
Abstract
Tumour progression and therapy resistance in squamous cell carcinoma of the skin (SCC) is strongly associated with resistance to intrinsic mitochondrial apoptosis. We thus investigated the role of various anti-apoptotic Bcl-2 proteins for apoptosis protection in SCC using the BH3 agonist ABT737 that can overcome multidomain Bcl-2 protein protection. Sensitive SCC cells underwent rapid loss of mitochondrial membrane potential (MMP), subsequent apoptosis concomitant with caspase-3 activation and an early release of mitochondria-derived cytochrome c and smac/DIABLO. In contrast, ABT737 resistance in subsets of SCC cells was not explained by XIAP, important for protection from DR-induced apoptosis in SCC. Of note, ABT737 did not prime SCC cells to DR-induced apoptosis. Interestingly, the ratio of Mcl-1 and Noxa determined sensitivity to ABT737: loss of Mcl-1 rendered resistant cells sensitive to ABT737, whereas loss of Noxa promoted resistance in sensitive cells. In line, suppression of Mcl-1 by the pan-Bcl-2 inhibitor Obatoclax or overexpression of Noxa rendered resistant SCC cells sensitive to BH3 mimetics. Our data indicate that targeting of the Mcl-1/Noxa axis is important to overcome resistance to mitochondrial apoptosis in SCC. Therefore, combination treatment of ABT737 or derivatives with Mcl-1 inhibitors, or inducers of Noxa, may represent a novel option of targeted therapy in metastatic SCC of the skin.
Similar content being viewed by others


Main
Apoptosis is an indispensible process to maintain cellular homeostasis, in particular in highly dynamic tissues. Apoptosis can be induced by activation of death receptors (DRs; such as TRAIL-R1/R2 or cluster of differentiation 95 (CD95)) or by intrinsic disturbance of mitochondria.1 Death ligands (DLs; TNF-related apoptosis-inducing ligand (TRAIL) or CD95L), when bound to their respective DRs, induce apoptosis by activation of procaspase-8 within the death-inducing signalling complex (DISC).2 Caspase-8 activation is followed by proteolytic cleavage of caspase-3.3 Extrinsic and intrinsic cell death is negatively controlled by caspase inhibitors such as X-linked inhibitor of apoptosis protein (XIAP)4 or by B-cell lymphoma 2 (Bcl-2) proteins that suppress the mitochondria outer membrane permeability (MOMP) by limiting Bax (Bcl-2-associated X protein)/Bak (Bcl-2 homologous antagonist/killer) translocation into the mitochondrial outer membrane.5 The extrinsic signalling cascade communicates with the intrinsic death pathway by cleavage of Bid (BH3 interacting-domain death agonist), a pro-apoptotic member of the BH3 (Bcl-2 homology domain 3)-only subfamily of Bcl-2 proteins.1 Other stimuli such as genotoxic stress allow for translocation and pore formation of pro-apoptotic multidomain Bcl-2 proteins Bax and Bak in the outer mitochondrial membrane.6, 7, 8 This process promotes release of mitochondria-derived apoptogenic proteins, in particular cytochrome c,9 or smac/DIABLO (second mitochondria-derived activator of caspases/direct IAP binding protein with low pI).10 Within the apoptosome,11 active caspase-9 finally leads to activation of caspase-3,12 and subsequent cell death.
Anti-apoptotic multidomain Bcl-2 proteins (Bcl-2, Bcl-2-like protein 2 (Bcl-w), B-cell lymphoma-extra large (Bcl-XL), induced myeloid leukaemia cell differentiation protein (Mcl-1) and Bcl-2-related protein A1 (A1)) with four Bcl-2 homology domains (BH1, BH2, BH3 and BH4) suppress the pro-apoptotic function of Bax-like proteins such as Bax, Bak and Bok (that contain BH1–BH3 domains) or the BH3-only proteins Bad (Bcl-2-associated death promoter), Bim (Bcl-2-like protein 11), Bid, Noxa (phorbol-12-myristate-13-acetate-induced protein 1) and Puma (p53 upregulated modulator of apoptosis).13 Regulation of mitochondria-mediated apoptosis is determined by the balance between pro- and anti-apoptotic Bcl-2 proteins.14
In a variety of cancer types, a decrease of BH3-only protein or upregulation of pro-survival Bcl-2 proteins is associated with poor prognosis.15 In metastatic squamous cell carcinoma (SCC) of the skin or the so-called ‘head and neck SCC’ (HNSCC), high expression of pro-survival Bcl-2 proteins conferred radio- and chemotherapy resistance.16, 17 These findings mark Bcl-2 proteins as regulators of SCC apoptosis and indicate that BH3 mimetics may hold therapeutic potential for metastatic SCC. The BH3 mimetics navitoclax (ABT263) and ABT199 are currently under investigation in clinical studies.18, 19, 20 Mechanistically, their lead compound ABT737 suppresses Bcl-2 activity by binding to the hydrophobic groove of Bcl-2, Bcl-w and Bcl-XL.18 As ABT263 upregulates Mcl-1, resistance to a number of Bcl-2 inhibitors (ABT737 and ABT263) has been described.21 Another compound, Obatoclax, was developed to block all anti-apoptotic Bcl-2 proteins including Mcl-1.22 Obatoclax blocks the interaction of Bim or Bax with Mcl-1.23 In this report, we have studied the effect of ABT737 for cell death in SCC of the skin and investigated the molecular mechanisms of resistance to different BH3 mimetics.
