
Abstract
Purpose:
Genome-wide sequencing of cell-free (cf)DNA of pregnant women aims to detect fetal chromosomal imbalances. Because the largest fraction of cfDNA consists of maternal rather than fetal DNA fragments, maternally derived copy-number variants (CNVs) are also measured. Despite their potential clinical relevance, current analyses do not interpret maternal CNVs. Here, we explore the accuracy and clinical value of maternal CNV analysis.
Methods:
Noninvasive prenatal testing was performed by whole-genome shotgun sequencing on plasma samples. Following mapping of the sequencing reads, the landscape of maternal CNVs was charted for 9,882 women using SeqCBS analysis. Recurrent CNVs were validated retrospectively by comparing their incidence with published reports. Nonrecurrent CNVs were prospectively confirmed by array comparative genomic hybridization or fluorescent in situ hybridization analysis on maternal lymphocytes.
Results:
Consistent with population estimates, 10% nonrecurrent and 0.4% susceptibility CNVs for low-penetrant genomic disorders were identified. Five clinically actionable variants were reported to the pregnant women, including haploinsufficiency of RUNX1, a mosaicism for segmental chromosome 13 deletion, an unbalanced translocation, and two interstitial chromosome X deletions.
Conclusion:
Shotgun sequencing of cfDNA not only enables the detection of fetal aneuploidies but also reveals the presence of maternal CNVs. Some of those variants are clinically actionable or could potentially be harmful for the fetus. Interrogating the maternal CNV landscape can improve overall pregnancy management, and we propose reporting those variants if clinically relevant. The identification and reporting of such CNVs pose novel counseling dilemmas that warrant further discussions and development of societal guidelines.
Genet Med 19 3, 306–313.
Similar content being viewed by others

Introduction
In recent years, massively parallel shotgun sequencing of cell-free (cf) plasma DNA from pregnant women has become the method of choice for noninvasive prenatal testing (NIPT) of common fetal aneuploidies. The main methodologies in use are either targeted sequencing or genome-wide random massively parallel sequencing.1,2,3,4,5,6,7,8 Although the implementation of NIPT was highly valued because of its high accuracy for detection of fetal trisomy 21, 18, and 13, several studies have now demonstrated that genome-wide analysis also enables the detection of other fetal aneuploidies, segmental imbalances, and even submicroscopic copy-number variations (CNVs).9,10,11,12,13,14,15,16,17 Because genome-wide cfDNA profiling interrogates not only fetal DNA but also maternal DNA, maternal genetic variation can be detected as well. Such maternal CNVs can be either of constitutional origin or derived from acquired disorders in which apoptotic tumor cells shed their DNA in the maternal blood. The presence of those maternal CNVs has been shown to confound NIPT interpretation by implying the aberration to be of fetal origin. However, we and others have shown that by analyzing a genome-wide profile, both maternal tumor-derived as well as constitutional CNVs can be distinguished from fetal CNVs because of the distinct copy-number profiles.6,16,18,19,20,21 By analyzing sequencing reads, one can differentiate between the fetal and maternal origin of subchromosomal CNVs, which enables accurate maternal CNV detection. Here, we identified five maternal CNVs deemed of clinical relevance that were reported to the pregnant women and their physicians.
Materials and Methods
Noninvasive prenatal testing
Peripheral blood samples from 9,882 and 500 pregnant women from Leuven and Antwerp, respectively, were collected from 11 weeks of gestation onward. Blood sampling, cfDNA extraction and library preparation were performed as described.13 Massively parallel sequencing was performed on the HiSeq2500 or NextSeq500 sequencer (Illumina) in fast mode, producing 50-bp or 75-bp single end reads, respectively. The results are part of the routine clinical work-up and paid-for-service. The implementation of NIPT and the interpretation of both fetal and maternal incidental findings were approved by the institutional ethics review board and are consented for by both the referring physician and the pregnant women.
