
Abstract
Chromatin assembly factor 1 (CAF1) consisting of p150, p60 and p48 is known to assemble histones onto newly synthesized DNA and thus maintain the chromatin structure. Here, we show that CAF1 expression was induced in human cytomegalovirus (HCMV)-infected cells, concomitantly with global chromatin decondensation. This apparent conflict was thought to result, in part, from CAF1 mislocalization to compartments of HCMV DNA synthesis through binding of its largest subunit p150 to viral immediate-early protein 2 (IE2). p150 interaction with p60 and IE2 facilitated HCMV DNA synthesis. The IE2Q548R mutation, previously reported to result in impaired HCMV growth with unknown mechanism, disrupted IE2/p150 and IE2/histones association in our study. Moreover, IE2 interaction with histones partly depends on p150, and the HCMV-induced chromatin decondensation was reduced in cells ectopically expressing the p150 mutant defective in IE2 binding. These results not only indicate that CAF1 was hijacked by IE2 to facilitate the replication of the HCMV genome, suggesting chromatin assembly plays an important role in herpesviral DNA synthesis, but also provide a model of the virus-induced chromatin instability through CAF1.
Similar content being viewed by others


Introduction
Chromatin assembly factor 1 (CAF1) consisting of p150, p60 and p48 assembles histones H3 and H4 onto newly synthesized DNA during replication and repair (1, 2 and references in 3, 4, 5). p150 utilizes its carboxyl-terminal third to bind p60 6, and knocking down either subunit disrupts the chromatin assembly activity, delays cell cycle progression and leads to cell death 7, 8, 9. p48, an Rb-interacting partner 10, is thought to escort various histone-metabolizing enzymes to their target sites 11. CAF1 physically associates with DNA replication forks in S phase 12, 13, 14. It facilitates DNA synthesis depending on the binding of p150's N-terminal 31 residues to the proliferating cell nuclear antigen (PCNA), which acts as a sliding clamp to stimulate the processivity of the DNA polymerase 12, 15, or the binding of p150 to the Werner syndrome protein in response to DNA damage 16.
CAF1 was initially identified as a complex in vitro to reconstitute the nucleosomal structure on the plasmid containing the DNA replication origin of simian virus 40 (SV40) 1, 17. Whether CAF1 plays a role in viral genome replication is the focus of the current study. The human cytomegalovirus (HCMV), a member of the herpesvirus family, is used as a model. HCMV contains a double-stranded DNA genome, which does not integrate into human chromosomes in infected cells. The genome with greater than 229 000 base pairs has a potential to encode more than 230 open reading frames 18. HCMV is an important human pathogen that maintains life-long latency in healthy individuals but causes serious diseases when reactivated in immunocompromised individuals, such as AIDS patients and organ transplant recipients 19. HCMV infection in permissive cells induces synthesis of S-phase proteins, such as cyclins, PCNA, thymidine kinases and so on. However, infected cells arrest in late G1 or early S, and cellular DNA replication ceases 20, 21, 22, 23. Thus, it is speculated that HCMV establishes an environment in which it hijacks the cellular DNA replication machinery to benefit its own genome replication.
HCMV DNA replication initiates from the replication origin oriLyt 24, 25, 26 and is thought to take place at sites near the nuclear domain 10 (ND10), which is disrupted as the replication progresses 27, 28. Subsequently, HCMV replication sites are enlarged to form globular replication compartments that eventually fill the nucleus at late stage 19, 29, 30. Viral proteins involved in HCMV DNA replication at least include UL44 (the DNA polymerase processivity factor), UL84 (likely a replication initiator interacting with the RNA/DNA hybrid within oriLyt) and the immediate-early 2 (IE2) protein 29, 31, 32, 33, 34. HCMV with IE2 gene deleted is not viable 35. IE2 is a well studied transactivator essential for the induction of a cascade of HCMV lytic genes 36, some of which directly participate in viral DNA replication. Alone or in combination with UL84 that binds to a conserved RNA stem-loop within oriLyt 31, 37, 38, IE2 stimulates the expression of several genes within oriLyt essential for oriLyt activation 39. In addition, Petrik et al. 40 found that a recombinant HCMV carrying IE2 gene with an amino acid substitution at residue 548 from glutamine to arginine (Q548R) replicates slowly and loses the ability to arrest cell cycle. Interestingly the mutation does not impair IE2's transactivation function 40, suggesting IE2 participates in HCMV viral DNA replication, in part, through a mechanism independent of transcriptional regulation.
Host proteins involved in HCMV DNA replication are not clearly understood. Recent studies show that, similar to herpes simplex virus type 1 (HSV-1), another member of the herpesvirus family, HCMV most likely assembles its genome with host histones into a dynamic nucleosomal structure in nucleus 41, 42, 43. Whether the host chromatin assembly activity plays a role in HCMV DNA replication is unanswered. Nitzsche et al. 43, 44 reported that PCNA, p48 of CAF1, and histone proteins, especially histone H2A, are colocalized with UL44, the viral DNA polymerase processivity factor, in HCMV-infected cells. However, it should be noted that the chromatin assembly activity is primarily determined by p150 and p60, but not p48 of CAF1. Thus, recruitment of p48 to the viral DNA replication center cannot ensure the involvement of chromatin assembly activity. In our previous yeast two-hybrid assay to screen for IE2-interacting proteins 45, one of positive clones identified using the C-terminal half (aa 291-579) of IE2 as bait is the p150 fragment containing amino acids 87 to 938 (full-length p150: 938 residues). Here, we show that CAF1 interacts with HCMV IE2 through p150 in HCMV-infected cells and by this interaction CAF1 is mislocated to viral DNA replication foci to facilitate HCMV genome replication. As a result, host chromatin was partially decondensed.
Results
CAF1 interaction with HCMV IE2
Our previous yeast two-hybrid screening identified that IE2 interacts with p150 (data not shown). The interaction of these two proteins in human cells was examined. The human lung adenocarcinoma cells H1299 expressing IE2 were subjected to immunoprecipitation and western blotting (IP-Western). As shown in Figure 1A, both p150 and p48 in IE2-transfected cells were present in the IE2 complex precipitated by an anti-IE antibody (IP: anti-IE, lane 2). As a negative control, IgG failed to pull down any of these two proteins (IP: IgG). These results demonstrate that transfected IE2 binds to CAF1 in cells.

HCMV IE2 interacts with CAF1. (A) IE2 interacts with the endogenous CAF1 in cells. H1299 cells were transfected with vector (lane 1) or the IE2-expressing plasmid (lane 2). Cell lysates were collected and subjected to IP with the mouse anti-IE antibody to pull down IE2-associated proteins. The precipitated proteins along with input controls were separated by SDS-PAGE and analyzed by western blotting with antibody against p150, p48 or IE2. IgG was used as a negative control for IP and the protein level of tubulin as the loading control for western blotting. (B) p150 colocalizes with IE2 in transfected cells. H1299 cells cultured on coverslips were cotransfected with HA-p150 and Flag-IE2 plasmids (a-d), GFP-p150 and Flag-IE2 plasmids (e-h), and GFP-p150 and Flag-IE1 plasmids (i-l). The localization of HA- (a) and Flag-tagged proteins (b, f and j) is visualized by immunostaining with the HA or Flag antibody. (m-p) U2OS cells cultured on coverslips were transfected with the IE2-expressing plasmid, followed by coimmunostaining with IE2 and p150 antibodies. Nucleus is visualized by 4′, 6-diamidino-2-phenylindole dihydrochloride (DAPI) incorporation (d, h, l and p). Images were obtained from Leica TCS NT confocal microscopy. All shown images represent the phenomenon occurring in more than 90% of the population.