Results
Expression profile of pro- and anti-apoptotic Bcl-2 proteins in primary and transformed keratinocytes
We first investigated the expression of different Bcl-2 proteins in a set of SCC cells and compared them with primary keratinocytes (PKs) (Figure 1a). A heterogeneous expression profile of Bcl-2 and Mcl-1 was detected. Bcl-XL was homogenously expressed in all cell lines studied. Bcl-2 was highly expressed in MET cells and in PKs. PKs are highly resistant to DR-mediated cell death, at least in part by high expression of cFLIP (CASP8 and FADD-like apoptosis regulator)24 or XIAP.25 Mcl-1 is more strongly expressed in transformed when compared with PKs. Thus, Mcl-1 could be an attractive therapeutic target in SCC. Bax and Bid were homogenously expressed, whereas Bim was strongly expressed in transformed but not in PKs. MET cells had high expression of Bak, and Bak levels were low in the HaCaT tumour progression model (IL4, IL4/7, A5, A5RT1 and A5RT3).26 Interestingly, Noxa is strongly expressed in HaCaT and HaCaT-derived SCC cells, but only weakly expressed in MET SCCs. In summary, heterogeneous expression of Bcl-2 proteins suggested that the mitochondrial signalling pathway is tightly regulated in SCC and merits a detailed analysis.

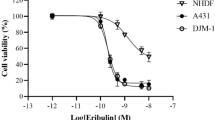
Expression analysis of Bcl-2 proteins and correlation with cell death sensitivity to ABT737 in primary and transformed keratinocytes. (a) Heterogeneous expression of pro- and anti-apoptotic Bcl-2 proteins in PKs and SCC cell lines. Three donors of PKs, HaCaT (K) and HaCaT-derived SCC, as well as DJM-1, SCC25 and cell lines from the intraindividual MET tumour progression model were analysed for expression of anti-apoptotic multidomain (Bcl-2, Bcl-XL and Mcl-1) and pro-apoptotic multidomain (Bax and Bak) or BH3-only Bcl-2 proteins (Bim, Bid and Noxa). Total protein lysates (5 μg) were separated with 4–12% NuPAGE gradient gels before detection of Bcl-2 proteins and β-tubulin as loading control by western blot analysis. One representative experiment of a total of three experiments is shown. (b and c) PKs and SCC cells show differential sensitivity to ABT737-induced cytotoxicity. PKs and different SCC cells were either stimulated with DMSO, different concentrations of ABT737 or its inactive Enantiomer (E, 10 μM) of ABT737 for 18–24 h followed by crystal violet assay as described in the Materials and Methods. (c) Summary of the differential sensitivity of PKs and SCC cell lines to 10 μM ABT737, 10 μM Enantiomer or diluents (DMSO) as compared with nontreated cells. S.E.M. of 3–5 independent experiments is shown (b and c)
Suppression of Bcl-2 activity by ABT737 promotes cell death in HaCaT-derived SCCs whereas MET cells or PKs are resistant
To examine the function of Bcl-2 and Bcl-XL, SCC lines and PKs were treated with different concentrations of ABT737 and cell survival was monitored. In contrast to PKs and MET cells, HaCaT and derived A5RT3 cells were sensitive to ABT737 (Figure 1b). The ABT737 Enantiomer was ineffective, indicating the specificity of the inhibitor (Figure 1b). Further analysis of other cell lines (Figure 1a) for ABT737-mediated cell death demonstrated reduced viability in all HaCaT-derived SCC, DJM and SCC25, but not in MET SCC or PKs (Figure 1c). Collectively, HaCaT-derived SCC critically required Bcl-2 proteins for survival, whereas PKs and MET cells tolerate ABT737-mediated suppression of Bcl-2, Bcl-w and Bcl-XL activity. This indicated that either other anti-apoptotic Bcl-2 proteins, such as A1 or Mcl-1, or the absence of BH3-only proteins such as Noxa in MET SCC cells are important for MOMP inhibition.
