
Abstract
Lysine-specific demethylase 5C (KDM5C) has been identified as an important chromatin remodeling gene, contributing to X-linked neurodevelopmental disorders (NDDs). The KDM5C gene, located in the Xp22 chromosomal region, encodes the H3K4me3-me2 eraser involved in neuronal plasticity and dendritic growth. Here we report 30 individuals carrying 13 novel and one previously identified KDM5C variants. Our cohort includes the first reported case of somatic mosaicism in a male carrying a KDM5C nucleotide substitution, and a dual molecular finding in a female carrying a homozygous truncating FUCA1 alteration together with a de novo KDM5C variant. With the use of next generation sequencing strategies, we detected 1 frameshift, 1 stop codon, 2 splice-site and 10 missense variants, which pathogenic role was carefully investigated by a thorough bioinformatic analysis. The pattern of X-chromosome inactivation was found to have an impact on KDM5C phenotypic expression in females of our cohort. The affected individuals of our case series manifested a neurodevelopmental condition characterized by psychomotor delay, intellectual disability with speech disorders, and behavioral features with particular disturbed sleep pattern; other observed clinical manifestations were short stature, obesity and hypertrichosis. Collectively, these findings expand the current knowledge about the pathogenic mechanisms leading to dysfunction of this important chromatin remodeling gene and contribute to a refinement of the KDM5C phenotypic spectrum.
Similar content being viewed by others


Introduction
Alterations in lysine-specific 5C demethylase (KDM5C; MIM 314690) affect brain development and function [1]. To date, 59 KDM5C variants have been identified in males and females with KDM5C-related disorder (OMIM *300534, Claes-Jensen type syndrome, MRXSCJ), characterized by Intellectual Disability (ID), spasticity, epilepsy, short stature, and behavioral disorders [2,3,4]. Phenotypic variability has been reported in KDM5C patients [5]; female carriers are asymptomatic or mildly affected [3, 6,7,8,9,10]. The difference in phenotypic spectrum between males and females has been recently delineated [11] and attributed to the X-inactivation (XCI) skewing [9, 12]. However, since KDM5C seems to partially escape XCI, additional mechanisms have been suggested [13, 14]. KDM5C is likely a dosage-sensitive gene, contributing to sex differences in brain [13,14,15] that correlate with hypothalamic Growth hormone (GH) expression [16] and sex differences in adiposity [17].
KDM5C (Xp11.22), containing 26 exons, encodes a ubiquitous 1560-aa protein catalyzing the removal of tri- and dimethyl groups from lysine 4 of histone 3 (H3K4) [1]. Members of the KDM5-gene family function as transcriptional co-repressor or enhancer modulators and contain highly-conserved domains: ARID, Jumonji-N (JmjN), Jumonji-C (JmjC), C5HC2 zinc finger, and two or more PHD domains [18]. While frameshift, stop codon, or splicing variants producing truncated KDM5C protein are distributed along the entire sequence, reported missense variants cluster in the N-terminal of the protein [4]. The KDM5C N-terminus seems sufficient for its catalytic activity, suggesting that predominant molecular mechanism underlying MRXSCJ could be loss of demethylase activity [19]. Few MRXSCJ missense variants reported in regions of the KDM5C C-terminus, with unknown function [12, 20,21,22,23], seem to be more frequently associated with autism spectrum disorders (ASDs) [24]. Two MRXSCJ missense variants, lying in two different protein regions, preserve its demethylase activity [1, 12, 19]; KDM5C variants may therefore be pathogenic by compromising non-enzymatic domains.
We report the identification of new KDM5C variants in 30 individuals, among which 13 female carriers. Five of the thirteen novel variants lie outside of the N-terminal catalytic part of the protein. The pathogenic role and phenotypic impact of both sequence alterations and pattern of X-chromosome inactivation have been thoroughly evaluated.
Methods
Cohort description and genetic analysis
We report seventeen males and thirteen females, carrier of novel or rare KDM5C variants, from fourteen families. Six families have been diagnosed with MRXSCJ by targeted-sequencing in 800 individuals with NDDs referred to the University Hospital of Padova. Eight families were enrolled through collaborations with European groups working on NDDs (Supplemental Methods). Targeted-sequencing was performed as described in Aspromonte et al. [25]. DNA sequencing and data analysis were performed according to standard/in-house protocols of each participating center (Supplemental Methods) [26,27,28,29]. American College of Medical Genetics (ACMG) guidelines were followed for variant classification [30]. To automatically assign a final variant interpretation, we used Intervar (https://wintervar.wglab.org/) and Varsome (https://varsome.com/), implementing ACMG criteria using statistically justified thresholds tuned by a calibration process [31, 32]. User-supplied criteria were manually adjusted to refine the automated variant classification. Transcript analysis was performed for PL-3 and PD-2616 probands as described [25]. For female carriers, XCI pattern was evaluated on the human androgen receptor (AR) gene locus; if not informative, the SLIT and NTRK-like family member 4 (SLITRK4) locus was evaluated [33]. To investigate variant possible pathogenic role, we performed in depth in silico analysis of structural and functional impact (Supplemental Methods). All the reported variants have been submitted to the LOVD database (https://databases.lovd.nl/shared/genes/KDM5C). Individual UK#G56706 had been reported in DECIPHER database (Patient code: 264839) [34].