Subsequently, we addressed the spatial relationship between IE2 and p150. Bromodeoxyuridine (BrdU), which can be incorporated into replicating DNA and marks DNA replication foci 9, 46, 47, was used to investigate whether IE2 and p150 colocalize with DNA replication foci in S-phase cells. The osteosarcoma cells U2OS expressing either HA-p150 (Supplementary information, Figure S1, a-f) or Flag-IE2 (g-l) were subjected to BrdU incorporation, followed by double immunostaining with the antibodies to BrdU (d-f and j-l) and the transfected protein (HA: a-c; Flag: g-i). As seen, HA-p150 in non-S cells (no BrdU uptake; a and d) was evenly distributed in the nucleus. Flag-IE2 in non-S cells showed a similar pattern (g and j). Consistent with the previous reports 9, 13, HA-p150 at early (b) and middle (c) S stage appeared as a pattern of loose dots and localized punctate dots, respectively, which resemble the BrdU staining (e and f). As expected, the IE2 pattern in S-phase cells was similar to that of p150 and BrdU (h and k, early S; i and l, mid S). Confocal microscopic analysis further confirmed the colocalization of IE2, but not IE1, and the transfected p150 in cells at middle S stage (Figure 1B, a to l). The IE2 staining also overlapped with that of the endogenous p150 (m-p). These results indicate that HCMV IE2 expressed in non-HCMV-infected cells associates with p150 at cellular DNA replication foci.
HCMV infection induces CAF1 expression, interaction and colocalization with IE2
The role of CAF1 during HCMV infection was investigated. First, we examined the expression levels of CAF1 subunits in infected cells. Human fibroblast cells MRC-5 were synchronized with serum starvation for 5 days, followed by infection of HCMV strain RC256 at a multiplicity of infection (MOI) of 1. After infection, cells were harvested at different time points for western analysis. The protein levels of p150 and p60 in HCMV-infected cells were clearly increased from 24 hour post infection (h.p.i.), reached to the highest level at 48 h.p.i. and dropped but remained induced at 72 h.p.i., Consistent with previous reports 21, 44, PCNA was upregulated at 12 h.p.i. (Figure 2A). Given that the cellular DNA replication slows down upon HCMV infection (see Introduction), the concomitant increase of p150, p60 and PCNA expression strongly suggests that the chromatin assembly activity is likely playing an important role in HCMV life cycle. We then analyzed the interaction between p150 and IE2 during HCMV infection by IP-Western. IE1, an alternatively spliced product of IE2 48, was used as a negative control. In agreement with our earlier studies in yeast and transfected cells (Figure 1 and data not shown), the anti-p150 antibody pulled down IE2, but not IE1, from lysates of HCMV-infected cells at 48 and 72 h.p.i. (Figure 2B, lanes 5 and 6). Consistently, only IE2 existed in the protein complex precipitated by anti-p60 or -p48 antibody (Figure 2C, lanes 3 and 4), indicating the association of IE2 with CAF1 in infected cells. Similar results were obtained from reciprocal IP experiments using anti-IE2 antibody (data not shown). Together these results demonstrate that CAF1 is induced and interacts with IE2 during HCMV infection.

CAF1 is upregulated and interacts with IE2 in HCMV-infected cells. (A) HCMV upregulates the expression of p150 and p60. Human fibroblast MRC-5 cells were serum-starved for 5 days and mock infected (lanes 1, 3, 5, 7, 9, 11 and 13) or infected with HCMV at an MOI of 1 (lanes 2, 4, 6, 8, 10, 12 and 14), followed by incubation with medium containing low serum (2%). At each indicated h.p.i., cell lysates were harvested and the proteins were separated by SDS-PAGE, followed by western blotting. The protein level of actin was used as the loading control. (B) IE2 interacts with p150 in HCMV-infected cells. Coimmunoprecipitation was performed with the lysates from HCMV-infected cells in (A). The anti-p150 antibody was used to precipitate p150-associated proteins, followed by western blotting using antibody against IE2 or IE1. IE1 was used as a negative control. (C) IE2 interacts with CAF1 complex in HCMV-infected cells. Experiments similar to (B) were performed. Anti-p150, -p60 and -p48 antibodies were used to pull down IE2 from lysates of cells at 48 h.p.i..
We further investigated the subnuclear localization of the endogenous CAF1 in HCMV-infected cells and whether it colocalizes with IE2 under this circumstance. Synchronized MRC-5 cells were infected, followed by double-immunostaining assays with specific antibodies to CAF1 subunit and IE2. The confocal microscopy analysis revealed that, in cells at 24 h.p.i., IE2 formed granule-like nuclear bodies in 36% of IE2-detectable cells (Figure 3A, b, yellow arrowhead). In this specific cell population, p150 aggregated into similar granules (a, yellow arrowhead). In contrast, p150 was punctated in non-infected cells (a, white arrowhead, IE2 negative). The merged image confirmed that the granule-like p150 colocalized with IE2 (c, yellow arrowhead). More significantly, in 92 % of infected cells at 48 h.p.i. (e-h), p150 remained associated with IE2, and the p150/IE2 granules became larger (yellow arrowhead). The phenomenon was even more striking in cells at 72 h.p.i. (i-l, yellow arrowhead).

CAF1 colocalizes with IE2 in specific nuclear compartments in HCMV-infected cells. MRC-5 cells were serum starved for 72 h and infected with HCMV at an MOI of 0.5, followed by incubation with serum-containing medium. At the indicated h.p.i., the localization of p150 and IE2 (A), and p48 and IE2 (B) was examined by double staining with corresponding antibodies. Images were obtained from Leica TCS NT confocal microscopy. White and yellow arrowheads indicate the uninfected and infected cells, respectively, based on the expression of HCMV IE2. The 4′, 6-diamidino-2-phenylindole dihydrochloride was used to stain the nucleus. All shown images represent the phenomenon occurring in more than 90% of the population. The exception has been indicated in main text.
Like p150, p48 also formed granule-like bodies and colocalized with IE2 after HCMV infection (Figure 3B). The cellular localization of p60 during HCMV infection was also analyzed. Since there was no appropriate antibody to stain the endogenous p60, U2OS cells expressing HA-p60 were infected with HCMV and doubly stained with the HA and IE2 antibodies. U2OS cells were used for their better transfection efficiency than fibroblasts and their permissiveness for HCMV infection 49. Consistently, HA-p60 aggregated and colocalized with IE2 at 48 h.p.i (Supplementary information, Figure S2A, yellow arrowhead). In contrast, in cells without IE2, HA-p60 displayed a uniform pattern of punctate staining throughout the nucleus (Supplementary information, Figure S2A, a, white arrowhead). Together these observations indicate that, in the nucleus of HCMV-infected cells, the CAF1 complex is mislocated to IE2 aggregates. Likewise, PCNA also displayed granule-like structures that overlapped with IE2 staining (Supplementary information, Figure S2B). Thus, HCMV infection induces expression of CAF1 and other important cellular DNA replication machinery components such as PCNA and their colocalization with IE2 in specific nuclear compartments.
CAF1, PCNA and IE2 colocalize at HCMV DNA replication centers
Whether the above aggregates containing CAF1, PCNA and IE2 in HCMV-infected cells are indeed sites for viral DNA synthesis remains to be determined. To this end, infected MRC-5 cells were doubly stained with antibodies to CAF1 subunit or PCNA and UL44. UL44 is an HCMV DNA polymerase processivity factor and thus commonly used as a marker for HCMV replication centers 34. As expected, both p150 and PCNA colocalized with UL44 (Figure 4A). The viral DNA replication centers were further verified by BrdU incorporation analysis. In mock-infected controls (Figure 4B, a and b) and HCMV non-infected cells (c-j, white arrowhead), the evenly distributed punctate staining of BrdU marked the cellular DNA replication foci. In sharp contrast, BrdU staining displayed a totally different pattern with localized granule structures in HCMV-positive cells (c-j, yellow arrowhead) and overlapped with that of IE2 (d). Further, we demonstrated that IE2-expressing granules were UL44 positive (e and f), and that p150 and p48 were also localized at these BrdU lump regions after HCMV infection (g-j).