Suppression of Bcl-2 activity by ABT737 leads to rapid loss of mitochondrial membrane potential (MMP, Φ) and DNA fragmentation
BH3 mimetics allow disintegration of mitochondria by loss of MMP and MOMP.18 To further dissect the impact of ABT737, MMP and DNA fragmentation was analysed. HaCaT and A5RT3 cells underwent MMP within 24 h, with further increase till 48 h of ABT737 treatment, whereas treatment with Enantiomer had no effect (Figures 2a and b). In contrast, PKs and MET SCC cells showed minimal ABT737-induced loss of MMP within 24 h, whereas treatment up to 48 h induced loss of MMP in PKs, and to a lesser extent in MET cells (Figures 2a and b). Kinetic analysis demonstrated loss of MOMP in HaCaT and A5RT3 as early as 4 h after addition of ABT737 (Figures 2c and d). These results demonstrate that anti-apoptotic Bcl-2 proteins maintain functional integrity of mitochondria in HaCaT-derived SCC. In turn, ABT737 rapidly disturbs MMP and allows overcoming Bcl-2-mediated protection of MOMP. We next examined ABT737-induced DNA fragmentation. Correlating with ABT737-induced MMP loss, we observed a time-dependent DNA fragmentation in ABT737-sensitive cell lines. Interestingly, MET cells accumulate in the G0/G1 cell cycle phase, as indicated by accumulation of cells with diploid DNA content (Figures 2e and f). These results argue that MOMP is critical for cell death induction by ABT737. Furthermore, ABT737 treatment resulted in cell cycle arrest in MET SCC cells by thus far unknown mechanisms. Taken together, our findings identify Bcl-2 proteins as indispensable inhibitors of mitochondria-mediated apoptosis in SCC.

Kinetics of ABT737-induced loss of mitochondrial membrane potential (MMP) promotes cell death in HaCaT and A5RT3 cells, but not in PKs or MET cells. (a and b) PKs (donors E and L), HaCaT, HaCaT-derived A5RT3 and MET1 or MET4 cells were either stimulated with 10 μM ABT737, the respective Enantiomer or with DMSO for 24 or 48 h (control). The loss of MMP was investigated by FACS using staining with TMRE as described in the Materials and Methods. Quantitative summary of three independent experiments is shown in (b). (c and d) HaCaT and A5-RT3 cells were stimulated with ABT737 (10 μM), inactive Enantiomer or DMSO (control) alone for the indicated times. Loss of MMP was analysed as described for (a). The quantitative summary of TMRE-positive cells of two independent experiments with stimulation with 10 μM of ABT737 is shown in (d). (e and f) Apoptotic cell death of the indicated cells was analysed by hypodiploidy analysis after stimulation with ABT737 (10 μM), Enantiomer (10 μM) or DMSO (control) for 24 and 48 h and the quantification of two independent experiments is depicted in (f)
ABT737-mediated cell death induces rapid early release of mitochondrial proteins and caspase activation irrelevant for clonogenic survival
MOMP results in the release of cytochrome c, smac/DIABLO and HtrA2/Omi from mitochondria that induces caspase-dependent or -independent cell death (for review, see Galluzzi et al.27 and Fulda et al.28). As a result of a caspase-dependent degradation of the respiratory complex, as previously described,29 activation of caspases may escalate mitochondrial dysfunction and could interfere with progression of MOMP. We thus next studied ABT737-mediated cell death, loss of MMP and caspase activation in the presence or absence of the pan-caspase inhibitor zVAD-fmk. ABT737-mediated loss of MMP (within 4 h of stimulation) was partially blocked by zVAD-fmk, indicating that the early loss of MMP is caspase dependent (Figure 3a). As inhibition of caspase activation does not necessarily reflect clonogenic survival, we performed clonogenicity assays of A5RT3 cells in the presence and absence of zVAD-fmk (Figure 3b). ABT737 repressed clonogenic survival irrespective of the presence of zVAD-fmk 4 and 7 days after initiation of treatment (Figure 3b). These results demonstrate that loss of ΔΦM is modulated by caspase activity, most likely because of escalating mitochondrial dysfunction by cleavage of respiratory complex components.29, 30 However, this does not alter the cell biological consequence, as clonogenic survival of SCC cells is not protected. We also assessed activation of caspases in the absence or presence of zVAD-fmk (Figure 3c). Treatment with ABT737 leads to rapid cleavage of caspase-3 and its substrate poly-ADP-Ribose polymerase-1 (PARP-1), whereas caspase-8 cleavage was only detected at later time points (4 h). These results show that ABT737 induces early and rapid caspase-3 activation and PARP-1 cleavage whereas caspase-8 activation occurs in a feedback loop, as previously described.31 Cleavage of caspase-3 is suppressed in the presence of zVAD-fmk (Figure 3c), indicating that the short-term inhibition of caspases ensures protection from early ABT737-induced MMP (Figure 3a). In contrast, cellular lysates of ABT737-resistant MET4 cells did not show evidence of caspase cleavage. Suppression of ABT737-mediated MMP by zVAD-fmk indicated that caspase inhibition also influenced the release of mitochondria-derived proteins. In A5-RT3 cells, the ABT737-initiated kinetic release of cytochrome c, smac/DIABLO and HtrA2/Omi started within 30 min, independent of caspase activation (Figure 3d). Importantly, caspase-3 activation and PARP-1 cleavage was found within 60 min, and the release of mitochondrial proteins preceded caspase-3 activation. Thus, ABT737 rapidly induced caspase-independent release of mitochondria-derived proteins and subsequent caspase activation in susceptible SCC cells.