Results
Molecular data
We report 14 novel and rare KDM5C variants in 14 unrelated families (Table 1): ten amino acid changes (p.Asp111Val; p.Leu420Pro; p.Pro531Leu; p.Gln566Leu; p.Arg599Cys; Ser1183Phe; p.Cys1190Trp; p.Leu1265Pro; p.Ala1292Ser and p.Arg1483Gln), one nonsense variant p.(Arg951*), a nucleotide insertion causing a frameshift with premature stop codon 24 residues downstream of codon 51 p.(Asp51Glyfs*24), and two donor splice-site variants (c.963 + 2T>C and c.2243 + 1G>T) affecting the donor splice consensus of exon 7 and 15, respectively (Supplementary Fig. 1). Both exons are included in all KDM5C alternative isoforms expressed in brain. Except for p.(Arg599Cys), reported in a MRXCSJ familial case [11], the identified variants, absent from the gnomAD database (https://gnomad.broadinstitute.org) (Table 1), are novel. These resulted to be de novo in four cases, maternally inherited in four cases, and segregating with the disease in multiple affected members in four families (Tables 2, 3, Fig. 1). The variant p.(Ala1292Ser), female PD-4009, was absent in the proband’s asymptomatic mother and brother; the deceased father was reported as completely asymptomatic. In case PD-3827, the de novo p.(Asp111Val) variant was detected in a mosaic state (50%) by targeted NGS on DNA extracted from peripheral blood leukocytes (PBLs); the mosaicism was confirmed by Sanger sequencing in PBLs and oral mucosal cells (Supplementary Fig. 2). Segregation was not possible in the adopted male proband PD-3535. XCI pattern was evaluated in nine of the thirteen KDM5C female carriers. A slightly unbalanced or random pattern of XCI was detected in three cases, while a moderately or complete skewed XCI pattern was found in six females, five of which reported as symptomatic (Table 3).

The red arrow indicates the MRXSCJ patients described in this study. The black square/circle indicate symptomatic subjects with KDM5C variants. KDM5C female carriers are indicated as gray circles when mildly affected or white circles when reported as asymptomatic. The stripes in the symbols of individuals II:1 and II:4 of pedigree PD-2616 indicate different clinical conditions. Variants have been named based on the following reference sequences: NM_004187.5 and NP_004178.2 for KDM5C; NM_000147.5 and NP_000138.2 for FUCA1; NM_000828.5 and NP_000819.4 for GRIA3; NM_031407.7 and NP_113584.3 for HUWE1. NT not tested, “wt” wild-type, “v” variant.
KDM5C structural organization
To analyse the impact of identified variants we performed in silico structural characterization of the KDM5C protein. The structure of a deleted N-terminal catalytic region (residues 6-769 with residues 76-388 deleted), including ARID and PHD1 domains, was recently crystallized [35]. Even though the structure of the KDM5C C-terminus, is not known, the overall architecture of the highly similar KDM5B paralogs has been investigated [35]. KDM5B and highly similar KDM5 members result to have an elongated shape with dumbbell-like architecture, composed of a globular N-terminal catalytic core, a rigid elongated tower region, and a C-terminal flexible domain (FLD) [35]. The tower region, corresponding to the central PLU-1 domain, of all KDM5 family members, consists of three spectrin-like (SPECL1-3) repeats (Fig. 2, Supplementary Fig. 3), each forming a three-helix bundle giving rise to a linear domain. In KDM5B, the downstream C-terminal region, between SPECL3 and the terminal PHD3 domain, has four helical regions (H1-4), separated by PHD2, an intrinsically disordered loop (IDL), and a putative IDL (PIDL) [35]. KDM5C alignment against the KDM5B sequence confirms the presence of three helical regions (H1-3), separated by PHD2, and an intrinsically disordered loop (IDL), while the H4 region is not conserved (Fig. 2, Supplementary Fig. 3). Downstream of the H3 region, the corresponding PIDL region in KDM5B presents a charged residue cluster, conserved across KDM5 family members, and containing a putative nuclear localization signal (NLS) (Fig. 2, Supplementary Fig. 3).

Gene exons numbered from 1 to 26 (GeneBank reference sequence NM_004187.3) are colored and approximately aligned with the corresponding domains. Nucleotide changes predicted to result in truncating protein products are mapped in the KDM5C exonic sequence and the truncated endpoints are indicated. Different geometric symbols are assigned to the various domain types and indicated by the abbreviations: Pink Trapezium: N-terminal (JmjN; aa 13-81) and C-terminal (JmjC; aa 389-618) Jumonji domains; Light blue hexagon: ARID, AT-rich interaction domain (aa 82-179); Cyan rhombus: Zinc Finger-PHD type, PHD-1 (aa 324-374) and PHD-2 (aa 1185-1250); Green pentagon: Zinc-finger C5HC2 type domain (aa 619-768); Yellow ovals: Spectrin-like domains, SPECL-1 (769-862), -2 (871-978), -3 (981-1090); Orange spheres: predicted helical-rich regions: H1 (1132-1168); H2 (1255-1329); H3 (1398-1428); IDL: putative intrinsically disorder loop (aa 1330-1397); Red: Arginine-rich region (1449-1483). KDM5C missense variants described in this study are mapped to the protein sequence (in bold, blue font, top panels). Other reported missense variants that locate on the same domains are indicated (black font, bottom panels).
In silico analysis of the identified KDM5C variants
Among the identified variants, p.(Arg951*) and p.(Asp51Glyfs*24) are expected to result in loss of function. The two splice-site alterations, c.963 + 2T>C and c.2243 + 1G>T, are predicted to cause skipping of part or of the entire exons 7 and 15, respectively, resulting in frameshift deletions (Supplementary Fig. 1). If these altered transcripts escaped nonsense mediated mRNA decay (NMD), they would result in prematurely truncated proteins with loss of the catalytic segment (Fig. 2). Six of the missense variants are predicted to create new acceptor/donor splice sites or to alter splicing enhancer/silencer sites (Table 1). The sequencing of cDNA, performed only in PD-2616, did not reveal altered splicing (data not shown).
Five of the identified missense variants map in the KDM5C N-terminal catalytic domain: one in the ARID domain (p.Asp111Val) and four in the JmjC domain, p.(Leu420Pro), p.(Pro531Leu), p.(Gln566Leu), and p.(Arg599Cys) (Fig. 2). Five variants have been identified in the KDM5C C-terminal flexible domain FLD: one in the PHD2 domain (p.Cys1190Trp) and four (p.Ser1183Phe; p.Leu1265Pro; p.Ala1292Ser; p.Arg1483Gln) in regions of unknown function (Figs. 2, 3).