CAF1 and PCNA colocalize with IE2 at HCMV viral DNA replication centers. (A) p150 and PCNA colocalize with UL44, the viral DNA polymerase processivity factor. MRC-5 cells were synchronized with serum-free medium for 72 h and infected with HCMV at an MOI of 0.5, followed by incubation with serum-containing medium. At 48 h.p.i., cells were doubly stained with antibodies to p150 and UL44 or PCNA and UL44. Images were obtained from Leica TCS NT confocal microscopy. (B) Aggregates formed by IE2, UL44, p150 and p48 during infection are BrdU-positive. MRC-5 cells were mock infected (a and b) or infected (c-j) with HCMV at an MOI of 0.5. At 48 h.p.i., cells were incubated with 20 μM BrdU for 2 h and doubly stained with antibodies to BrdU and IE2 (a-d), UL44 and IE2 (e and f), BrdU and p150 (g and h), and BrdU and p48 (i and j). Images were obtained from OLYMPUS IX71 fluorescence microscopy. White and yellow arrowheads indicate uninfected and infected cells, respectively. Similar observations were also obtained from HCMV-infected U2OS cells. (C) p48-and UL44-positive granules are disrupted by the viral DNA replication inhibitor PAA. MRC-5 cells were infected with HCMV at an MOI of 0.5 and cultured in the absence (a and b) or presence (c and d) of 200 μM PAA for 48 h, doubly stained for p48 (a and c) and UL44 (b and d). Images were obtained from OLYMPUS IX71 fluorescence microscopy. The exposure time for image collection of UL44 is 0.01 s (b) or 0.05 s (d). Yellow arrowheads indicate examples of infected cells. Similar observations were also obtained from HCMV-infected U2OS cells. All shown images represent the phenomenon occurring in more than 90% of the population. The exception has been indicated in main text.
Moreover, phosphonoacetic acid (PAA), a viral DNA polymerase inhibitor that blocks HCMV replication 50, totally disrupted the UL44- and p48-coexpressing aggregations (Figure 4C, c and d). Even though a longer exposure of the image (0.05 s compared with 0.01 s for control cells without PAA) was taken, UL44 granules were hardly observed in the presence of PAA (Figure 4C, c and d). p150 aggregations in HCMV-infected cells also disappeared after PAA treatment (Supplementary information, Figure S3). Similar results were obtained from HCMV-infected U2OS cells (data not shown). Thus far, these data clearly indicate that the cellular DNA replication machinery components such as CAF1 and PCNA are mislocated to centers of viral DNA synthesis during HCMV infection. The PAA experiment further implicates that viral DNA synthesis is prerequisite to aggregate CAF1 into the viral replication foci.
CAF1 promotes HCMV DNA replication
Whether CAF1 is indeed involved in HCMV DNA replication was examined. Human lung fibroblast HEL299 cells were transfected with si-scramble, siRNA against HTLV tax as a control or si-p150. Consistent with a previous report 7, the expression of p150 and p60 was simultaneously decreased when p150 was knocked down (Figure 5A, lane 3). These cells were then infected with HCMV at an MOI of 1, and the relative viral DNA copy number of cells 1 or 3 days post infection was measured using the quantitative real-time PCR with DNA primers recognizing a region within the UL44 promoter. As shown, HCMV DNA replication was greatly inhibited in cells with reduced p150/p60 expression (lane 3), indicating that the CAF1 complex is required for efficient HCMV genome synthesis. The finding was further supported by the following two observations. First, HCMV viral DNA replication was impaired in p150-constitutively knocked down MRC-5 (Supplementary information, Figure S4A) and U2OS (data not shown) cells. Second, HCMV genome synthesis was interfered in cells with p150C expression (Supplementary information, Figure S4B, lane 3). p150C, comprising the C-terminal aa 641-938 of p150, not only loses the chromatin assembly function but also dominant negatively inhibits the endogenous CAF1 activity 51. Same phenomenon was observed in two independent U373MG cell lines stably expressing HA-p150C (data not shown). Consistently, the viral DNA amount in HA-p150-stably expressing cells was increased both at day 3 post infection (p.i.) and day 5 p.i. compared with that in cells with vector only (Figure 5B). The enhancement of viral DNA synthesis by HA-150 overexpression was correlated with the increased total amount of p150 (Figure 5B, compare lanes 4-6 with 1-3). These results indicate that CAF1 is required, at least in part, for HCMV DNA replication.

CAF1 contributes to HCMV DNA replication. (A) HCMV DNA replication is decreased in human fibroblast HEL299 cells with reduced CAF1 expression. HEL299 cells transiently transfected with si-scramble, si-tax or si-p150 were infected with HCMV at an MOI of 1. Total DNAs were collected at the indicated day post infection (d.p.i.) for quantification by real-time PCR with DNA primers targeting the HCMV UL44 promoter. The DNA copy number obtained has been normalized to that of cellular GAPDH promoter. The protein level of p150 and p60 is shown below the bar chart. (B) HCMV viral DNA synthesis is increased in cells stably expressing p150. Experiments similar to (A) were performed, except that U373MG cells stably producing HA-p150 were used. The protein levels of HA-p150 and total p150 at each indicated d.p.i. are shown below the bar chart. Similar results were obtained from two independent experiments.
CAF1 facilitates HCMV DNA replication depending on p150 interaction with IE2
Given that p150 binds to IE2 and IE2 was previously shown to interact with members of the HCMV DNA replication machinery 52, we investigated if IE2 directs CAF1 to viral replication foci through interacting with p150. First, we characterized the IE2-interacting domain within p150 and asked if the p150 mutant without this specific region fails to stimulate HCMV DNA synthesis. A series of p150 deletion mutants were in vitro translated and incubated with GST or GST fused to the full-length IE2. As expected, the full-length p150 (1-938) bound to GST-IE2, but not GST alone (Figure 6A). Deletion of the PEST domain (aa 245-296) or one of the two highly charged domains KER (lysine, glutamic acid and arginine-rich, aa 311-445) and ED (clusters of glutamic acid and aspartic acid, aa 564-641) did not affect the interaction between p150 and IE2. Finally, all p150 fragments without aa 506-563 failed to be precipitated by GST-IE2 and, in contrast, fragments containing these residues associated with IE2. These include a fragment with only aa 445–619. We further analyzed if aa 506-563 mediates IE2 binding. The fragment containing aa 445-505 was used as a control. Since these two fragments are too small to be visualized, they were fused with the green fluorescent protein and two nuclear localization sequences from SV40 large T antigen (GFP/2LTNLS). As expected, GFP/2LTNLS alone (GFP) or GFP/2LTNLS fused to p150 (445-505) (GFP/445-505) failed to associate with IE2, whereas GFP/2LTNLS-p150 (505-564) (GFP/505-564) was pulled down by GST-IE2 (Figure 6A). Residues 505-564 were likely the only domain to interact with IE2, as p150 mutants lacking aa 506-563 lost the ability to interact with IE2 (Δ506-563 and Δ446-563). In contrast, deletion of regions outside this potential IE2-interacting domain did not impair IE2/p150 interaction (Δ446-504, Δ297-310 and Δ642-729). Together, we conclude that aa 506-563 constitute a single domain of p150 to bind IE2.

p150 defective in IE2 binding fails to localize to viral DNA replication foci to enhance viral DNA synthesis. (A) Residues 506-563 of p150 are critical for IE2 binding. GST or GST-IE2 on beads was used to pull down various in vitro translated 35S-labeled p150 fragments. Left panel shows the schematic diagram of p150 deletion mutants. The GFP sequence containing two nuclear localization sequences form SV40 large T protein (GFP/2LTNLS) was fused with aa 445-505 (GFP/445-505) or aa 505-564 (GFP/505-564). The results from autoradiography are presented at the right panel. (B) p150 defective in IE2 binding fails to colocalize with UL44. U373MG cells were transiently transfected with the plasmid encoding the p150 mutant missing aa 446-504 or aa 506-563, followed by HCMV infection at an MOI of 0.5. At 48 h.p.i., cells were doubly stained with antibodies against p150 and UL44 (left panel). Note that p150 antibody only recognizes the overexpressed p150 in this case. Cells with p150 and UL44 colocalizing together (white bar), distributing separately (black bar) or with the above two phenomena happening simultaneously (gray bar) were counted and plotted (right panel). (C) p150 defective in IE2 binding fails to enhance viral DNA synthesis. U373MG cells stably expressing the p150 mutant without aa 446-504 (lane 2) or aa 506-563 (lane 3) were infected with HCMV at an MOI of 1. Total DNAs were collected and quantified by real-time PCR as described in Figure 5A. The protein levels of p150 derivatives determined by western blotting are shown below the bar chart.
