foundation for Advancement of Medical Science (to YT).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplemental Information for this article can be found on the Human Genome Variation website .
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Akahira-Azuma, M., Tsurusaki, Y., Enomoto, Y. et al. Refining the clinical phenotype of Okur–Chung neurodevelopmental syndrome. Hum Genome Var 5, 18011 (2018). https://doi.org/10.1038/hgv.2018.11
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2018.11
This article is cited by
-
Protein kinase CK2: a potential therapeutic target for diverse human diseases
Signal Transduction and Targeted Therapy (2021)
-
Okur-Chung neurodevelopmental syndrome-linked CK2α variants have reduced kinase activity
Human Genetics (2021)
-
Dual molecular diagnosis of tricho-rhino-phalangeal syndrome type I and Okur-Chung neurodevelopmental syndrome in one Chinese patient: a case report
BMC Medical Genetics (2020)
-
Overrepresentation of genetic variation in the AnkyrinG interactome is related to a range of neurodevelopmental disorders
European Journal of Human Genetics (2020)
-
Identification of de novo CSNK2A1 and CSNK2B variants in cases of global developmental delay with seizures
Journal of Human Genetics (2019)