Cartoon representation of the N-terminal part of KDM5C model (residues 1-915) from AlphaFold with domains colored as indicated in Fig. 2. Since the C-terminal part contains long disordered regions that cannot be modeled and due to the low reliability of the helical domains arrangement, only the model of PHD2 domain has been shown in the picture. Positions of the six amino acid substitutions mapping in the JmjC, ARID, and PHD2 domain are shown as orange spheres in the models, and close-up views of their affected intramolecular bonding with surrounding residues are shown in zoom panels. For p.(D111V) in the ARID domain, the sequence alignment shows the conservation of this mutated position among KDM5 and other ARID-containing proteins.
The JmjC variants are predicted to disrupt intramolecular bonds in the JmjC core domain (p.Pro531Leu and p.Arg599Cys) or in surrounding α-helices (p.Leu420Pro and p.Gln566Leu) (Fig. 3). Interestingly, none of the variants impact residues that coordinate the cofactors (metal ion FeII and αKG) binding [36]. Of note, the Gln566 is located in a JmjC region (residues 554-569), highly conserved in all KDM5 family members, that seems to assume a dynamic conformation. In fact, this sequence lacks electron density in the KDM5C crystal structure (5FWJ); on the contrary, in KDM5B (5A1F) the corresponding region has a poorly defined electron density but shows a fast hydrogen/deuterium exchange, suggesting a disordered state [35]. Our in silico analysis suggests that the four JmjC variants may impact KDM5C demethylase activity by reducing the stability of the catalytic core or affecting its conformation dynamic.
We identified only one variant, p.(Asp111Val), in the ARID domain. Substitution of the negative charged Asp111 with the hydrophobic valine residue is predicted to disrupt intra-domain hydrogen bonds and alter the electrostatic surface, destabilizing ARID fold.
The c.332A>T nucleotide substitution is predicted to activate a cryptic exonic donor site with loss of 23 exon 3 nucleotides (Supplementary Fig. 1). This may result in the deletion of residues 111-117, part of a core ARID domain α-helix, causing its unfolding. The c.332A>T variant can anyway have an impact on the KDM5C-DNA binding mediated by the ARID domain.
Among KDM5C C-terminal variants, p.(Cys1190Trp) involves one cysteine interacting with the PHD2 domain Zn2+ atom (Fig. 3), which has a structural role in maintaining the domain stability; substitution of Cys1190 therefore potentially causes PHD2 unfolding (Fig. 3).
In the disordered region preceding the PHD2 domain (residues 1185-1250), we identified the missense variant p.(Ser1183Phe). This region, enriched with serine residues and conserved in KDM5D paralogs, contains a putative phosphorylation site (Supplementary Fig. 3). Other three C-terminal variants, p.(Leu1265Pro), p.(Ala1292Ser), and p.(Arg1483Gln), map downstream of PHD2 (Fig. 2).
The mutated positions Leu1265 and Ala1292 are conserved among other KDM5 members and involve two hydrophobic residues located in the helical H2 region (Fig. 2, Supplementary Fig. 3). Finally, the p.(Arg1483Gln) maps in the positively-charged region (residues 1448–1489) and alters a residue of the putative NLS (1479-PKRVRS-1484) suggesting a possible impact on the protein translocation to the nucleus (Fig. 2, Supplementary Fig. 3).
Phenotypic findings
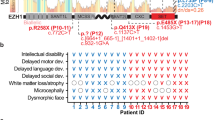
Thirty individuals from fourteen families, carrying novel or rare KDM5C variants were enrolled. For 28 (15 males and 13 females) detailed clinical information were available. Clinical features collected from male and female subjects are summarized in Tables 2, 3, respectively. A comparison of clinical features of males and females is summarized in Table 4. Detailed clinical descriptions for each family are reported in the Supplementary Material. Pedigrees and available pictures in Fig. 1.
Except for the grandfather transmitting the p.(Cys1190Trp) in family LA-MI, reported as affected by schizophrenia, all males carrying KDM5C variants (14/15) have a global developmental delay (DD) with severely compromised or absent language. The cognitive profile has been evaluated in 13 males; 62% (8/13) of them present a severe cognitive impairment, while four (4/13; 31%) have a mild ID (Tables 2, 4). Growth delay or short stature have been reported in seven individuals. Motor abnormalities, including fine and gross motor deficits, spasticity, altered fine motor coordination, dysarthria, and hypotonia have been reported in four males (4/15; 27%). Contractures of elbows and hands have been reported in two individuals from the same family (PL-1) (Supplementary Material). The presence of movement disorders, including hyperreflexia (3/15), ataxic gait (2/15), stereotypies (2/15), lingual dystonia (1/15), were reported in eight individuals (53%) from six families. Behavioral alterations were frequently observed among males (80%; 12/15), including ADHD (53%; 8/15), tantrums (40%; 6/15), autistic features (53%; 8/15), aggressive behavior (40%; 6/15), anxiety (40%; 6/15) and mood disorder (40%; 6/15). Six individuals presented a happy demeanor (40%; 6/15). Frequently reported sleep disturbances included disrupted sleep pattern, snoring, nocturnal awakening, reduced sleep duration (53%; 8/15) (Supplementary Material). In five cases the sleep disorder was associated with an abnormal EEG, or related to hypermotor seizures. A total of eight males (53%; 8/15) presented EEG abnormalities, including abnormal activity of the parietal lobes and multifocal epileptic anomalies. Eight cases (53%) developed epilepsy with multifocal, generalized, sleep-related seizures that where drug-resistant in two subjects (Supplementary Material). Brain MRI, performed in eleven males, detected abnormalities in four individuals (36%, 4/11). These included vascular lesions and gliosis in the white matter of the frontal lobes, hippocampal malrotation, T2 hyper-intensity in the periventricular white matter, dilated Virchow-Robin spaces and thin corpus callosum, a pineal dermoid cyst. Abnormalities of the skull observed in three males included microcephaly, brachycephaly, and dolichocephaly; relative macrocrania was described in three cases. A high forehead was observed in six males (40%; 6/15), while two presented a small forehead. Other dysmorphic features included deep-set eyes (5/15), long face (4/15), strabismus (3/15), thin upper lip (3/15), epicanthus (2/15), hypotelorism (2/15), sparse eyebrows (2/15). Six individuals (40%) presented teeth abnormalities: widely-spaced teeth, diastema and malocclusion. Gastrointestinal problems were reported in three cases. Finally, endocrine/metabolic anomalies, such as hypothyroidism, hypertrichosis, obesity, or overweight with abnormal response to growth hormone, were described in six males (40%) of our cohort.