ABT737 induces the release of mitochondrial proteins and reduces clonogenic survival in a caspase-independent manner, whereas caspase activity accelerates the early loss of MMP. (a and c) ABT737-sensitive A5-RT3 and ABT737-resistant MET4 cell lines were analysed for the loss of MMP (a) or the cleavage of caspase-8, caspase-3 and PARP-1 (c) in the presence or absence of zVAD-fmk. A5RT3 and MET4 cells were either separately or in combination prestimulated with 10 μM of zVAD-fmk (1 h) or ABT737 (5 μM) (6 h in a; and 1 h or 4 h in c) and analysed for MMP by FACS (a) or caspase and PARP-1 cleavage by western blotting (c). (b) Clonogenic survival of A5-RT3 cells was determined in the presence and absence of zVAD-fmk (prestimulation 1 h, 10 μM) and/or ABT737 (5 μM, for 6 h). After the respective incubation times, medium was exchanged with fresh medium and clonogenic survival was investigated 4 to 7 days later. Colony formation was visualized with crystal violet. One representative experiment of a total of three independent experiments is shown. (d) Release of mitochondrial proteins into the cytoplasm was kinetically determined after prestimulation with or without zVAD-fmk and ABT737 (5 μM) for the indicated times by cellular subfractionation. Cleavage of caspase-3 and PARP-1 in total cellular lysates as well as release of cytochrome c, smac/DIABLO, HtrA-2/Omi and of COXIV or β-tubulin as control for purity of the cell fractionation was determined by western blot analysis
XIAP is critical for CD95L- and TRAIL-induced apoptosis, but not necessary to maintain cell death resistance to ABT737 in MET1 cells
A synergistic toxicity upon costimulation of tumour cells with ABT737 and DL such as TRAIL or chemotherapeutics was suggested.32, 33 These observations raised the possibility that HaCaT-derived or MET SCC undergo synergistic cell death when combining ABT737 and DLs (e.g., CD95L, TRAIL or tumour necrosis factor (TNF)). ABT737-sensitive and -resistant cell lines were prestimulated with a nontoxic concentration of ABT737 and then treated with CD95L (Figures 4a and d), TRAIL (Supplementary Figures 1a–d) or TNF (Supplementary Figure 1E). Neither ABT737-sensitive cell lines (HaCaT and A5RT3) nor ABT737-resistant cells (MET1 and MET4) were sensitized to DL-induced apoptosis by this pretreatment (Figures 4a and d and Supplementary Figures 1a–e). These results demonstrate that ABT737 is not priming SCC to apoptosis, as suggested previously for other cell lines.34 This effect was independent of Bcl-2, as Bcl-2 overexpression in HaCaT cells did not alter DL sensitivity (Figures 4e and f). This suggests that HaCaT are able to activate DISC-mediated signals sufficient for caspase-3 activation, independent from the mitochondrial amplification loop, and can be classified as type I cells35 that do not require mitochondria-mediated caspase activation for DR-induced apoptosis.