Next, we used competition assays to demonstrate that IE2 indeed interacts with the endogenous p150 at aa 506–563. IE2 was expressed in H1299 cells in the presence of GFP, GFP/445-505 or GFP/505–564. The IE2 complex was immunoprecipitated with IE2 antibody, followed by western analysis using the antibody against IE2, GFP, or the endogenous p150. As seen in Supplementary information, Figure S5, the IE2 antibody pulled down both IE2 and the endogenous p150 in the presence of GFP (lane 1). IE2/p150 interaction remained unchanged when GFP/445-505 was added (lane 2) but was reduced by expressing GFP/505-564 (lane 3), likely due to direct interaction of GFP/505-564 and IE2 as GFP/505-564, but not GFP/445-505, was present in the IE2-pulled-down complex (lane 3). Thus, GFP/505-564 successfully competes with the endogenous p150 for IE2 binding.
Whether the p150 mutant defective in IE2 interaction is also unable to localize to HCMV DNA replication foci was analyzed. U373MG cells transiently expressing p150 mutants were infected with HCMV, followed by double-immunostaining assays with antibodies to UL44 and p150. As a control, 75.8% of p150 (Δ446-504) aggregated at viral DNA replication centers positively stained with UL44 (Figure 6B, left panel, a and b, and right panel, Δ446-504, white bar), while 16.1% did not (right panel, Δ446-504, black bar). Around 8% showed both phenomena simultaneously (right panel, Δ446-504, gray bar). In sharp contrast, less than 10 % of p150 (Δ506-563) colocalized with UL44 (right panel, Δ506-563, white bar). The majority of the population (83.3%) displayed distinct staining from UL44 (left panel, c and d, and right panel, Δ506-563, black bar) and around 11% showed both patterns together (right panel, Δ506-563, gray bar). These results indicate that the IE2 interaction domain within p150 is critical to localize p150 to HCMV DNA replication foci.
We then determined whether p150 (Δ506-563) fails to facilitate viral DNA replication. U373MG cells stably expressing p150 (Δ446-504) or p150 (Δ506-563), tagged with a C-terminal V5, were infected with HCMV and directly quantified for the viral DNA copy number. As shown in Figure 6C, only p150 (Δ446-504), but not p150 (Δ506-563), stimulated viral DNA replication, strongly indicating that CAF1 requires IE2/p150 interaction to facilitate viral DNA synthesis. One might argue that p150 (Δ506-563) might have dramatic structure defects and lost its physiological function, therefore it fails to increase HCMV DNA replication. This was unlikely the case, as p150 (Δ506-563) still formed a complex with p60 and colocalized with p60 at cellular DNA replication foci (Supplementary information, Figure S6). These results indicate that the failure of p150 (Δ506-563) to increase HCMV viral DNA synthesis is mainly due to its inability to interact with IE2.
Finally, to address whether the chromatin assembly activity of CAF1 is required for p150 to facilitate HCMV DNA replication, HCMV-infected U373MG cells stably expressing the p150 fragment (aa 1-641), which only includes the IE2-binding motif but is unable to assemble with p60 (Supplementary information, Figure S6A, lane 3), or p150 fragment (aa 505-938), which contains the binding domains for IE2 and p60, were directly analyzed for the viral DNA copy number. As seen in Supplementary information, Figure S7, p150 (aa 505-938) enhanced the viral DNA synthesis (Supplementary information, Figure S4C, lane 3), while p150 (aa 1-641) failed to do so (lane 2). Consistently, in HEL299 cells with HCMV infection at a low MOI (0.01), the efficiency of viral DNA replication was also increased in the presence of p150 (Δ446-504) and p150 (aa 505-938), but not p150 (Δ506-563) (data not shown). Taken together, these results indicate that both IE2 interaction and chromatin assembly activity are required for CAF1 to facilitate HCMV genome replication.
IE2Q548R is defective in p150 interaction
To understand if any mutation within IE2 sequence that disrupts p150 binding indeed decreases HCMV viral DNA replication, the p150 interaction domain within IE2 was mapped. The in vitro translated p150 was incubated with GST fused to IE1 or IE2 derivative, followed by GST pull-down assays. As shown in Figure 7A, both the GST-fused N-terminal fragments of IE2, aa 1-98 and 1-290, failed to precipitate p150. GST-IE1 did not pull down p150 either. Thus, the p150 interaction domain was unlikely located in the N-terminal half of IE2. In contrast, all IE2 fragments containing the most C-terminal residues 491-579 pulled down p150. Together we conclude that the C terminus of IE2 is essential for p150 binding. Supplementary information, Figure S8 shows SDS gels demonstrating the purity of all Escherichia coli-expressed proteins used here. Consistently, in IP-western assays, the endogenous p150 failed to be coprecipitated with the IE2 fragment without aa 490 to 579 (data not shown).

The Q548R mutation within IE2 disrupts IE2 binding to p150. (A) The C-terminal 89 residues of IE2 bind p150. In vitro GST pull-down experiments similar to Figure 6A were performed, except that the in vitro translated 35S-labeled p150 was incubated with indicated GST-IE derivatives on beads. A schematic diagram of IE2 domain structure and deletion mutants used is shown below the autoradiography. Zn, Zn finger domain; HLH, helix-loop-helix motif. Right column indicates the binding ability of each GST-IE derivative to p150. (B) IE2Q548R has impaired binding to ectopically expressed HA-p150. Coimmunoprecipitation was performed with H1299 cells transiently coexpressing HA-p150 and Flag-tagged wild-type IE2 or IE2 with the glutamine at amino-acid position 548 mutated to an arginine (IE2Q548R). Anti-HA antibody was used to precipitate HA-p150 and its associated IE2. Tubulin was used as a loading control. (C) IE2Q548R and IE1 do not bind the endogenous p150. H1299 cells were transfected with plasmid-encoding Flag-tagged wild-type IE2, IE2Q548R or IE1, followed by IP with anti-IE antibody and western blotting to analyze the pulled-down proteins. Actin was used as a loading control for input and a negative control for IP. (D) IE2Q548R does not colocalize with p150 in cells. H1299 cells cultured on coverslips were transfected with Flag-tagged wild-type IE2 (a-c) or IE2Q548R (d-f), followed by coimmunostaining with the anti-p150 antibody (a and d) and the anti-IE antibody (b and e). The fluorescence images were obtained from OLYMUS IX71 fluorescence microscopy. All shown images represent the phenomenon occurring in more than 90% of the population.
As mentioned in Introduction, the recombinant HCMV with Q548R mutation in IE2 sequence replicates much slower than wild-type HCMV. Since Q548 happens to locate within the p150-binding domain (aa 490-579) identified above, we investigated whether this IE2 mutation disrupts IE2 interaction with p150 and thus leads to impaired HCMV viral DNA synthesis. This was proved to be likely the case as the binding of Flag-IE2Q548R to HA-p150 was obviously less efficient compared with wild-type IE2 (Figure 7B, compare lane 2 with 1). The endogenous p150 was only coprecipitated with Flag-IE2, but not Flag-IE2Q548R or Flag-IE1 (Figure 7C). As a negative control, actin was not present in the complex pulled down by the anti-IE antibody (Figure 7C). Moreover, Flag-IE2Q548R failed to colocalize with the endogenous p150 in S-phase cells (Figure 7D, d-f). These results indicate that the previously reported recombinant HCMV with the Q548R substitution within the IE2 sequence likely generates an IE2 mutant defective in CAF1 recruitment and thus shows impaired viral DNA replication and growth.
CAF1 couples histones to IE2
Since CAF1 is a histone chaperone for depositing H3/H4 onto replicating DNA 1, 2, 4, we next investigated whether CAF1 likely facilitates HCMV viral DNA synthesis by coupling histones to IE2. As demonstrated in Figure 8A, H3 and H4 associated with the transfected Flag-IE2 (lane 2), but the association was reduced when p150 was depleted (lane 3). Interestingly, Flag-IE2Q548R failed to interact with H3 and H4 (lane 4), indicating that CAF1 is the major mediator for IE2 association with histones. To check if ectopically expressed Flag-IE2 interacts with free histones or DNA-associated histones, we probed for histone H3 monomethylated at Lys 4 (H3K4me1) in IE2 complex. H3K4me1 is only present in chromatin-associated H3 53. It was found that the IE2-bound H3 was not monomethylated at this residue, suggesting that IE2 binds to DNA-free histones. To further verify this, the lysates from transfected cells were separated into chromatin-bound and -unbound fractions (Figure 8B, lanes 1-4) and the latter was used to perform IP assays (Figure 8B, lanes 5 and 6). Very few histone H3 existed in DNA-free condition (lanes 3 and 4), which is consistent with the evidence showing that 99% of H3 is in chromatin 53. The protein level of p150 was nearly equal in both chromatin-bound and -unbound fractions (compare lane 3 with 1). Most IE2 did not associate with chromatin (compare lane 4 with 2). In agreement with our data in Figure 8A, IE2 indeed formed complex with chromatin-free H3 (Figure 8B, lane 6). Given that CAF1 functions to deposit the newly synthesized histones H3 and H4 onto replicating DNA, all these results agree with our hypothesis that IE2 recruits CAF1 and its associated histones to assist the chromatin formation on replicating viral DNA.