We identified 13 females carrying KDM5C variants: 10 familial variants (5 simplex and 5 multiplex families), and 3 sporadic cases (Table 3), among which, two carry de novo variants. In the third sporadic case, the variant was found to be absent in the mother and the healthy brother but segregation analysis could not be performed in the father, deceased before the molecular characterization of his daughter. We speculated that the father, referred as completely asymptomatic, could not carry such variant. Among individuals with familial variants, five are asymptomatic (50%; 5/10) and four have mild ID (20%; 4/10). The mother transmitting the pathogenic p.(Arg951*) variant (family PD-3021), referred as not having learning disabilities, showed short stature and hypertrichosis. The three sporadic cases (PD-3597, PD-4009, PD-2616) presented psychomotor delay and an overall more severe phenotype, with moderate to severe ID. Autistic features were reported in PD-3597: mild behavioral disturbances, such as anxiety, mood swings and irritability, were described in PD-4009. Sleep disorders were reported only in PD-2616. Other MRXSCJ neurological features, such as hyperreflexia, spasticity, and epilepsy, have been observed only in sporadic cases (Table 3 and Supplementary Material).
Co-occurrence of KDM5C disorder and a second genetic finding
In three cases of our cohort (PD-2616, PD-4009, UK#G56706) additional likely pathogenic variants were detected in genes associated with phenotypes partially overlapping with the KDM5C-related disorder.
The female proband PD-2616, carrying the de novo KDM5C p.(Leu1265Pro) variant, had a dual diagnosis due to the concurrent presence of a homozygous alteration in the FUCA1 gene (NM_000147.5:c.564G>A; NP_000138.2:p.Trp118*), further complicating her clinical condition. Alterations of FUCA1 are responsible for fucosidosis (OMIM *230000), an autosomal recessive lysosomal storage disease that shares clinical features of MRXSCJ, such as ID, short stature, growth retardation and spastic paraplegia; all these features are present in our patient. The regression in language abilities seen in this individual, compatible with the neurodegeneration of fucosidosis type I, distinguishes this patient from the other reported MRXSCJ cases. Brain MRI anomalies, such as hypo-myelination and abnormal intensities of the globus pallidum and the medullary lamina, detected in this individual are the hallmarks of fucosidosis. In turn, this girl presented hyperreflexia, epilepsy, strabismus and hypertrichosis, typical of the KDM5C-related disorders. She also suffered from sleep disturbances, not observed in patients with fucosidosis, but frequently reported in our KDM5C cohort (Fig. 1, Table 3, Supplementary Material). The KDM5C variant of this patient is new, it is de novo, it insists on a functional domain which is a mutational hot spot (Fig. 2) and our bioinformatic analysis suggests a likely pathogenicity (Table 1).
The female proband PD-4009, in addition to the KDM5C p.(Ala1292Ser) variant, presented a new variant (NM_000828.5:c.573G>T; NP_000819.4:p.Arg191Ser) in the Glutamate Receptor Ionotropic Ampa 3 (GRIA3; OMIM 305915) gene. According to the ACMG-AMP guidelines the GRIA3 variant is classified as of uncertain significance (Intervar: PM1, PM2; Varsome; PP3, PM2). GRIA3 is associated with the Wu-type X-linked syndromic intellectual developmental disorder (MRXSW; OMIM 300699), characterized by moderate to severe ID, macrocephaly, epilepsy, autistic features, self-injury and aggressive behavior [37]. Further clinical features are short stature, asthenic body habitus, hyporeflexia, and sleep disorders [38]. Our case never manifested epileptic seizures, frequent in GRIA3 disorders [37]. In addition, she presents focal dystonia, increased mixed muscular tone (spasticity and rigidity), pyramidal signs and underdeveloped language, more consistent with a KDM5C-related disorder.
The male proband UK#G56706, in addition to the de novo KDM5C splicing variant c.963 + 2T>C, carried the maternally inherited variant NM_031407.7:c.8945G>A (NP_113584.3:p.Arg2982Gln) in the HUWE1 gene (Xp11.22, OMIM 300697). According to the ACMG-AMP guidelines the HUWE1 variant is classified as of uncertain significance (Intervar; PM1, PM2; Varsome: PM2, BP4). Alterations of the HUWE1 gene are associated with non-syndromic ID and Turner Type X-linked Syndromic Intellectual developmental disorder (MRXST; OMIM *309590). In addition, variants in HUWE1 have been found in an individual with ID, ASD, ADHD, sleep and gastrointestinal problems [39]. Carrier females are usually described as normal or less symptomatic. Our patient presents global DD, severe language impairment, seizures, and short stature; all features shared by MRXSCJ and MRXST. However, behavioral disturbances, including ASD, aggressivity, anxiety, ADHD, commonly reported in KDM5C-related disorders, were the major problems in this patient. A role of the HUWE1 variant in modifying the phenotype of this patient, although not certain, is not excludable.
Discussion
We present a clinical evaluation and molecular characterization of 30 unreported individuals carrying KDM5C variants, among which thirteen females. Our cohort includes the first case of somatic mosaicism in a male carrying a KDM5C nucleotide substitution, and a dual molecular finding in a female carrying a homozygous truncating FUCA1 alteration together with a de novo KDM5C variant. Five of the thirteen novel variants cluster in the uncharacterized KDM5C C-terminus, where, to date, few alterations have been reported. Our findings confirm reported aspects of KDM5C phenotypes such as obesity and hypertrichosis, which can be explained by the relationship between KDM5C and GH expression in the hypothalamus [16, 17]. In fact, pathological GH serum levels produce profound clinical effects on hair [40]. This mechanism might be responsible of the hypertrichosis observed in KDM5C patients.