DL-mediated cell death is not facilitated by ABT737 in SCC cell lines. (a–d) Inhibition of Bcl-2 family proteins by ABT737 does not synergize with CD95L-induced cell death. A5-RT3, HaCaT, MET1 and MET4 cells were analysed for CD95L-induced cell death in the presence of nontoxic or low toxic concentration of ABT737. Cells were either left unstimulated, prestimulated for 1 h with ABT737 (0.1 μM for A5RT3 and HaCaT; 1 μM for MET cells) or with the respective diluents (DMSO) or inactive compound (Enantiomer) followed by stimulation with CD95L for 18–24 h with the indicated concentrations. Cytotoxic effect of DMSO, Enantiomer or ABT737 alone was normalized as 100% as indicated in the Materials and Methods. (e and f) Bcl-2 overexpression does not confer resistance to CD95L- or ABT737-induced cell death. HaCaT cells were transduced with Bcl-2 or control retroviruses. Bcl-2 expression was analysed by western blotting. β-Tubulin serves as internal loading control (e; inlet). For analysis of cell death, the respective cell lines were either stimulated with CD95L (e) or with 10 μM of ABT737, Enantiomer or DMSO alone for 18–24 h. Surviving attached cells were quantified with crystal violet assay. Summary of three to five independent experiments is shown and S.E.M. is depicted
XIAP is a potent inhibitor of active caspase-3, and the high expression of XIAP24, 25, 36 in MET cells prompted us to study whether XIAP is the decisive protein protecting MET cells from ABT737-induced cell death. To interrogate this hypothesis, we performed loss-of-function studies for XIAP in MET cells. Stable knockdown of XIAP did not sensitize to ABT737-mediated cell death in either MET cell line (Figures 5a and b), whereas cells were readily sensitized to DL-induced cell death (Figures 5c and d, open squares and triangles). These results argue for an XIAP-independent mechanism of resistance to ABT737. Interestingly, transduction with retroviruses sensitized MET1 cells to ABT737, but this nonspecific effect was independent of XIAP expression (Figures 5c and d, black squares and triangles). However, knockdown of XIAP increased DL-mediated cell death (Figures 5c and d, curves with black squares and triangles), indicating the functional relevance of the knockdown. We next studied the impact of XIAP for caspase-3 activation during DL treatment (Figures 5e and f). In control cells, CD95L (Figure 5e) or TRAIL (Figure 5f) stimulation allowed for predominant caspase-3 cleavage to p20 (indicative of XIAP-protected prodomain cleavage),25 and to a lesser extent p17. In contrast, prestimulation with ABT737 allowed stronger cleavage to the p17 fragment concomitant with increased PARP-1 cleavage (Figures 5e and f, left panels). In line with our previous report,25 XIAP knockdown demonstrated increased cleavage of caspase-3 to the p17 fragment, and PARP-1 cleavage was increased. A similar increase in full cleavage (p17/p15) was detected in the presence of ABT737/CD95L or ABT737/TRAIL (Figures 5e and f, right panel). These results demonstrate that XIAP not only protects from DL-induced apoptosis, but also ABT737 sensitizes to CD95L- and TRAIL-induced cell death by increased caspase-3 activation. However, our data indicate that XIAP is not crucial to protect from BH3 mimetic-initiated apoptosis in SCC.

XIAP expression does not explain ABT737 resistance in MET cells, whereas XIAP suppresses TRAIL- or CD95L-induced caspase-3 activation and cell death. (a) Knockdown of XIAP in MET cell was performed as described in the Materials and Methods. Expression of XIAP, cIAP1, cIAP2 and β-tubulin was analysed by western blotting in parental MET 1 cells, or in cells transduced with either XIAP shRNA-containing vector or hyper random sequence (HRS)-shRNA expressing vector. (b–d) Analysis of ABT737-mediated (b), CD95L-induced (c) or TRAIL-induced (d) cell death in control and XIAP knockdown cells was analysed. Transduced MET cells were either stimulated for 18–24 h (b; MET1, MET2, MET4) or prestimulated for 1 h with 10 μM ABT737 or 10 μM Enantiomer (Enan.) followed by stimulation with indicated concentrations of TRAIL or CD95L (MET1) for additional 18–24 h. Cell survival was monitored using crystal violet assay. Summary of three to five independent experiments is shown and S.E.M. was determined. (e and f) Following 1 h of prestimulation with ABT737 (5 μM) or Enantiomer (5 μM), stimulation with CD95L (25 U/ml) (e) or TRAIL (100 ng/ml) (f) alone or in combination with ABT737 or Enantiomer for 3 h was performed. Cleavage of caspase-3 and PARP-1 was analysed in 5 μg of total cellular lysates that were separated with 4–12% NuPAGE gradient gels before detection of indicated proteins, and β-tubulin as loading control, by western blot analysis