CNV analysis
An overview of the study design is depicted in Supplementary Figure S1 online. Routine diagnostic analysis of chromosomal Z, ZZ, bin median (BM), and other median (OM) scores in combination with a visual inspection of the genomic representation profiles was performed as described.13 In this way, clinically relevant maternal aberrations were identified. In addition, copy-number profiling of the Leuven cohort (n = 9,882) was performed by SeqCBS analysis using sequencing data compared to a reference pool of 100 normal samples.13 SeqCBS is a method based on nonhomogeneous Poisson processes with a change point model.22 To chart the landscape of nonrecurrent CNVs, copy-variable regions were blacklisted as described.13 Hereafter, the data were filtered by fold change (FC) and maximum number of CNVs per sample. Specifically, only deletions with FC <0.7 and duplications with FC >1.3 were accepted. Moreover, all samples with more than 20 CNVs across the entire genome were discarded from the analysis. This filtering methodology reduced the number of samples to 9,289 and the total number of CNVs to 76,853. Of those, 1,061 CNVs had a size exceeding 500 kb. For accurate detection of recurrent CNVs, i.e., risk factors for developmental disorders detected in the normal population as listed in Table 1 , copy-variable regions were not masked in the analysis.
Microarray and FISH
Following plasma separation, maternal blood samples were temporarily stored in case further analysis was needed. Genomic DNA from maternal lymphocytes was extracted using the QIAamp DNA Blood Mini QIAcube Kit on the Qiacube robotic workstation according to the manufacturer’s instructions. Array analysis was performed using either the 8 × 60K CytoSure ISCA v2 microarray (AMADID 020040, Oxford Gene Technology, OGT, Oxford, UK) for array comparative genomic hybridization (CGH) (cases 1 to 4) or the HumanCytoSNP-12 v2.1 DNA Analysis Kit (Illumina) for the single-nucleotide polymorphism (SNP) array (case 5). Visualization of results and data analysis were performed using the CytoSure Interpret Software (Oxford Gene Technology) and Cartagenia BENCH software (Cartagenia N.V., Leuven, Belgium) in the case of array CGH and GenomeStudio software (Illumina) and using CNV Webstore v2.023 in the case of SNP array. Fluorescent in situ hybridization (FISH) was performed according to standard procedures.
Results
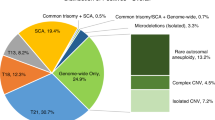
Using NIPT-derived genome-wide sequencing data of 9,289 pregnant women, we charted the incidence and landscape of rare CNVs present in the normal population by combining routine diagnostic NIPT analysis with SeqCBS analysis (Supplementary Figure S1 online).13,22 Two groups of CNVs can be distinguished.
First, recurrent CNVs mediated by nonallelic homologous recombination between flanking low copy repeats are, for the majority, proven risk factors for developmental disorders.24,25,26,27 In a retrospective validation, we compared the incidence of the most frequently detected recurrent CNVs (>500 kb) in a healthy control population with that in the Leuven cohort ( Table 1 ). Based on our data, the incidence of such CNVs is almost 0.4%, which is in line with published reports.27 Not surprisingly, these CNVs are low-penetrant and/or risk factors for late-onset disorders such as hereditary neuropathy with liability to pressure palsies and Charcot-Marie-Tooth.
Second, nonrecurrent CNVs exceeding 500 kb in size (n = 1,061), for which no disease association is currently known in the majority of cases, occurred in approximately 10% of the pregnant women. This incidence is also consistent with current population estimates.27 The specificity of SeqCBS-based CNV detection has been evaluated previously.22 As a proof of concept in the present study, and as part of a prospective validation, array CGH was performed on DNA extracted from maternal lymphocytes of 15 samples to confirm the presence of 19 suspected CNVs. In all instances, the maternal origin of the CNV was confirmed. No CNVs over 500 kb were missed by SeqCBS analysis, except for one deletion and one duplication that overlapped with a blacklisted region.
Because the aim of NIPT is to determine the genetic composition of the fetus and not of the supposedly normal pregnant woman, those incidentally detected maternal CNVs are, in principle, not reported. In addition, using the current NIPT analysis pipeline, carriership of the maternal CNV in the fetus cannot be determined. However, in the five cases described here, the detection of the maternal CNV was deemed clearly relevant for the mother, the fetus, or both.
Haploinsufficiency of RUNX1 (case 1)
NIPT was performed for a 41-year-old pregnant woman because of advanced maternal age and maternal anxiety. The genome-wide representation profile resulted in an overall chromosome 21 Z-score of −3.3, which is a value compatible with the presence of a fetal monosomy 21. However, the median value of all chromosome 21 subchromosomal bins, referred to as bin median (BM), was −0.2, a value well within the normal range, thereby flagging this potential monosomy as undetermined but excluding a fetal trisomy ( Figure 1a ). Visual inspection of the plot identified a localized segmental monosomy ( Figure 1a ), and SeqCBS analysis (FC = 0.53) suggested this aberration to be a maternal event. The size was estimated to be approximately 730 kb and the predicted karyotype was nipt[hg19] 21q12(36,378,448-37,110,033)×1. The presence of a maternal deletion 712 kb in size was confirmed by array CGH performed on DNA extracted from maternal lymphocytes (karyotype: arr[hg19] 21q12(36,381,575–37,094,277)×1; Figure 1b ). The deletion was shown to be intragenic in the RUNX1 gene.