IE2 associates with histones depending on CAF1. (A) p150 is required for histone H3/H4 association with IE2. U2OS cells were transfected with si-scramble (lanes 1, 2 and 4) or si-p150 (lane 3) twice, followed by transfection of plasmid-encoding Flag-tagged wild-type IE2 (lanes 2 and 3) or IE2Q548R (lane 4). Cell lysates were collected for IP with anti-Flag antibody and western blotting to analyze the pulled-down proteins. Lanes were run on the same gel but were non-contiguous (white lines). (B) IE2 interacts with free histone H3. U2OS cells transfected with vector (lanes 1 and 3) or plasmid-encoding Flag-tagged wild-type IE2 (lanes 2 and 4) were lysed with NP-40 lysis buffer to release chromatin-unbound proteins (lanes 3 and 4). After centrifugation, the cell pellets were sonicated to fragmentize chromatin DNA and release the chromatin-bound proteins (lanes 1 and 2). Equal volume of each lysate was separated by SDS-PAGE, followed by western blotting to analyze the amount of indicated proteins. Flag-IE2 in the chromatin-unbound fractions was then immunoprecipitated with anti-Flag antibody. The pulled-down proteins were analyzed by western blotting (lanes 5 and 6). PCNA was used as a loading control for input and a negative control for IP.
p150 defective in IE2 association partially resists HCMV-induced nuclease hypersensitivity of host chromatin
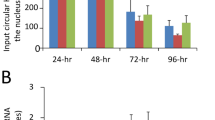
Given that CAF1 and PCNA are mislocated to viral genome replication centers (Figures 3 and 4), the cellular chromatin structure is likely altered in HCMV-infected cells. To address this possibility, the bulk chromatin of mock- or HCMV-infected MRC-5 cells was collected and digested with micrococcal nuclease (MNase), followed by protein removal and DNA separation with agarose gel electrophoresis. As shown in Figure 9A, the chromatin DNAs from HCMV-infected cells became more sensitive to MNase digestion than that from mock-infected cells (compare lanes 3, 5, 7, 9 and 11 with 2, 4, 6, 8 and 10, respectively), as evident by the appearance of the DNA fragment corresponding to mono-, di-, or trinucleosomes. This indicates that after HCMV infection, the cellular chromatin is not packed as tightly as in uninfected cells. We then examined whether p150 defective in IE2 binding is able to protect cells from HCMV-induced chromatin decondensation. Consistent with the results shown in Figure 9A, HCMV infection of cells transfected with vector alone (Figure 9B, upper panel, lane 10) or p150-V5 (lane 11) induced nuclease hypersensitivity, as more low-molecular-weight DNA was accumulated in virally infected cells after MNase digestion (lower panel, left and middle graphs). Importantly, enforced expression of the p150 mutant lacking IE2 interaction domain led to partial resistance to HCMV-induced nuclease hypersensitivity (right graph). Taken together, these results support our hypothesis that HCMV induces alterations in host-chromatin structure, in part, by interfering with CAF1 function by IE2-mediated mislocation of CAF1 to viral DNA replication centers.

p150 defective in IE2 binding partially reduces the HCMV-induced nuclease hypersensitivity of host chromatin. (A) Cellular chromatin is more sensitive to nuclease digestion after HCMV infection. MRC-5 cells were mock infected (lanes 2, 4, 6, 8 and 10) or infected with HCMV at an MOI of 1 (lanes 3, 5, 7, 9 and 11). At 72 h.p.i., the chromatin DNAs from equal amount of cells were digested with 10 U micrococcus nuclease (MNase; lanes 2-11) for indicated time, followed by separation with a 2 % agarose gel (upper panel). Lane 1: the 100-bp ladder of the DNA molecular weight marker (Fermentas). The DNA fragment corresponding to mono-, di- or trinucleosomes is indicated in the right. The intensity of the MNase products from mock-infected cells at lane 8 or HCMV-infected cells at lane 9 was quantified with Metamorph software (Universal Imaging) and normalized to the total intensity of each lane. The relative intensity is shown in the right graph. (B) p150 defective in IE2 binding diminishes the HCMV-induced nuclease hypersensitivity of host chromatin. U373 cells stably expressing vector alone (lanes 1, 4, 7 and 10), p150-V5 (lanes 2, 5, 8 and 11) or the p150 mutant lacking aa 506-563 (lanes 3, 6, 9 and 12) were mock infected (lanes 1-3 and 7-9) or infected with HCMV at an MOI of 1 (lanes 4-6 and 10-12). At 72 h.p.i., the chromatin DNAs from equal amounts of cells were mock treated (lanes 1-6) or digested with 10 U MNase (lanes 7-12) for 20 min, followed by separation with a 2 % agarose gel. Lane 13: the 100-bp ladder of the DNA molecular weight marker (Fermentas). The DNA fragment corresponding to mono-, di- or trinucleosomes is indicated in the right. The intensity of the MNase products from mock-infected cells (upper panel: lanes 7-9) or HCMV-infected cells (upper panel: lanes 10-12) was quantified with Metamorph software (Universal Imaging) and normalized to the total intensity of each lane (lower panel).
Discussion
Possible roles of CAF1 in HCMV life cycle
CAF1 function in viral genome synthesis has never been demonstrated in virus-infected cells. Here, we provide the first evidence that CAF1 facilitates HCMV DNA replication. We show that (I) HCMV infection upregulates the protein levels of p150 and p60 of CAF1, as well as PCNA and (II) HCMV IE2 binds to p150 and through this interaction locates the CAF1 complex, possibly with histones, to centers of viral genome synthesis to promote HCMV DNA replication. In addition, we unexpectedly found p150, independently of p60, binds to and activates the HCMV major immediate-early promoter that drives IE2 synthesis 54. Based on all these, it is hypothesized that an IE2-CAF1 autoregulation loop might facilitate HCMV viral DNA replication.
Next key question remained to be answered is how CAF1 promotes HCMV genome synthesis. Although HCMV virion contains no histone proteins 55, 56, a number of reports indicate that host histones with different modifications associate with HCMV DNA in infected cells cultured in labs or isolated from humans 49, 57, 58, 59, 60, 61, 62. Nitzsche et al. 43 further indicate that HCMV DNA is chromatinized in infected cells; histones are localized with UL44 at replication foci and there is a robust increase of histone occupancy coupled with viral DNA replication. All these data agree nicely with our current finding in which CAF1 is found involved in HCMV DNA synthesis (Figure 5) and the chromatin assembly activity is required for CAF1 to function in this aspect (Supplementary information, Figure S7). Thus, it is very likely that HCMV genome undergoes CAF1-mediated chromatin assembly during replication, and, through an unknown mechanism, is devoid of histones when packed into virion. The proposed model is further supported by our finding that histones associate with IE2 depending on CAF1 (Figure 8). In addition, the large aggregation of CAF1 is disrupted by PAA (Figures 4C and S3). Since PAA inhibits viral DNA polymerase activity, it is speculated that the CAF1 aggregation forms after the onset of viral DNA synthesis, which fits the function of CAF1 in transferring H3/H4 dimer onto newly synthesized DNA. Nevertheless, the model is likely specific to HCMV, as its close neighbors, the HSVs, replicate in the compartments that exclude histones, even though their genome associates with histones in infected cells 63.