The ratio of Mcl-1 and Noxa determines resistance to ABT737-induced cell death in SCC cells
Previous studies in cancer models indicated that Mcl-1 is critical for mitochondrial cell death suppression.37 The Mcl-1-interacting Noxa is a decisive factor to overcome Mcl-1 protection.38 We thus compared Mcl-1 and Noxa expression levels in the different SCC lines exhibiting sensitivity or resistance to ABT737 (Figure 1a). Western blot analysis showed largely homogenous expression level of Mcl-1 in ABT737-sensitive HaCaT and HaCaT-derived SCC cells (IL4, I4/7, A5, A5RT1 and A5-RT3) as compared with ABT737-resistant MET1, MET2 and MET4 cell lines. Thus, a direct role of Mcl-1 expression levels to predict ABT737-mediated apoptosis is not obvious (Figure 1a). In contrast, Noxa is highly expressed in ABT737-sensitive HaCaT and HaCaT-derived SCC cells, but lacks in ABT737-resistant MET cells (Figure 1a). These results suggest that a high Mcl-1/Noxa ratio predicts ABT737 resistance. The four previously characterized SCC cell lines were also treated with Obatoclax, a pan-Bcl-2 inhibitor that also targets Mcl-1 activity (Supplementary Figures 2a–d).23 In all cell lines, including ABT737-resistant MET cells, Obatoclax dose-dependently induced cell death (Supplementary Figures 2a–d). Thus, pan-Bcl-2 inhibitors overcome Mcl-1-mediated cell death resistance in SCC. When we combined ABT737 and sublethal concentrations of Obatoclax, viability assays showed that prestimulation with Obatoclax sensitizes MET cells to ABT737-induced cell death and early loss of MMP (Supplementary Figures 2e and f). Thus either Mcl-1 or A1 may act as critical resistance proteins to protect from ABT737-mediated apoptosis. We next tested the functional impact of Mcl-1 in more detail (Figures 6a–c). Successful repression of Mcl-1 in MET cells (Figure 6a) by two different siRNAs led to a minor reduction of cell viability as compared with control-transfected cells. However, loss of Mcl-1 dramatically sensitized MET1 or MET4 cells for ABT737-mediated loss of MMP and cell death (Figures 6b and c). Gratifyingly the protection of MMP from ABT737 correlated to the efficiency of siRNA-mediated downregulation of Mcl-1 (Figure 6a). These findings demonstrate the indispensible role of Mcl-1 for the maintenance of functional mitochondrial integrity in SCC.

The ratio of Mcl-1 and Noxa determines ABT737 resistance in SCC cells. Transient knockdown of Mcl-1 in MET1 and MET4 cells (a–c) or knockdown of Noxa in A5RT3 and HaCaT cells (d–f) was performed with different Mcl-1-specific (a–c) or Noxa-specific siRNA (d–f) and control siRNA as described in Materials and Methods section. The successful knockdown of Mcl-1 (a) or Noxa (d) was determined by western blot analysis using the respective Noxa- and Mcl-1-specific antibodies. β-Tubulin serves as control for equal loading. One experiment of total of three independent experiments (b and e) or the quantitative summary of three independent experiments+S.E.M. is shown (a and d, above the western blot analysis). (c and f) One representative experiment of a total of three independent experiments (b and e) or the quantitative summary of three independent experiments together with S.E.M. is shown (c and f). (c) For the analysis of ABT737-induced cell death, MET1 and MET4 cells with knockdown of Mcl-1 (a and c) or HaCaT and A5RT3 cells with knockdown of Noxa (d and f) were stimulated with diluents (DMSO), Enantiomer (10 μM) or ABT737 (10 μM) for 18–24 h. Surviving attached cells were quantified with crystal violet assay. Summary of three to five independent experiments is shown+S.E.M.
To corroborate the findings for the Mcl-1/Noxa axis, we investigated the functional role of Noxa for ABT737 sensitivity. We thus suppressed Noxa in HaCaT and A5RT3 cells using four independent siRNAs (Figure 6d). Importantly, ABT737-mediated cell death (Figure 6f) and MMP (Figure 6e) were strongly repressed in a Noxa dose-dependent manner in these cell lines (Figure 6d). In turn, the low expression level of Noxa in MET cells could be a critical reason for their ABT737 resistance. Therefore, we next generated MET1 and MET4 cells that constitutively overexpress Noxa (Figure 7a). When we tested these cells for their sensitivity to ABT737, survival of Noxa-expressing cells was lower as compared with vector controls (Figure 7b). In line, ABT737-mediated loss of MMP (Figures 7c and d) was strongly enhanced in Noxa-expressing MET1 and MET4 cells. Our experiments firmly establish that the Mcl-1/Noxa ratio is of critical importance for regulation of ABT737 sensitivity in SCCs.