Case 1: Maternal interstitial deletion on chromosome 21. (a) Noninvasive prenatal testing (NIPT) profiling of chromosome 21 showed an undetermined chromosome call based on combined analysis of Z, ZZ, bin median (BM), and other median (OM) scores. Dot plots of Z-score vs. ZZ-score (Y and X axis, respectively) are shown with undetermined zones (2.5–3) marked with grey lines. Z-scores of 5 Mb sliding windows are plotted across the chromosome to visualize the aberrations. Dotted lines represent ±1.5 Z-scores, and the areas above and below these cut-off values are green and red, respectively. Violin plots of BM and OM with their respective median indicated with a single dot represent the distribution of all the bin-level Z-scores. Further details regarding the NIPT data analysis pipeline are available elsewhere.13 (b) Array CGH performed on genomic DNA extracted from maternal lymphocytes confirmed the NIPT results and showed an intragenic deletion in RUNX1. The upper panel shows an overview of Log ratios of probes on chromosome 21. The deleted region on 21q22.12, with Log values of −1, is highlighted in the lower panel.
Haploinsufficiency for RUNX1 has been shown to cause autosomal-dominant familial platelet disorder with associated myeloid malignancy (FPDMM, OMIM 601399), which is characterized by qualitative and quantitative defects of blood platelets and a predisposition for the development of myeloid malignancies.28 Given the clinical relevance of this deletion and the direct consequences for delivery management (i.e., avoiding vacuum extraction), it was reported to the pregnant woman. She appeared to have been followed from childhood for an unknown disorder of thrombocytopenia and platelet dysfunction. However, invasive prenatal testing was not recommended for this indication. Postnatal follow-up of the newborn boy confirmed the inheritance of the RUNX1 deletion from his mother, and he presented a comparable platelet dysfunction with disturbed ATP secretion and thrombocyte aggregation. Given the increased risk for development of myeloid malignancies, clinical follow-up was recommended for both mother and son.
Maternal mosaic segmental deletion of chromosome 13 (case 2)
NIPT was indicated because of maternal age for a 43-year-old woman. The Z-score for chromosome 13 dropped to −17, suggesting the presence of fetal monosomy 13, which, owing to the normal BM, was overruled to an undetermined chromosome call excluding a fetal trisomy (Supplementary Figure S2a online). The latter, in combination with visual inspection of the genome-wide profile, suggested a segmental loss rather than monosomy 13 (Supplementary Figure S2a online). Whereas SeqCBS analysis did not indicate the presence of a maternal deletion in this region, array CGH on maternal lymphocytes demonstrated a partial deletion of approximately 35 Mb on chromosome 13 to be present in mosaic form (Supplementary Figure S2b online). Additional FISH analysis to determine the grade of mosaicism confirmed chromosome 13q deletion to be present in approximately 20% of maternal blood cells (data not shown). The molecular karyotype of the fetus was normal upon invasive testing (data not shown). The pregnant woman was informed through her treating obstetrician and genetic counseling is pending.
Maternal unbalanced translocation (case 3)
NIPT was performed at the personal request of a 19-year-old pregnant woman. For this male pregnancy, a very low Z-score of −11.6 for chromosome X was detected, suggesting a maternal monosomy X ( Figure 2a ). However, visual inspection of the segmental Z-scores along chromosome X suggested the presence of a terminal deletion of the long arm of chromosome X rather than monosomy X ( Figure 2a ). Further inspection of other chromosome-specific Z-score profiles revealed the presence of a terminal duplication of the long arm of chromosome 3, which was also indicated by SeqCBS analysis (FC = 1.51). Array CGH performed on DNA extracted from maternal lymphocytes showed both aberrations to be of maternal origin ( Figure 2b ). The presence of both a terminal duplication and a terminal deletion can be indicative of an unbalanced translocation. Metaphase FISH analysis and conventional karyotyping confirmed the presence of an unbalanced translocation t(Xq;3q) in the mother (data not shown). Follow-up invasive testing was performed to determine whether the fetus inherited these abnormalities because it concerned a male fetus, a large proportion (25.2 Mb) of the X chromosome was deleted, and the 1.37 Mb duplication on chromosome 3 overlapped in part with the 3q29 duplication syndrome region. Array CGH of DNA from amniotic fluid cells revealed no unbalanced translocation involving these chromosomes and no known pathogenic CNVs (data not shown). The pregnant woman was informed through her treating obstetrician and genetic counseling is pending.