Our work expands the repertoire of KDM5C mutations, contributes to refining a phenotypic spectrum that, lacking a typical gestalt, remains difficult to identify on solely clinical grounds, and points to the apparent relative frequency of KDM5C variants in NDDs.
Expanding the phenotypic spectrum of KDM5C-related disorder
In males, KDM5C pathogenic variants have been associated with moderate to severe ID, growth delay/short stature, seizures, happy demeanor, aggressive behavior and motor or movement disorders. These features are common in our male cohort: 62% presenting severe cognitive deficit, 60% motor or movement disorders, 53% epilepsy, and 47% growth delay (Table 4). Four individuals (31%) showed a mild ID. Behavioral and psychiatric disturbances have been found at a greater rate (73%) than previously reported (41%) [11]. One male, interestingly the first case of KDM5C somatic mosaicism, is mildly affected, showing good tolerance to frustration and good interaction skills. The mosaicism can justify the milder phenotype observed in our patient with respect to MRXSCJ males. We observed in our cohort a higher prevalence of ADHD (53%), ASD (40%), and sleep disorders (53%) than previous reports [4, 7, 24]. A higher frequency of ADHD and ASD has been highlighted by caregivers of KDM5C individuals [41]. While ADHD and ASD have been already reported, sleep disturbances, described in a few cases [5, 12, 42, 43], have not been explicitly associated with KDM5C-related disorder. The KDM5 protein family is involved in the circadian molecular machinery [42]. KDM5C acts a repressor of CLOCK-BMAL1-mediated activation of Per2, a core component of the circadian clock [42]. In the wild-type mouse brain, KDM5C alternative spliced isoforms follow a circadian oscillation pattern of expression [43]; the appropriate ratio among multiple KDM5C alternative spliced isoforms may be pivotal for the proper function of the circadian system [43]. Our findings suggest that sleep disturbances have been under-evaluated in previous clinical descriptions and may be a characteristic feature in KDM5C-related disorders.
Phenotype variability in females with KDM5C-related disorder
The series of 13 females we describe confirm the main findings of a recent review of the KDM5C female phenotype [11]. In our cohort 38% of female carriers are asymptomatic while 52% present DD; in general, the phenotype is milder in females than in males; 57% of the cognitively tested females presenting a mild ID. Nevertheless, we report three cases of females carrying KDM5C variants, two documented and one putative de novo, with severe phenotypes including characteristic MRXSCJ behavioral, neurological and endocrine features (Table 3). To date, only seven sporadic affected females reported in literature have been found to be more severely affected than female carriers with KDM5C family history [11, 44, 45]. The molecular mechanisms that explain the more severe phenotype in these females have not yet been completely elucidated. In our study, the XCI analysis performed in nine of the females carrying KDM5C variants, revealed an unbalanced or skewed pattern in six individuals (67%); five of these females are reported as symptomatic, in two cases severely affected. One of these females with skewed XCI, carried the de novo KDM5C variant, the p.(Arg599Cys), previously reported in a female with learning disability and growth delay [11]. Our patient presented a moderate ID and ASD. In X-linked disorders, the phenotypic variability in females may be attributed to the X-inactivation pattern, since a preferential activation of the wild-type allele can attenuate the pathogenic variant impact. On the other hand, the preferential activation of the mutated allele may explain the full phenotypic expression of an X-linked disorder in females. Despite the limited number of tested individuals, our findings support a definite role for XCI in the KDM5C phenotypic expression. Even though a partial XCI escape has been hypothesized [13, 14], more recent data suggest a possible XCI modulating effect in the KDM5C phenotype [9, 12, 45, 46]. On the other hand, it is always possible to hypothesize that phenotypic variability in monogenic disorders be, at least partially, due to additional modifying factors [12, 43]; this cannot be excluded in KDM5C individuals. Two of the severely affected females of our cohort presented additional variants in genes, FUCA1 and GRIA3, associated with phenotypes partially overlapping with the KDM5C-related disorder. The female carrying the KDM5C de novo p.(Leu1265Pro) variant had a dual diagnosis for the concurrent presence of a homozygous pathogenic FUCA1 variant, further complicating her clinical condition. Our findings further evidence that atypical clinical expression of monogenic conditions, like the KDM5C-related disorder, may be driven by additional genetic alterations in genes associated with partially overlapping phenotypes.
KDM5C variant clusters
We describe 13 novels and one previously reported KDM5C variants. Four are likely to result in a truncated protein or be subjected to NMD, causing a loss of protein function.
For newly identified missense variants further studies, such as demethylase activity assay or evaluation of their impact on DNA methylation profile, is warranted to finally define functional consequences [4, 47]. However, our in silico analysis allowed to predict possible protein effects and observed two distinct variant clusters along the KDM5C sequence. Five of the missense variants map in the N-terminal catalytic region, where most reported MRXSCJ variants cluster, and are predicted to compromise DNA binding mediated by ARID domain or JmiC enzymatic activity. The other five missense variants (p.Cys1190Trp, p.Ser1183Phe; p.Leu1265Pro; p.Ala1292Ser; p.Arg1483Gln)map in the KDM5C C-terminus which function is still largely unknown. Our in silico analysis allowed to better characterize the structural properties of the C-terminal KDM5C region; information was derived by aligning the KDM5 family members, and using annotation inferred from the recent characterization of KDM5B [35]. We found that the characteristic central PLU-1 region is composed of three spectrin-like repeats (SPECL1-3) acting as a rigid linker between the N-terminal catalytic core and a C-terminal flexible domain (FLD). The five C-terminal missense variants cluster in the FLD and are predicted to affect diverse structural/functional motifs of this region. The p.(Cys1190Trp) variant affects the PHD2 folding, p.(Ser1183Phe) alters a phosphorylation site, and p.(Arg1483Gln) could impact the protein nuclear localization. Two variants, p.(Leu1265Pro) and p.(Ala1292Ser), map in the helical region H2, downstream of the PHD2 domain, highlighting the importance of this structural region. To date, only a few missense variants have been reported in the C-terminal region [12, 20,21,22,23]. Functional characterization of p.(Arg1115His), mapping in the spectrin domain, showed that, although partially mis-localized in the cytoplasm, the protein maintains stability and retains the histone demethylase activity [12]. In addition, compared to the wild-type and other mutants causing a reduced enzymatic activity, the p.(Arg1115His) mutant affects the expression of a distinct set of genes [12]. These data suggest that C-terminal variants might affect distinct KDM5C regulatory functions utilizing enzymatic-independent molecular mechanisms [4, 12]. It has been suggested that the KDM5C C-terminus mediates the initial scanning of the nucleosome DNA, before demethylation occurs [35] implying that variants in the FLD domain may alter the recognition of KDM5C target genes. Although functional characterizations are needed to establish the pathogenicity of KDM5C C-terminal variants, our findings do contribute to strengthen the functional role of the flexible domain at the C-terminus of the protein.