Overexpression of Noxa sensitizes MET1 and MET4 cells to ABT737-induced cell death. (a) Overexpression of Noxa in MET1 and MET4 cells was performed as described in the Materials and Methods section. The successful overexpression of Noxa (a) was determined by western blot analysis using Noxa-specific antibodies; β-tubulin serves as control for equal loading. (b) For analysis of ABT737-induced cell death, cells characterized in (a) were stimulated with diluents (DMSO), Enantiomer (10 μM) or ABT737 (10 μM) for 18–24 h. Surviving attached cells were quantified with crystal violet assay. Summary of three to five independent experiments is shown and S.E.M. is depicted. (c and d) For determination of loss of MMP, the respective genetically manipulated cells were stimulated for 8 h with Enantiomer (10 μM) or ABT737 (10 μM); loss of MMP was visualized by TMRE staining and FACS analysis. One representative experiment (c) or the quantitative summary of three independent experiments together with S.E.M. is shown (a and d)
Discussion
Defects in the execution of mitochondrial apoptosis are often involved in cancer development and progression.28 As pro-survival Bcl-2 proteins block stress-induced apoptosis in cancer, a plethora of therapeutics that mimic BH3-only proteins have been developed.39 ABT737 and the respective oral derivative ABT263 (Navitoclax) are powerful inhibitors of Bcl-2 proteins that suppress Bim/Bcl-2 complexes and, to a lesser extent, Bim/Bcl-XL or Bim/Bcl-w complexes.40 In this study, we investigated the therapeutic potential of BH3 mimetics for SCC of the skin. Using ABT737, we identified a number of SCC cell lines (HaCaT, A5RT3, DJM, SCC25 and others, compare Figure 1) that respond to single-agent ABT737 therapy by MOMP-mediated cell death induction (Figures 2a–d and 3a), caspase-independent release of mitochondria-derived proteins (Figure 3c), activation of caspase-3 (Figures 3c and d) and finally execution of cell death (Figures 2e and f). These findings show the indispensable function of Bcl-2, Bcl-XL and Bcl-w as resistance factors that protect from intrinsic pathway-induced cell death in a subset of SCC cells.
Combined treatment with ABT737 has previously been described as a successful strategy to overcome resistance and to restore cell death sensitivity to chemotherapeutic agents. For example, ABT737 sensitized ovarian cancer to carboplatin,41 glioblastoma to ionizing radiation42 and pancreatic, prostate, renal, lung and melanoma cells to TRAIL.43 Moreover, ABT737 sensitized SCC cells to cisplatin and etoposide.33 Based on these observations, we initially investigated the impact of ABT737 for DR-induced apoptosis in SCC cells (Figure 4 and Supplementary Figure 1). Surprisingly, and in contrast to previous reports, we were unable to increase cell death sensitivity to CD95L (Figures 4a–d), TRAIL or TNF (Supplementary Figure 1) by ABT737. These findings, together with the lack of protection by Bcl-2 in HaCaT cells, indicate that at least in a subset of SCC, protection of MOMP by Bcl-2 may not alter DL-induced cell death. If all SCC cell lines undergo DR-induced apoptosis independent from mitochondrial protection by Bcl-2, defining SCC as so-called type I cells35 requires a more extensive study. Nonetheless, our data indicate that combination therapies of Bcl-2 inhibitors and TRAIL receptor agonists32 need to be tested more vigorously. Our data indicate the need for suitable biomarkers that predict DR sensitivity in SCC. Examples of viable biomarkers, as identified by our study and previous work, could be XIAP24 or the caspase-8 inhibitor cFLIP.44
XIAP affects cell death sensitivity when triggering of either the intrinsic or extrinsic cell death pathway occurs.1 We found a dominant XIAP expression in ABT737-resistant MET cells, but not in HaCaT and A5RT3 cells.24, 25 Thus, XIAP was a likely candidate for apoptosis resistance to ABT737. However, suppression of XIAP in MET cells did not overcome ABT737 resistance, whereas it was critical to maintain resistance to DL-induced apoptosis (compare Figure 5). With these results, we demonstrate that mitochondria-mediated apoptosis is predominantly regulated upstream of caspase-3 in SCC. In contrast, DR-induced apoptosis is regulated by XIAP independent from a mitochondrial amplification loop. Therefore, we conclude that ABT737 does not prime SCC cells to DR-mediated cell death. Consequently, sensitivity to BH3 agonists must be regulated by other factors directly linked to mitochondrial signalling pathways.