Case 3: Maternal unbalanced translocation t(Xq;3q). (a) Noninvasive prenatal testing profile for chromosomes 3 and X indicative for a terminal duplication on the long arm of chromosome 3 and a terminal deletion on the long arm of the X chromosome. (b) Array CGH performed on DNA extracted from maternal lymphocytes confirmed these imbalances.
Maternal interstitial chromosome X deletions (cases 4 and 5)
In case 4, NIPT was performed at 12 weeks of gestation for advanced maternal age (35 years old). Whereas all parameters for chromosomes 21, 18, and 13 for this female fetus were within the normal range, a Z-score of −3.8 and ZZ-score of −6.6 on chromosome X were detected ( Figure 3a ). A normal BM of −0.7 suggested the presence of a subchromosomal abnormality rather than a full monosomy. Visual inspection of the genome-wide representation profile indicated an interstitial deletion on the long arm of chromosome X ( Figure 3a ). To confirm the probable maternal origin of this large deletion, array CGH on DNA extracted from maternal lymphocytes was performed and confirmed the presence of an 8.7 Mb maternal deletion on chromosome X with the following karyotype: arr[hg19] Xq23q25(113,852,760–122,559,857)×1 ( Figure 3b ). The deletion contained several genes, among which UBE2A, GRIA3, LAMP2, AGTR2, UPF3B, and CUL4B have been shown to be associated with several well-known X-linked intellectual disability (ID) syndromes.29,30 Given the clinical significance of the deletion, the family was seen for genetic counseling. Family history was negative with regard to ID. The carrier mother was asymptomatic, as expected, because carriers of an UBE2A mutation are clinically normal and have highly skewed X-inactivation patterns.31 Because the ultrasound at 15.5 weeks of gestation confirmed the female gender of the fetus, the parents opted not to proceed with an invasive test. They were informed about the occurrence of ID in a boy inheriting this deletion. The indication for carrier testing of other female relatives at risk was also discussed.

Case 4: Maternal interstitial deletion on Xq23q25. (a) Noninvasive prenatal testing genomic representation profile for chromosome X showing Z-scores and ZZ-scores in the monosomy range. However, a normal value for bin median and the visualization of the Z-cores across subchromosomal bins indicate the presence of an interstitial deletion on Xq23q25. (b) Array CGH on genomic DNA extracted from maternal lymphocytes shows Log ratios of −1 on Xq23q25, thus confirming the presence of a maternal deletion of 8.7 Mb on the long arm of chromosome X.
In the fifth case, NIPT was performed in a 37-year-old woman because of advanced maternal age. For this female fetus, the genome-wide representation profile showed a Z-score of −5.3 and a ZZ-score of −5.9 for chromosome X, indicative of monosomy X ( Figure 4a ). However, upon visual inspection, a segmental deletion on the long arm of chromosome X was observed. The presence of the deletion was verified by SNP array on DNA extracted from the maternal lymphocytes. SNP array results confirmed the presence of a deletion of 8 Mb with the following karyotype: arr[hg19] Xq21.1q21.31(81,180,501–89,178,028)×1 ( Figure 4b , c ). The deletion contains several genes causing disease upon loss of function, i.e., CHM, ZNF11, and POU3F4. Deletion of the entire coding region is consequently expected to have the same effect. Nonsense, frameshift, or splice-site mutations in CHM cause choroideremia (OMIM 303100), which is characterized by degeneration of the choriocapillaris, retinal pigment epithelium, progressive vision loss, choroidal sclerosis, choroidoretinal degeneration, night blindness, and atrophy around the optic disc. The disease is inherited in an X-linked fashion, with onset in the second to third decade of life. Female carriers are much more mildly affected, explaining why the mother at 37 years of age might not have developed any symptoms. Truncating mutations in ZNF11 cause a recessive, nonsyndromal form of intellectual disability (OMIM 300803).32 Loss-of-function mutations in POU3F4 cause a recessive form of sensorineural deafness with perilymphatic gusher (OMIM 304400). Mild hearing loss can occur in carrier females. Deletion of at least both CHM and POU3F4 has been described to cause chromosome Xq21 deletion syndrome or choroideremia, deafness, and mental retardation syndrome (OMIM 303110), combining clinical features described for the single genes.