KDM5C C-terminal variants and their associated phenotypes
It has been observed that KDM5C variations near the C-terminus are more frequently found in patients with autistic features [20, 23, 24]. Of our reported patients with C-terminal variants, two individuals of the same family, carrying the p.(Ser1183Phe), presented ASD in association with severe ID. However, we report five individuals with autistic features who carry alterations in the KDM5C N-terminal catalytic: p.(Arg599Cys) and p.(Gln566Leu) in the JmjC domain and the c.963 + 2T>C located in the Zn-finger domain. Furthermore, in one family segregating the p.(Cys1190Trp) located in the C-terminal PHD2 domain, one of the carrier males was reported as affected by schizophrenia. Several findings have indicated that ASD and schizophrenia are connected with impaired epigenetic regulation of gene transcription [48]. Future studies are needed to unravel the precise mechanisms connecting KDM5C with psychiatric disorders and to establish better the genotype-phenotype correlation in KDM5C C-terminal variants. The identification of novel disease-causing mechanisms implicated in KDM5C disorders will contribute to elucidate the neuronal epigenetic network; this is particularly important in view of the recent applications of strategies to reverse epigenetic modifications caused by KDM5C alterations with the prospect of specific therapeutic interventions [49, 50].
Conclusions
We report thirteen novel KDM5C variants, five of which clustering at the C-terminal non-catalytic part of the protein, supporting the existence of distinct KDM5C regulatory functions that utilize enzymatic-independent molecular mechanisms. We found sleep disorders to be a frequent feature of the KDM5C phenotype. As reported in other neurodevelopmental conditions, even in our cohort, KDM5C de novo variants tend to be associated with a more severe phenotype. Our data support the role played by XCI in modifying the KDM5C female phenotype. Future identification of additional genetic modifiers will contribute to a better understanding of the KDM5C phenotype, improving diagnosis and clinical care.
Web resources
GenBank, https://www.ncbi.nlm.nih.gov/genbank/, OMIM, https://www.omim.org/, UCSC, https://genome.ucsc.edu/, ENSEMBL, https://www.ensembl.org/index.html, HSF, https://hsf.genomnis.com/home, SIFT, (http://blocks.fhcrc.org/sift/SIFT.html), Polyphen, http://genetics.bwh.harvard.edu/pph2/, CADD (https://cadd-sv.bihealth.org, PDB, https://www.rcsb.org/, RING (http://protein.bio.unipd.it/ring), UniprotKB database (https://www.uniprot.org/, ClustalO (https://www.ebi.ac.uk/Tools/msa/clustalo/), Espript (https://espript.ibcp.fr), MobiDB (https://mobidb.org/, ELM resource (http://elm.eu.org/), VarSome (https://varsome.com/).
Data availability
All data generated or analysed during this study are included in this published article and its Supplementary Information files. Genetic variants reported in this study have been submitted to the “Global Variome shared LOVD” and they can be accessed using the url databases.lovd.nl/shared/genes/KDM5C. (Individual IDs: 405782, 405784, 405804, 405805, 405807, 408280, 408281, 408282, 408285, 408297, 408298, 408300, 408317, 408318, 408319, 408320).
References
Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, et al. The X-Linked Mental Retardation Gene SMCX/JARID1C Defines a Family of Histone H3 Lysine 4 Demethylases. Cell. 2007;128:1077–88.
Claes S, Devriendt K, Van Goethem G, Roelen L, Meireleire J, Raeymaekers P, et al. Novel syndromic form of X-linked complicated spastic paraplegia. Am J Med Genet. 2000;94:1–4.
Jensen LR, Amende M, Gurok U, Moser B, Gimmel V, Tzschach A, et al. Mutations in the JARID1C Gene, Which Is Involved in Transcriptional Regulation and Chromatin Remodeling, Cause X-Linked Mental Retardation. Am J Hum Genet. 2005;76:227–36.
Hatch HAM, Secombe J. Molecular and cellular events linking variants in the histone demethylase KDM5C to the intellectual disability disorder Claes-Jensen syndrome. FEBS J. 2021 https://doi.org/10.1111/febs.16204. Epub ahead of print.
Adegbola A, Gao H, Sommer S, Browning M. A novel mutation in JARID1C/SMCX in a patient with autism spectrum disorder (ASD). Am J Med Genet Part A. 2008;146A:505–11.
Tzschach A, Lenzner S, Moser B, Reinhardt R, Chelly J, Fryns JP, et al. Novel JARID1C/SMCX mutations in patients with X-linked mental retardation. Hum Mutat. 2006;27:389.
Abidi FE, Holloway L, Moore CA, Weaver DD, Simensen RJ, Stevenson RE, et al. Mutations in JARID1C are associated with X-linked mental retardation, short stature and hyperreflexia. J Med Genet. 2008;45:787–93.