IE2 facilitates HCMV DNA replication independently of its transactivator function
Our finding that IE2, independently of its roles in transcriptional regulation, served as an important mediator in directing cellular DNA replication machinery to compartments of HCMV genome synthesis is especially intriguing. CAF1-dependent association of histone proteins with IE2 (Figure 8) further supports the idea that IE2 controls chromatin assembly of viral genome during DNA replication. A previous study found that the cellular transcriptional coactivator HCF1 couples with the histone assembly factor ASF1b to HSV-1 replication compartments for regulating viral DNA replication 64. Together with our data in the current study, it is believed that, at least for herpesviruses, transcriptional regulators essential for viral lytic gene expression can link histone chaperons to viral DNA replication machinery. In IE2-transfected cells, either exogenously added or the endogenous p150 exhibits a similar dot-like structure to that of BrdU (Supplementary information, Figure S1), indicating that IE2 alone does not affect p150's subnuclear localization. In contrast, in HCMV-infected cells, IE2 no longer associates with the cellular DNA replication foci but instead aggregates into viral replication centers together with CAF1 (Figures 3 and 4). We suspect that viral factors are required to mediate the specific localization of IE2-CAF1 complex. Sourvinos et al. 33 proposed that viral DNA likely serves as a determinant of IE2-granule location. It has also been reported that IE2 uses N-terminal 215 residues to interact with the leucine zipper domain of UL84 that binds to the RNA/DNA hybrid region of oriLyt 37, 52. Thus, it seems that both DNA-binding and protein-protein interaction are able to target IE2 to viral DNA replication foci. IE2 may use its N-terminal portion to bind viral replication complex and its C-terminal end to tether the cellular DNA synthesis machinery. The fact that IE2 could modulate HCMV replication independently of its transactivation function is further supported by a recent finding in which IE2 is shown to control UL84 protein stability rather than mRNA level 65.
Host effects caused by HCMV hijacking of CAF1
The current study shows that HCMV infection induces host chromatin decondensation, as evidenced by the nucleosomal ladder generated by MNase digestion (Figure 9). This is consistent with an earlier observation using electron microscopy by Kierszenbaum and Huang 66 which clearly indicates a loosen host chromatin containing less nucleosomes in HCMV-infected fibroblasts. Our result of chromatin decondensation also resembles the previously published effect caused by depleting p150 or p60 or overexpressing the dominant-negative form of p150 8, 9, 51. Given that p150 and IE2 colocalize with UL44 and BrdU (Figure 4) and that ectopically expressing p150 (Δ506-563), a p150 mutant defective in IE2 interaction, partially restores the host-chromatin integrity during HCMV infection (Figure 9B), we propose that the HCMV-induced chromatin decondensation is likely due to viral IE2-mediated CAF1 mislocation to viral replication compartments. Nevertheless, the incomplete restoration also suggests that other histone chaperons, such as ASF1a 43 and HIRA (unpublished data), might be involved through an IE2-independent manner. In addition, IE2-mediated CAF1 mislocation could contribute to the reported cell cycle arrest and block in host DNA synthesis during HCMV infection 21, 22, 67, 68, 69. CAF1 also functions in DNA repair and heterochromatin silencing 70, 71, which can both be affected once CAF1 is mislocated by IE2. In summary, our current data provide important evidence to show that viruses such as HCMV have evolved an efficient way to take over the essential cellular chromatin assembly machinery for their own benefit.
Materials and Methods
Plasmids and antibodies
Please refer to the Supplementary information, Data S1.
Cell culture, transfection and virus infection
H1299, U2OS and MRC-5 cells were obtained from and maintained as instructed by ATCC. The human glioblastoma astrocytoma U373MG cells were obtained from ECACC and cultured in Minimal Essential Medium Alpha Medium (Invitrogen) containing 10% fetal bovine serum, 100 U/ml penicillin G sodium (Invitrogen) and 100 μg/ml streptomycin sulfate (Invitrogen). All transfections were carried out by using lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. In all, 5×105 and 3×106 cells were seeded onto 6-well plates or 10-cm plates, respectively, one day before transfection. At 24 h after transfection, cells were harvested for further analyses. To generate stable clones, 700 μg/ml G418 (Sigma) was used to enrich cells acquiring the plasmid. In transfection/infection experiments, cells were transfected with indicated plasmids, incubated for 4 h, followed by viral infection immediately. HCMV strain RC256 72 was obtained from ATCC and maintained according to the manufacturer's instructions. Infections were conducted at the indicated MOI for 2 h. Cells were then washed with phosphate-buffered saline (PBS) three times and harvested at each indicated time point for further analyses.
Yeast two-hybrid, GST pull-down, IP, western blotting and luciferase assay
Yeast two-hybrid assays were performed as described 45. GST pull-down, IP and western analyses were all carried out as indicated 54, 73.
Immunostaining
Cells were grown on glass coverslips (22×22 mm), followed by transfection or infection as described above. At each designated time point after transfection or infection, cells were fixed with 2% (v/v) formaldehyde in PBS. In BrdU incorporation analyses, cells were incubated with 100 μM BrdU (Sigma) for 15 min or 20 μM BrdU for 2 h before fixation, and the fixed cells were then treated with 2 N HCl at room temperature for 30 min to expose the epitope, followed by neutralization with 0.1 M boric acid (Na2B7O4). All fixed cells were quenched with Quench Buffer (PBS containing 0.1% Tritox-100 and 150 mM solution acetate, pH 7.4). Cells were then washed with wash buffer (PBS containing 0.1% Tritox-100), followed by blocking with blocking buffer (wash buffer containing 3% bovine serum albumin). For analyzing the localization of exogenous proteins, cells were incubated with the corresponding primary antibody at room temperature for 1 h. To detect the endogenous proteins or viral proteins, cells were incubated with the primary antibody at 4 °C overnight, washed and then incubated with the species-specific fluorophore-conjugated secondary antibody (Jackson ImmunoResearch or Invitrogen) at room temperature for 1 h. Finally, cells were incubated with 300 nM 4′, 6-diamidino-2-phenylindole dihydrochloride (Sigma) for nuclear staining and mounted with Biomeda Gel Mount (for traditional fluorescence microscopy) or with Molecular Probe ProLong Antifade Kit (for confocal microscopy). Images from traditional fluorescence microscopy were collected using an OLYMPUS IX71 fluorescence microscopy fitted with an UPlanFl 60×/NA 1.25 oil objective. Images from confocal microscopy were collected using a Leica TCS NT confocal microscopy fitted with a Plan-APOCHROMAT 63×/NA 1.32 oil objective. The final composite images were created using Adobe Photoshop 7.0.
Real-time PCR
At each designated time point after infection, cells were lysed using lysis buffer (50 mM Tris-HCl, pH 8.1, 10 mM EDTA, 1% SDS, 1% deoxycholic acid, 1 mM DTT and 1 mM PMSF) and sonicated with Misonix sonicator 3000. Cell lysates were then extracted by phenol-chloroform-isoamyl alcohol (Sigma). Total DNAs were then precipitated by ethanol, followed by dissolving into TE buffer (10 mM Tris-HCl, pH 7.5, 1 mM EDTA). The real-time PCR were performed using LightCycler (Roche) and QuantiTect SYBR Green PCR Kit (Qiagen) according to the manufacturer's instructions. PCR Primers used are: UL44 sense, 5′-CGACCGAGTTTTCTGGCATGG-3′ and UL44 antisense, 5′-GATTCCCTGTGCCTATTTCAC-3′; GAPDH sense, 5′-GTATTCCCCCAGGTTTACAT-3′ and GAPDH antisense, 5′-TTCTGTCTTCCCTCACTCC-3′; and MIEP sense, 5′- GGGACTTTCCTACTTGGCAG -3′ and MIEP antisense, 5′-GACGGTTCACT AAACGAG -3′.
MNase analysis
At each designated time point after infection, cells grown on 6-well plates were washed with PBS three times, fixed with 1% formaldehyde at room temperature for 5 min and then placed on ice for 5 min, followed by permeablization with ice-cold NP-40 lysis buffer (10 mM Tris-HCl, pH7.4, 10 mM NaCl, 3 mM MgCl2, 0.5% Nonidet P-40, 0.15 mM spermine and 0.5 mM spermidine) on ice for 10 min. After this, cells were treated with 10 U MNase (Worthington) in digestion buffer (10 mM Tris-HCl, pH7.4, 15 mM NaCl, 60 mM KCl, 1 mM CaCl2, 0.15 mM spermine and 0.5 mM spermidine) at 37 °C for indicated time. The reaction was stopped by adding the same volume of fresh MNase stop buffer (20 mM EDTA, 2 mM EGTA, 0.75 mg/ml proteinase K and 2% SDS). After overnight incubation at 37 °C, the cell lysates were extracted by phenol-chloroform-isoamyl alcohol (Sigma). Total DNAs were then precipitated by isopropanol, dissolved into TE buffer (10 mM Tris-HCl, pH 7.5, 1 mM EDTA), followed by separation with 2% agarose gel. The intensity of the digested products was quantified with Metamorph software (Universal Imaging).