Case 5: Maternal interstitial deletion on Xq21.1q21.31. (a) Noninvasive prenatal testing profile for chromosome X. The Z-scores and ZZ-scores are both below −3, which is the threshold to call a monosomy. However, the bin median of 0 points in the direction of a subchromosomal copy-number variant that is confirmed by the graphic representation of the Z-scores along the X chromosome. (b) SNP array profile of chromosome X of the mother. The SNP array profile, with B allele frequency on top and Log ratio below, confirms the presence of a maternal deletion of 8 Mb on the X chromosome: arr[hg19] Xq21.1q21.31(81,180,501–89,178,028)×1. (c) Detail of the SNP array profile of the Xq21.1q21.31 region in the mother.
After the mother was informed of the fetal trisomy risk, it was decided to also report this deletion to the mother, despite her carrying a female fetus, for the following reasons: (i) the mother might develop symptoms of choroiderema or hearing loss in the future; (ii) in another pregnancy with a male fetus, there is an indication for invasive prenatal diagnosis for this deletion; and (iii) postnatal microarray testing of the female fetus or of other female relatives is warranted in the case of a planned pregnancy. Invasive prenatal testing was not recommended because the symptoms for carrier females are generally mild. Postnatal follow-up of the newborn girl using SNP array confirmed the presence of the maternal deletion on chromosome X. The girl and her mother were referred to an outpatient otorhinolaryngologist and ophthalmologist.
Discussion
Genome-wide sequencing of cfDNA from plasma of pregnant women enables the identification of fetal aneuploidies. Even though detection of fetal trisomy 21, 18, and 13 can be performed with high sensitivity and specificity, false-positive and false-negative results still occur. Most discordant results between NIPT and fetal karyotype can be explained by biological events that may occur during gestation, such as placental mosaicism, true fetal mosaicism, and co-twin demise.10,33,34 Because no distinction is made between maternal or fetal sequences in shotgun sequencing, imbalances in the maternal genome usually skew the statistical parameters and scoring system, thereby impairing the accuracy of fetal trisomy detection. NIPT profiles have been shown to be confounded by circulating maternal tumor DNA or by maternal constitutional CNVs, which can result in false-positive results.6,16,18,19,20,21 For instance, age-related loss of the maternal X chromosome in blood cells has been shown to cause false-positive monosomy X results.10,12 Also, maternal mosaicism for chromosome segments can be a source of false-positive (case 2 in this article) or false-negative NIPT results.
CNV is one of the most prevalent forms of genetic variation among individuals, and it is known to significantly contribute to phenotypical heterogeneity.35 The majority of CNVs are considered benign polymorphic variations in healthy individuals. Here, we demonstrate that the analysis and interpretation of those maternal constitutional CNVs are warranted because some turn out to be clinically relevant incidental findings.36 Often, these incidental findings are looked at as negative aspects of genome-wide screening, similar to the search for secondary variants in exome sequencing.37 We consider these to provide an opportunity to obtain clinically important information, which can be used to promote the health of the fetus and/or the mother.