Rujirabanjerd S, Nelson J, Tarpey PS, Hackett A, Edkins S, Raymond FL, et al. Identification and characterization of two novel JARID1C mutations: suggestion of an emerging genotype–phenotype correlation. Eur J Hum Genet. 2010;18:330–5.
Ounap K, Puusepp-Benazzouz H, Peters M, Vaher U, Rein R, Proos A, et al. A novel c.2T > C mutation of the KDM5C/JARID1C gene in one large family with X-linked intellectual disability. Eur J Med Genet. 2012;55:178–84.
Brookes E, Laurent B, Õunap K, Carroll R, Moeschler JB, Field M, et al. Mutations in the intellectual disability gene KDM5C reduce protein stability and demethylase activity. Hum Mol Genet. 2015;24:2861–72.
Carmignac V, Nambot S, Lehalle D, Callier P, Moortgat S, Benoit V, et al. Further delineation of the female phenotype with KDM5C disease causing variants: 19 new individuals and review of the literature. Clin Genet. 2020;98:43–55.
Vallianatos CN, Farrehi C, Friez MJ, Burmeister M, Keegan CE, Iwase S. Altered Gene-Regulatory Function of KDM5C by a Novel Mutation Associated With Autism and Intellectual Disability. Front Mol Neurosci. 2018;11:104.
Li N, Carrel L. Escape from X chromosome inactivation is an intrinsic property of the Jarid1c locus. Proc Natl Acad Sci USA. 2008;105:17055–60.
Xu J, Deng X, Disteche CM. Sex-Specific Expression of the X-Linked Histone Demethylase Gene Jarid1c in Brain. PLoS ONE. 2008;3:e2553.
Grafodatskaya D, Chung BH, Butcher DT, Turinsky AL, Goodman SJ, Choufani S, et al. Multilocus loss of DNA methylation in individuals with mutations in the histone H3 Lysine 4 Demethylase KDM5C. BMC Med Genomics. 2013;6:1.
Bonthuis PJ, Rissman EF. Neural growth hormone implicated in body weight sex differences. Endocrinology. 2013;154:3826–35.
Link JC, Wiese CB, Chen X, Avetisyan R, Ronquillo E, Ma F, et al. X chromosome dosage of histone demethylase KDM5C determines sex differences in adiposity. J Clin Investig. 2020;130:5688–702.
Wang Z, Liu D, Xu B, Tian R, Zuo Y. Modular arrangements of sequence motifs determine the functional diversity of KDM proteins. Brief Bioinform. 2021;22:bbaa215.
Tahiliani M, Mei P, Fang R, Leonor T, Rutenberg M, Shimizu F, et al. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature. 2007;447:601–5.
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–21.
Ahmed Alfares. A multicenter clinical exome study in unselected cohorts from a consanguineous population of Saudi Arabia demonstrated a high diagnostic yield | Elsevier Enhanced Reader. 2017. https://reader.elsevier.com/reader/sd/pii/S1096719217300276?token=A2A12F7BD123C04F06ACBE994EA1544D30DA155603B4A72FFD4E1FE9E982856995CC8667C923167AF41C8BDEC4306890&originRegion=eu-west-1&originCreation=20211021131846.
Faundes V, Newman WG, Bernardini L, Canham N, Clayton-Smith J, Dallapiccola B, et al. Histone Lysine Methylases and Demethylases in the Landscape of Human Developmental Disorders. Am J Hum Genet. 2018;102:175–87.
Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020;180:568–84.
Wu PM, Yu WH, Chiang CW, Wu CY, Chen JS, Tu YF. Novel Variations in the KDM5C Gene Causing X-Linked Intellectual Disability. Neurol Genet. 2021;8:e646.
Aspromonte MC, Bellini M, Gasparini A, Carraro M, Bettella E, Polli R, et al. Characterization of intellectual disability and autism comorbidity through gene panel sequencing. Hum Mutat. 2019;40:1346–63.
Hu H, Haas SA, Chelly J, Van Esch H, Raynaud M, de Brouwer APM, et al. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol Psychiatry. 2016;21:133–48.
Musacchia F, Ciolfi A, Mutarelli M, Bruselles A, Castello R, Pinelli M, et al. VarGenius executes cohort-level DNA-seq variant calling and annotation and allows to manage the resulting data through a PostgreSQL database. BMC Bioinform. 2018;19:477.
Piard J, Hu JH, Campeau PM, Rzońca S, Van Esch H, Vincent E, et al. FRMPD4 mutations cause X-linked intellectual disability and disrupt dendritic spine morphogenesis. Hum Mol Genet. 2018;27:589–600.
Pippucci T, Licchetta L, Baldassari S, Marconi C, De Luise M, Myers C, et al. Contribution of ultrarare variants in mTOR pathway genes to sporadic focal epilepsies. Ann Clin Transl Neurol. 2019;6:475–85.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med: Off J Am Coll Med Genet. 2015;17:405–24.
Li Q, Wang K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am J Hum Genet. 2017;100:267–80.
Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. VarSome: the human genomic variant search engine. Bioinformatics. 2019;35:1978–80.
Bertelsen B, Tümer Z, Ravn K. Three New Loci for Determining X Chromosome Inactivation Patterns. J Mol Diagn. 2011;13:537–40.
Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519:223–8.
Dorosz J, Kristensen LH, Aduri NG, Mirza O, Lousen R, Bucciarelli S, et al. Molecular architecture of the Jumonji C family histone demethylase KDM5B. Sci Rep. 2019;9:4019.
Horton JR, Engstrom A, Zoeller EL, Liu X, Shanks JR, Zhang X, et al. Characterization of a Linked Jumonji Domain of the KDM5/JARID1 Family of Histone H3 Lysine 4 Demethylases. J Biol Chem. 2016;291:2631–46.
Wu Y, Arai AC, Rumbaugh G, Srivastava AK, Turner G, Hayashi T, et al. Mutations in ionotropic AMPA receptor 3 alter channel properties and are associated with moderate cognitive impairment in humans. Proc Natl Acad Sci USA. 2007;104:18163–8.