References
Smith S, Stillman B . Purification and characterization of CAF-I, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell 1989; 58:15–25.
Li Q, Zhou H, Wurtele H, et al. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell 2008; 134:244–255.
Takami Y, Ono T, Fukagawa T, Shibahara K, Nakayama T . Essential role of chromatin assembly factor-1-mediated rapid nucleosome assembly for DNA replication and cell division in vertebrate cells. Mol Biol Cell 2007; 18:129–141.
Corpet A, Almouzni G . Making copies of chromatin: the challenge of nucleosomal organization and epigenetic information. Trends Cell Biol 2009; 19:29–41.
Klapholz B, Dietrich BH, Schaffner C, et al. CAF-1 is required for efficient replication of euchromatic DNA in Drosophila larval endocycling cells. Chromosoma 2009; 118:235–248.
Kaufman PD, Kobayashi R, Kessler N, Stillman B . The p150 and p60 subunits of chromatin assembly factor I: a molecular link between newly synthesized histones and DNA replication. Cell 1995; 81:1105–1114.
Hoek M, Stillman B . Chromatin assembly factor 1 is essential and couples chromatin assembly to DNA replication in vivo. Proc Natl Acad Sci USA 2003; 100:12183–12188.
Nabatiyan A, Krude T . Silencing of chromatin assembly factor 1 in human cells leads to cell death and loss of chromatin assembly during DNA synthesis. Mol Cell Biol 2004; 24:2853–2862.
Quivy JP, Gerard A, Cook AJ, Roche D, Almouzni G . The HP1-p150/CAF-1 interaction is required for pericentric heterochromatin replication and S-phase progression in mouse cells. Nat Struct Mol Biol 2008; 15:972–979.
Qian YW, Wang YC, Hollingsworth RE Jr, Jones D, Ling N, Lee EY . A retinoblastoma-binding protein related to a negative regulator of Ras in yeast. Nature 1993; 364:648–652.
Parthun MR, Widom J, Gottschling DE . The major cytoplasmic histone acetyltransferase in yeast: links to chromatin replication and histone metabolism. Cell 1996; 87:85–94.
Shibahara K, Stillman B . Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 1999; 96:575–585.
Krude T . Chromatin assembly factor 1 (CAF-1) colocalizes with replication foci in HeLa cell nuclei. Exp Cell Res 1995; 220:304–311.
Marheineke K, Krude T . Nucleosome assembly activity and intracellular localization of human CAF-1 changes during the cell division cycle. J Biol Chem 1998; 273:15279–15286.
Probst AV, Dunleavy E, Almouzni G . Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell Biol 2009; 10:192–206.
Jiao R, Harrigan JA, Shevelev I, et al. The Werner syndrome protein is required for recruitment of chromatin assembly factor 1 following DNA damage. Oncogene 2007; 26:3811–3822.
Stillman B . Chromatin assembly during SV40 DNA replication in vitro. Cell 1986; 45:555–565.
Murphy E, Shenk T . Human cytomegalovirus genome. Curr Top Microbiol Immunol 2008; 325:1–19.
Mocarski ES, Shenk T, Pass RF . Cytomegaloviruses. In: Knipe DM, Howley PM, eds. Fields Virology. Philadelphia: Lippincott Williams and Wilkins, 2007:2701–2772.
Wiebusch L, Hagemeier C . Human cytomegalovirus 86-kilodalton IE2 protein blocks cell cycle progression in G(1). J Virol 1999; 73:9274–9283.
Dittmer D, Mocarski ES . Human cytomegalovirus infection inhibits G1/S transition. J Virol 1997; 71:1629–1634.
Lu M, Shenk T . Human cytomegalovirus infection inhibits cell cycle progression at multiple points, including the transition from G1 to S. J Virol 1996; 70:8850–8857.
Estes JE, Huang ES . Stimulation of cellular thymidine kinases by human cytomegalovirus. J Virol 1977; 24:13–21.
Masse MJ, Karlin S, Schachtel GA, Mocarski ES . Human cytomegalovirus origin of DNA replication (oriLyt) resides within a highly complex repetitive region. Proc Natl Acad Sci USA 1992; 89:5246–5250.
Hamzeh FM, Lietman PS, Gibson W, Hayward GS . Identification of the lytic origin of DNA replication in human cytomegalovirus by a novel approach utilizing ganciclovir-induced chain termination. J Virol 1990; 64:6184–6195.
Borst EM, Messerle M . Analysis of human cytomegalovirus oriLyt sequence requirements in the context of the viral genome. J Virol 2005; 79:3615–3626.
Crough T, Khanna R . Immunobiology of human cytomegalovirus: from bench to bedside. Clin Microbiol Rev 2009; 22:76–98, Table of Contents.
Maul GG . Initiation of cytomegalovirus infection at ND10. Curr Top Microbiol Immunol 2008; 325:117–132.
Ahn JH, Jang WJ, Hayward GS . The human cytomegalovirus IE2 and UL112-113 proteins accumulate in viral DNA replication compartments that initiate from the periphery of promyelocytic leukemia protein-associated nuclear bodies (PODs or ND10). J Virol 1999; 73:10458–10471.
Roizman B, Knipe DM, Whitley RJ . Hepes Simplex Viruses. In: Knipe DM, Howley PM, eds. Fields Virology. Philadelphia: Lippincott Williams and Wilkins, 2007:2501–2601.
Pari GS . Nuts and bolts of human cytomegalovirus lytic DNA replication. Curr Top Microbiol Immunol 2008; 325:153–166.
Mercorelli B, Sinigalia E, Loregian A, Palu G . Human cytomegalovirus DNA replication: antiviral targets and drugs. Rev Med Virol 2008; 18:177–210.
Sourvinos G, Tavalai N, Berndt A, Spandidos DA, Stamminger T . Recruitment of human cytomegalovirus immediate-early 2 protein onto parental viral genomes in association with ND10 in live-infected cells. J Virol 2007; 81:10123–10136.
Sarisky RT, Hayward GS . Evidence that the UL84 gene product of human cytomegalovirus is essential for promoting oriLyt-dependent DNA replication and formation of replication compartments in cotransfection assays. J Virol 1996; 70:7398–7413.
Marchini A, Liu H, Zhu H . Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J Virol 2001; 75:1870–1878.
Stinski MF, Petrik DT . Functional roles of the human cytomegalovirus essential IE86 protein. Curr Top Microbiol Immunol 2008; 325:133–152.
Colletti KS, Smallenburg KE, Xu Y, Pari GS . Human cytomegalovirus UL84 interacts with an RNA stem-loop sequence found within the RNA/DNA hybrid region of oriLyt. J Virol 2007; 81:7077–7085.
Spector DJ, Tevethia MJ . Protein-protein interactions between human cytomegalovirus IE2-580aa and pUL84 in lytically infected cells. J Virol 1994; 68:7549–7553.
Xu Y, Cei SA, Rodriguez Huete A, Colletti KS, Pari GS . Human cytomegalovirus DNA replication requires transcriptional activation via an IE2- and UL84-responsive bidirectional promoter element within oriLyt. J Virol 2004; 78:11664–11677.
Petrik DT, Schmitt KP, Stinski MF . Inhibition of cellular DNA synthesis by the human cytomegalovirus IE86 protein is necessary for efficient virus replication. J Virol 2006; 80:3872–3883.
Kent JR, Zeng PY, Atanasiu D, Gardner J, Fraser NW, Berger SL . During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J Virol 2004; 78:10178–10186.
Deshmane SL, Fraser NW . During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J Virol 1989; 63:943–947.
Nitzsche A, Paulus C, Nevels M . Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J Virol 2008; 82:11167–11180.