Different categories of incidental findings can be distinguished, as demonstrated by the examples presented in this article, as follows: (i) clinically actionable maternal aberrations and (ii) maternal chromosomal abnormalities that, if inherited, are potentially harmful for the fetus. In the first category, the RUNX1 deletion (case 1) can be classified as a congenital disorder that may remain undiagnosed, but with clinical utility of early diagnosis. This disorder is associated with thrombocytopenia and platelet dysfunction. Knowledge of this condition has direct consequences for delivery management. Moreover, the disorder has been associated with myeloid malignancies, and knowledge of the condition will influence follow-up of the patient and his mother.38 A second category includes chromosomal abnormalities that are asymptomatic in the mother but might be of clinical relevance in the fetus. Upon identification of maternal segmental imbalances by NIPT, mainstream sequencing and analysis methods are inaccurate for determining whether the fetus is a carrier as well and, as a consequence, often warrant reporting if clinically relevant. In the case of mosaic segmental chromosomal 13 deletion in the blood (case 2), chances are high that the imbalance originated during early embryonic development, with an increased chance for germline mosaicism.39 Hence, the fetus could have inherited this deletion. Such imbalances would either be embryonic lethal or cause severe developmental disorders. For X-autosomal balanced translocation carriers, it is likely that the derivative X chromosome is preferentially active due to the process of X-inactivation. However, in case 3, the mother carries an unbalanced X-autosomal translocation. Because she did not present any developmental anomalies, we presumed that the derivative X chromosome is preferentially inactivated. Because the male fetus has a 50% chance of inheriting the derivative chromosome X, resulting in a segmental nullisomy X, an invasive test was recommended. Nevertheless, for future pregnancies, the unbalanced translocation might affect not only male fetuses but also females. Therefore, even in case of a female pregnancy, the unbalanced translocation would have been reported to the patient. In cases 4 and 5, with an 8.7 Mb deletion and an 8 Mb deletion on the chromosome X, respectively, no invasive tests were recommended because the fetus was female. However, reporting the carriership to the mother could help explain (future) symptoms present in the mother and allows the opportunity for invasive testing in future (male) pregnancies. Moreover, testing carriership of female relatives is warranted for the same reasons.
Broadening the interpretation scope of NIPT to include maternal CNVs raises the question of which CNVs to report. In general, we adhered to the principles put forward in a Belgian consensus statement for invasive prenatal genome-wide screening.40 Nevertheless, it should be noted that these guidelines are based on invasive prenatal tests that interrogate only the fetal DNA. In contrast, NIPT profiles both fetal and maternal CNVs, and not all recommendations valid for fetal aberrations were or could be applied to maternal incidental findings. The latter are reported if they concern highly penetrant monogenic disorders and have clinical utility. Susceptibility CNVs are genetic risk factors with reduced penetrance and/or variable expression and are associated with phenotypes such as intellectual disability, autism spectrum disorder, epilepsy, and psychiatric disorders. The predictability of the future phenotype resulting from such susceptibility CNVs remains very poor. Although the degree of penetrance has been estimated for several CNVs, large confidence intervals exist.
The current consensus for invasive prenatal chromosomal array testing is to report only fetal CNVs for which the risk for a severe phenotype is sufficiently large and/or that are associated with structural malformations for which ultrasound follow-up is warranted. In the present study, a selection of recurrent imbalances frequently occurring in a healthy control population was identified in approximately 0.4% of pregnant women ( Table 1 ), which is in close agreement with the estimated cumulative incidence of these CNVs at birth.27 We decided not to report these maternal CNVs for two reasons. First, the mother reached reproductive age and thus has to be considered normal or at low risk, and she might or might not know about the presence of the CNV. If the mother knows of her carriership, then it would not change pregnancy management at all. If the mother is unaware, then reporting the CNV during pregnancy will probably induce unnecessary anxiety. Second, NIPT cannot (yet) determine whether the fetus is a carrier of the maternal CNV. Hence, reporting those CNVs has the risk of instigating unnecessary invasive testing. If the fetus is positive, then the mother would be faced with the dilemma of whether to continue her pregnancy for a variant she is carrying herself. However, it can be argued that reporting such incidental findings might, for some CNVs, trigger expert ultrasound follow-up.
In conclusion, identification of maternal CNVs enhances pregnancy management and can be considered an incidental benefit of genome-wide cfDNA profiling. Nevertheless, the identification and reporting of such CNVs pose novel counseling dilemmas that warrant further discussions and the development of societal guidelines.
Disclosure
J.R.V. reports being the founder of and stockholder in Cartagenia, a company that provides software for clinical analysis of genomics data. The analysis used in this study has been licensed to Cartagenia, for which J.R.V.’s laboratory receives license fees. The other authors declare no conflict of interest.
References
Liao GJ, Lun FM, Zheng YW, et al. Targeted massively parallel sequencing of maternal plasma DNA permits efficient and unbiased detection of fetal alleles. Clin Chem 2011;57:92–101.
Norton ME, Brar H, Weiss J, et al. Non-Invasive Chromosomal Evaluation (NICE) Study: results of a multicenter prospective cohort study for detection of fetal trisomy 21 and trisomy 18. Am J Obstet Gynecol 2012;207:137.e1–137.e8.