Philips AK, Sirén A, Avela K, Somer M, Peippo M, Ahvenainen M, et al. X-exome sequencing in Finnish families with Intellectual Disability—four novel mutations and two novel syndromic phenotypes. Orphanet J Rare Dis. 2014;9:49.
Cerminara M, Spirito G, Pisciotta L, Squillario M, Servetti M, Divizia MT, et al. Case Report: Whole Exome Sequencing Revealed Disease-Causing Variants in Two Genes in a Patient With Autism Spectrum Disorder, Intellectual Disability, Hyperactivity, Sleep and Gastrointestinal Disturbances. Front Genet. 2021;12:625564.
Horesh EJ, Chéret J, Paus R. Growth Hormone and the Human Hair Follicle. Int J Mol Sci. 2021;22:13205.
Hatch HAM, O’Neil MH, Marion RW, Secombe J, Shulman LH. Caregiver-reported characteristics of children diagnosed with pathogenic variants in KDM5C. Am J Med Genet Part A. 2021;185:2951–8.
DiTacchio L, Le HD, Vollmers C, Hatori M, Witcher M, Secombe J, et al. Histone Lysine Demethylase JARID1a Activates CLOCK-BMAL1 and Influences the Circadian Clock. Science 2011;333:1881–5.
Talebizadeh Z, Shah A, DiTacchio L. The potential role of a retrotransposed gene and a long noncoding RNA in regulating an X-linked chromatin gene (KDM5C): Novel epigenetic mechanism in autism. Autism Res. 2019;12:1007–21.
McMichael G, Bainbridge MN, Haan E, Corbett M, Gardner A, Thompson S, et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol Psychiatry. 2015;20:176–82.
Fieremans N, Van Esch H, Holvoet M, Van Goethem G, Devriendt K, Rosello M, et al. Identification of Intellectual Disability Genes in Female Patients with a Skewed X-Inactivation Pattern. Hum Mutat. 2016;37:804–11.
Brand BA, Blesson AE, Smith-Hicks CL. The Impact of X-Chromosome Inactivation on Phenotypic Expression of X-Linked Neurodevelopmental Disorders. Brain Sci. 2021;11:904.
Coursimault J, Goldenberg A, Nicolas G, Saugier-Veber P, Coutant S, Vincent A, et al. Contribution of DNA methylation profiling to the reclassification of a variant of uncertain significance in the KDM5C gene. Eur J Med Genet. 2022;65:104556.
Vallianatos CN, Iwase S. Disrupted intricacy of histone H3K4 methylation in neurodevelopmental disorders. Epigenomics. 2015;7:503–19.
Poeta L, Padula A, Attianese B, Valentino M, Verrillo L, Filosa S, et al. Histone demethylase KDM5C is a SAHA-sensitive central hub at the crossroads of transcriptional axes involved in multiple neurodevelopmental disorders. Hum Mol Genet. 2019;28:4089–102.
Vallianatos CN, Raines B, Porter RS, Bonefas KM, Wu MC, Garay PM, et al. Mutually suppressive roles of KMT2A and KDM5C in behaviour, neuronal structure, and histone H3K4 methylation. Commun Biol. 2020;3:278.
Acknowledgements
We would like to thank all the families involved in this study for their generous participation. We are grateful to “SPECIALmente Noi Onlus Foundation” for promoting research in epilepsy and autism spectrum disorder (ASD). We also thank Francesco Benedicenti (Department of Pediatrics, Regional Hospital of Bolzano, Bolzano, Italy), Vincenzo Nigro (Department of Precision Medicine, University “Luigi Vanvitelli”, Naples, Italy; Telethon Institute of Genetics and Medicine TIGEM, Pozzuoli, Italy) and Tommaso Pippucci (University of Bologna, Bologna, Italy) for their generous collaboration in recruiting KDM5C cases. This study makes use of DECIPHER (http://www.deciphergenomics.org), which is funded by Wellcome. AM, AT, CC, SD and EK are members of the European Reference Network for Rare malformation syndromes, intellectual and other neurodevelopmental disorders (ERN-ITHACA; https://ern-ithaca.eu/); LL is member of the European Reference Network for Rare and Complex Epilepsies (ERN-EPICARE; https://epi-care.eu/).
Funding
This work was supported by Italian Ministry of Economic Development (Grant F/050011/02/X32 to MGM); Italian Ministry of Health Young Investigator (Grant GR-2011-02347754 to EL); Fondazione Pierfranco e Luisa Mariani (Grant CM22 to CC); Polish National Science Center (Grant 2012/07/B/NZ4/01764); Telethon Foundation, Telethon Undiagnosed Diseases Program (Grant GSP15001) and Maria Rosaria Maglione Onlus Foundation (Grant MRM#2020 to DD).
Author information
Authors and Affiliations
Contributions
AM, MGM, MCA and EL conceived, wrote, and reviewed the work; EL, MCA, SRN and MG performed experiments, analysed and interpreted the data; EL and MCA designed the computational framework and curated collected data; EB, RP (Roberta Polli), MCA, and EL carried out the NGS methodological set-up; DD, LV, MM, LL, RD, SD, CC, SE, VL, AT, DB, FL, GB, SP, FS, RP, E.K., MC, SRN, provided clinical data. All authors revised and approved the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
According to approved protocols of each referring clinical center, written informed consent was obtained from the probands or their legal representatives for specimen collection and genetic analysis. All individuals recruited provided informed consent for their participation in the study and publication of relevant findings.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Leonardi, E., Aspromonte, M.C., Drongitis, D. et al. Expanding the genetics and phenotypic spectrum of Lysine-specific demethylase 5C (KDM5C): a report of 13 novel variants. Eur J Hum Genet 31, 202–215 (2023). https://doi.org/10.1038/s41431-022-01233-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-022-01233-4
This article is cited by
-
Genetic architecture of childhood speech disorder: a review
Molecular Psychiatry (2024)
-
The value of exomes across the ages
European Journal of Human Genetics (2023)