Mate JL, Ariza A, Muñoz A, Molinero JL, López D, Navas-Palacios JJ . Induction of proliferating cell nuclear antigen and Ki-67 expression by cytomegalovirus infection. J Pathol 1998; 184:279–282.
Huang CF, Wang YC, Tsao DA, Tung SF, Lin YS, Wu CW . Antagonism between members of the CNC-bZIP family and the immediate-early protein IE2 of human cytomegalovirus. J Biol Chem 2000; 275:12313–12320.
Nakayasu H, Berezney R . Mapping replicational sites in the eucaryotic cell nucleus. J Cell Biol 1989; 108:1–11.
van Dierendonck JH, Keyzer R, van de Velde CJ, Cornelisse CJ . Subdivision of S-phase by analysis of nuclear 5-bromodeoxyuridine staining patterns. Cytometry 1989; 10:143–150.
Castillo JP, Kowalik TF . Human cytomegalovirus immediate early proteins and cell growth control. Gene 2002; 290:19–34.
Reeves M, Murphy J, Greaves R, Fairley J, Brehm A, Sinclair J . Autorepression of the human cytomegalovirus major immediate-early promoter/enhancer at late times of infection is mediated by the recruitment of chromatin remodeling enzymes by IE86. J Virol 2006; 80:9998–10009.
Huang ES . Human cytomegalovirus. IV. Specific inhibition of virus-induced DNA polymerase activity and viral DNA replication by phosphonoacetic acid. J Virol 1975; 16:1560–1565.
Ye X, Franco AA, Santos H, Nelson DM, Kaufman PD, Adams PD . Defective S phase chromatin assembly causes DNA damage, activation of the S phase checkpoint, and S phase arrest. Mol Cell 2003; 11:341–351.
Colletti KS, Xu Y, Cei SA, Tarrant M, Pari GS . Human cytomegalovirus UL84 oligomerization and heterodimerization domains act as transdominant inhibitors of oriLyt-dependent DNA replication: evidence that IE2-UL84 and UL84-UL84 interactions are required for lytic DNA replication. J Virol 2004; 78:9203–9214.
Loyola A, Bonaldi T, Roche D, Imhof A, Almouzni G . PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state. Mol Cell 2006; 24:309–316.
Lee SB, Ou DS, Lee CF, Juan LJ . Gene-specific transcriptional activation mediated by the p150 subunit of the chromatin assembly factor 1. J Biol Chem 2009; 284:14040–14049.
Baldick CJ, Jr, Shenk T . Proteins associated with purified human cytomegalovirus particles. J Virol 1996; 70:6097–6105.
Varnum SM, Streblow DN, Monroe ME, et al. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J Virol 2004; 78:10960–10966.
Groves I, Reeves M, Sinclair J . Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic "pre-immediate early" repression of viral gene expression mediated by histone post-translational modification. J Gen Virol 2009; 90:2364–2374.
Liu XF, Yan S, Abecassis M, Hummel M . Establishment of murine cytomegalovirus latency in vivo is associated with changes in histone modifications and recruitment of transcriptional repressors to the major immediate-early promoter. J Virol 2008; 82:10922–10931.
Cuevas-Bennett C, Shenk T . Dynamic histone H3 acetylation and methylation at human cytomegalovirus promoters during replication in fibroblasts. J Virol 2008; 82:9525–9536.
Ioudinkova E, Arcangeletti MC, Rynditch A, et al. Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 2006; 384:120–128.
Reeves MB, MacAry PA, Lehner PJ, Sissons JG, Sinclair JH . Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc Natl Acad Sci USA 2005; 102:4140–4145.
Murphy JC, Fischle W, Verdin E, Sinclair JH . Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J 2002; 21:1112–1120.
Knipe DM, Cliffe A . Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol 2008; 6:211–221.
Peng H, Nogueira ML, Vogel JL, Kristie TM . Transcriptional coactivator HCF-1 couples the histone chaperone Asf1b to HSV-1 DNA replication components. Proc Natl Acad Sci USA 107:2461–2466.
Sanders RL, Del Rosario CJ, White EA, Spector DH . Internal deletions of IE2 86 and loss of the late IE2 60 and IE2 40 proteins encoded by human cytomegalovirus affect the levels of UL84 protein but not the amount of UL84 mRNA or the loading and distribution of the mRNA on polysomes. J Virol 2008; 82:11383–11397.
Kierszenbaum AL, Huang ES . Chromatin pattern consisting of repeating bipartite structures in WI-38 cells infected with human cytomegalovirus. J Virol 1978; 28:661–664.
Salvant BS, Fortunato EA, Spector DH . Cell cycle dysregulation by human cytomegalovirus: influence of the cell cycle phase at the time of infection and effects on cyclin transcription. J Virol 1998; 72:3729–3741.
Bresnahan WA, Boldogh I, Thompson EA, Albrecht T . Human cytomegalovirus inhibits cellular DNA synthesis and arrests productively infected cells in late G1. Virology 1996; 224:150–160.
Jault FM, Jault JM, Ruchti F, et al. Cytomegalovirus infection induces high levels of cyclins, phosphorylated Rb, and p53, leading to cell cycle arrest. J Virol 1995; 69:6697–6704.
Huang S, Zhou H, Tarara J, Zhang Z . A novel role for histone chaperones CAF-1 and Rtt106p in heterochromatin silencing. EMBO J 2007; 26:2274–2283.
De Koning L, Corpet A, Haber JE, Almouzni G . Histone chaperones: an escort network regulating histone traffic. Nat Struct Mol Biol 2007; 14:997–1007.
Spaete RR, Mocarski ES . Insertion and deletion mutagenesis of the human cytomegalovirus genome. Proc Natl Acad Sci USA 1987; 84:7213–7217.
Lee CF, Ou DS, Lee SB, et al. hNaa10p contributes to tumorigenesis by facilitating DNMT1-mediated tumor suppressor gene silencing. J Clin Invest 2010; 120:2920–2930.
Acknowledgements
We thank Drs Paul D Kaufman (University of Massachusetts Medical School) and Bruce Stillman (Cold Spring Harbor Laboratory) for providing plasmids, Dr Mark F Stinski (University of Iowa) for providing BAC construct and Dr Sheng-Chung Lee (National Taiwan University) for providing anti-rabbit antibody. We also thank Y-T Yang (NHRI) for constructing pSEP7-HA-p150C, C-H Lin (NHRI) and Dr C-H Hsu (Academia Sinica) for preparing GST fusion proteins, and Dr. C-W Wu (NHRI) for providing partial funding. This work was primarily supported by grants from Academia Sinica and NHRI to L-J Juan.
Author information
Authors and Affiliations
Corresponding author
Additional information
(Supplementary information is linked to the online version of the paper on the Cell Research website)
Supplementary information
Supplementary information, Figure S1
Both p150 and IE2 localize at BrdU-positive foci in transfected cells. (PDF 232 kb)
Supplementary information, Figure S2
Co-localization of p60 and PCNA with IE2 in HCMV-infected cells. (PDF 423 kb)
Supplementary information, Figure S3
p150 and UL44-positive granules are disrupted by the viral DNA replication inhibitor PAA. (PDF 125 kb)
Supplementary information, Figure S4
CAF1 activity is required for HCMV DNA replication. (PDF 200 kb)
Supplementary information, Figure S5
The p150 fragment containing only amino acids 505 to 564 interacts with IE2 and competes for the binding of the endogenous p150 to IE2 in transfected cells. (PDF 131 kb)
Supplementary information, Figure S6
The p150 mutant lacking aa 506-563 forms a complex with HA-p60 and is localized to cellular DNA replication foci. (PDF 60 kb)
Supplementary information, Figure S7
GST-IE1 and GST-IE2 derivatives used in Figures 6A and 7A are shown by coo-massie blue staining. (PDF 114 kb)
Rights and permissions
About this article
Cite this article
Lee, SB., Lee, CF., Ou, DC. et al. Host-viral effects of chromatin assembly factor 1 interaction with HCMV IE2. Cell Res 21, 1230–1247 (2011). https://doi.org/10.1038/cr.2011.53
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2011.53
Keywords
This article is cited by
-
Human cytomegalovirus downregulates SLITRK6 expression through IE2
Journal of NeuroVirology (2017)