Bianchi DW, Platt LD, Goldberg JD, Abuhamad AZ, Sehnert AJ, Rava RP ; MatErnal BLood IS Source to Accurately diagnose fetal aneuploidy (MELISSA) Study Group. Genome-wide fetal aneuploidy detection by maternal plasma DNA sequencing. Obstet Gynecol 2012;119:890–901.
Palomaki GE, Kloza EM, Lambert-Messerlian GM, et al. DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation study. Genet Med 2011;13:913–920.
Ehrich M, Deciu C, Zwiefelhofer T, et al. Noninvasive detection of fetal trisomy 21 by sequencing of DNA in maternal blood: a study in a clinical setting. Am J Obstet Gynecol 2011;204:205.e1–205.11.
Zhang H, Gao Y, Jiang F, et al. Non-invasive prenatal testing for trisomies 21, 18 and 13: clinical experience from 146,958 pregnancies. Ultrasound Obstet Gynecol 2015;45:530–538.
Boon EM, Faas BH. Benefits and limitations of whole genome versus targeted approaches for noninvasive prenatal testing for fetal aneuploidies. Prenat Diagn 2013;33:563–568.
McCullough RM, Almasri EA, Guan X, et al. Non-invasive prenatal chromosomal aneuploidy testing–clinical experience: 100,000 clinical samples. PLoS One 2014;9:e109173.
Srinivasan A, Bianchi DW, Huang H, Sehnert AJ, Rava RP. Noninvasive detection of fetal subchromosome abnormalities via deep sequencing of maternal plasma. Am J Hum Genet 2013;92:167–176.
Lau TK, Jiang FM, Stevenson RJ, et al. Secondary findings from non-invasive prenatal testing for common fetal aneuploidies by whole genome sequencing as a clinical service. Prenat Diagn 2013;33:602–608.
Wang Y, Chen Y, Tian F, et al. Maternal mosaicism is a significant contributor to discordant sex chromosomal aneuploidies associated with noninvasive prenatal testing. Clin Chem 2014;60:251–259.
Wang S, Huang S, Ma L, et al. Maternal X chromosome copy number variations are associated with discordant fetal sex chromosome aneuploidies detected by noninvasive prenatal testing. Clin Chim Acta 2015;444:113–116.
Bayindir B, Dehaspe L, Brison N, et al. Noninvasive prenatal testing using a novel analysis pipeline to screen for all autosomal fetal aneuploidies improves pregnancy management. Eur J Hum Genet 2015;23:1286–1293.
Flowers N, Kelley J, Sigurjonsson S, Bruno DL, Pertile MD. Maternal mosaicism for a large segmental duplication of 18q as a secondary finding following non-invasive prenatal testing and implications for test accuracy. Prenat Diagn 2015;35:986–989.
Wapner RJ, Babiarz JE, Levy B, et al. Expanding the scope of noninvasive prenatal testing: detection of fetal microdeletion syndromes. Am J Obstet Gynecol 2015;212:332.e1–332.e9.
Brady P, Brison N, Van Den Bogaert K, et al. Clinical implementation of NIPT - technical and biological challenges. Clin Genet 2016;89:523–530.
Helgeson J, Wardrop J, Boomer T, et al. Clinical outcome of subchromosomal events detected by whole-genome noninvasive prenatal testing. Prenat Diagn 2015;35:999–1004.
Osborne CM, Hardisty E, Devers P, et al. Discordant noninvasive prenatal testing results in a patient subsequently diagnosed with metastatic disease. Prenat Diagn 2013;33:609–611.
Amant F, Verheecke M, Wlodarska I, et al. Presymptomatic identification of cancers in pregnant women during noninvasive prenatal testing. JAMA Oncol 2015;1:814–819.
Bianchi DW, Chudova D, Sehnert AJ, et al. Noninvasive prenatal testing and incidental detection of occult maternal malignancies. JAMA 2015;314:162–169.
Vandenberghe P, Wlodarska I, Tousseyn T, et al. Non-invasive detection of genomic imbalances in Hodgkin/Reed-Sternberg cells in early and advanced stage Hodgkin’s lymphoma by sequencing of circulating cell-free DNA: a technical proof-of-principle study. Lancet Haematol 2015;2:e55–e65.
Shen J, Zhang NR. Change-point model on nonhomogeneous poisson processes with application in copy number profiling by next-generation DNA sequencing. Ann Appl Stat 2012;6:476–496.
Vandeweyer G, Reyniers E, Wuyts W, Rooms L, Kooy RF. CNV-WebStore: online CNV analysis, storage and interpretation. BMC Bioinformatics 2011;12:4.
Sahoo T, Theisen A, Rosenfeld JA, et al. Copy number variants of schizophrenia susceptibility loci are associated with a spectrum of speech and developmental delays and behavior problems. Genet Med 2011;13:868–880.
Grayton HM, Fernandes C, Rujescu D, Collier DA. Copy number variations in neurodevelopmental disorders. Prog Neurobiol 2012;99:81–91.
Coe BP, Girirajan S, Eichler EE. The genetic variability and commonality of neurodevelopmental disease. Am J Med Genet C Semin Med Genet 2012;160C:118–129.
Coe BP, Witherspoon K, Rosenfeld JA, et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet 2014;46:1063–1071.
Béri-Dexheimer M, Latger-Cannard V, Philippe C, et al. Clinical phenotype of germline RUNX1 haploinsufficiency: from point mutations to large genomic deletions. Eur J Hum Genet 2008;16:1014–1018.
de Leeuw N, Bulk S, Green A, et al. UBE2A deficiency syndrome: Mild to severe intellectual disability accompanied by seizures, absent speech, urogenital, and skin anomalies in male patients. Am J Med Genet A 2010;152A:3084–3090.
D’souza RS, Levandowski C, Slavov D, et al. Danon disease: clinical features, evaluation, and management. Circ Heart Fail 2014;7:843–849.
Czeschik JC, Bauer P, Buiting K, et al. X-linked intellectual disability type Nascimento is a clinically distinct, probably underdiagnosed entity. Orphanet J Rare Dis 2013;8:146.
Tarpey PS, Smith R, Pleasance E, et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet 2009;41:535–543.
Benn P, Cuckle H, Pergament E. Non-invasive prenatal testing for aneuploidy: current status and future prospects. Ultrasound Obstet Gynecol 2013;42:15–33.
Curnow KJ, Wilkins-Haug L, Ryan A, et al. Detection of triploid, molar, and vanishing twin pregnancies by a single-nucleotide polymorphism-based noninvasive prenatal test. Am J Obstet Gynecol 2015;212:79.e1–79.e9.
Pinto D, Marshall C, Feuk L, Scherer SW. Copy-number variation in control population cohorts. Hum Mol Genet 2007;16 Spec No. 2:R168–R173.
Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST ; Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 2011;13:680–685.
Green RC, Berg JS, Grody WW, et al.; American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 2013;15:565–574.
Osato M, Yanagida M, Shigesada K, Ito Y. Point mutations of the RUNx1/AML1 gene in sporadic and familial myeloid leukemias. Int J Hematol 2001;74:245–251.
Conrad DF, Keebler JE, DePristo MA, et al.; 1000 Genomes Project. Variation in genome-wide mutation rates within and between human families. Nat Genet 2011;43:712–714.
Vanakker O, Vilain C, Janssens K, et al. Implementation of genomic arrays in prenatal diagnosis: the Belgian approach to meet the challenges. Eur J Med Genet 2014;57:151–156.
Acknowledgements
We sincerely thank Kristien Philippe and Sofie Verbeke for contributing to the genetic counseling of the patients. This work was made possible by grants from the University of Leuven (KU Leuven): GOA (GOA/12/015 to J.R.V., K.D., and H.V.E.) and SymBioSys (PFV/10/016 to J.R.V.).
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Information
(ZIP 347 kb)
Rights and permissions
About this article

Cite this article
Brison, N., Van Den Bogaert, K., Dehaspe, L. et al. Accuracy and clinical value of maternal incidental findings during noninvasive prenatal testing for fetal aneuploidies. Genet Med 19, 306–313 (2017). https://doi.org/10.1038/gim.2016.113
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2016.113
Keywords
This article is cited by
-
Population screening for 15q11-q13 duplications: corroboration of the difference in impact between maternally and paternally inherited alleles
European Journal of Human Genetics (2024)
-
A placental trisomy 2 detected by NIPT evolved in a fetal small Supernumerary Marker Chromosome (sSMC)
Molecular Cytogenetics (2021)
-
Eliciting women’s preference for prenatal testing in China: a discrete choice experiment
BMC Pregnancy and Childbirth (2020)
-
Non-invasive prenatal testing leading to a maternal diagnosis of Charcot–Marie–Tooth neuropathy
Journal of Human Genetics (2020)
-
Noninvasive prenatal testing: from aneuploidy to single genes
Human Genetics (2020)