In contrast to HaCaT and A5RT3 cells, MET cells and PK were resistant to ABT737-induced loss of MMP, caspase-3 activation and apoptosis. Resistance to apoptosis in general or specifically to ABT737-induced apoptosis was previously linked to Mcl-1 function in melanoma45 or SCC cells.37 To overcome Mcl-1 inhibition, Obatoclax was developed to break resistance to more selective BH3 mimetics.23 In our analysis, low concentrations of Obatoclax were sufficient to overcome ABT737 resistance in MET cells, as indicated by rapid loss of MMP and cell death (Supplementary Figure 2). These findings indicated an important role for Mcl-1 or A1. When we scrutinized the role of Mcl-1 for ABT737-induced apoptosis in SCC, we identified Noxa as a potential candidate for resistance, whereas Mcl-1 expression did not correlate with ABT737 sensitivity. Our knockdown experiments for Mcl-1 (Figures 6a–c) or for overexpression of Noxa in ABT737-resistant cells (Figure 7) corroborated these data and further established that the expression ratio of Mcl-1 and Noxa determines the sensitivity to ABT737. This was – gratifyingly – further supported by our data that demonstrated increased ABT737 resistance in cells with suppressed Noxa levels (Figures 6d–f). When Mcl-1 was lost or Noxa was reconstituted, cell death was dramatically increased in combination with ABT737, in line with recent studies in melanoma using bortezomib46 or in SCC using chemotherapeutics.33 Other have reported a mouse model that provoked suppression of Mcl-1 by caloric restriction (induced by 2-deoxyglycose). This was sufficient to sensitize to ABT737.47 S1, another BH3 mimetic that targets Mcl-1 and Bcl-2, also promotes upregulation of Noxa and thereby overcomes ABT737 resistance via increased endoplasmic reticulum stress.48 Together, Noxa-dependent inactivation of Mcl-1,49 suppression of Mcl-1 expression,50 p53-dependent regulation of Mcl-142 or caspase-dependent degradation of Mcl-151 are possible triggers that confer sensitivity to ABT737-induced apoptosis. Therefore, compounds that not only target multidomain Bcl-2 proteins but also reduce the Mcl-1/Noxa ratio could represent innovative therapeutic strategies.52
As outlined above, combination therapies may represent the most promising approach for cancer therapy with BH3 mimetics, as previously suggested.28 Inhibition of Bcl-2 and Bcl-XL in vitro and in vivo by gossypol (a naturally occurring Bcl-2 inhibitor) correlated with apoptosis induction.53 However, BH3 mimetics not only influence cell death, but also senescence,54 autophagy55 or ER stress responses.48 ABT263 has a serious dose-limiting side effect that restricts its clinical utility, as severe thrombocytopenia occurs as on-target effect of BCL-XL neutralization.20 ABT199, which selectively inhibits Bcl-2 while sparing Bcl-XL, has recently emerged as therapeutic advance in therapy-resistant haematological malignancies.20 As our study highlights the role of the Mcl-1/Noxa axis for SCC cell death resistance, other combination therapies appear promising. We suggest using BH3 mimetics with strong Mcl-1 inhibitory function. Examples of such BH3 mimetics include MIM156 or maritoclax (Marinopyrrol A).57 Alternatively, combination therapies that upregulate Noxa (such as Bortezomib) could overcome Mcl-1 inhibition. As indicated at least in a subset of our SCC cell lines, SCCs are strongly protected by Mcl-1 from BH3 mimetics. Thus agents such as MIM1 or maritoclax appear more promising than ABT737 or navitoclax, respectively. Other combination therapies may utilize AT101 (a gossypol isomer) for inhibition of both Bcl-2 and Mcl-1.58 Therefore, treatment strategies in SCCs will likely depend on the differential expression profile of Bcl-2 proteins and call for a more systematic study of the Bcl-2 proteins in patient-derived xenograft (PDX) models of SCC of the skin.
Materials and Methods
Materials
The following antibodies (Abs) were used for western blot analysis: Bak, Bcl-XL and cytochrome c (7H8.2C12; BD Bioscience Pharmingen, San Diego, CA, USA), Bcl-2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), Bax (B73520) and XIAP (H62120; Transduction Laboratories, San Diego, CA, USA), cytochrome c oxidase subunit II (part of COX IV complex, 12 C4F12; Life Technologies, Darmstadt, Germany), Mcl-1 (D35A5; New England Biolabs, Frankfurt, Germany) and NOXA (114C307; Abcam, Cambridge, UK). Polyclonal Abs to human Bid was kindly provided by X Wang (MD Anderson Cancer Center, Houston, TX, USA). Antiserum recognizing human HtrA2/Omi was kindly provided by Suzuki et al.,59 and Abs to Smac/DIABLO were kindly provided by Marion McFarlane (MRC, Leicester, UK). Abs to caspase-8 (C-15; kindly provided by PH Krammer, DKFZ, Heidelberg, Germany), caspase-3 (CPP32; generously provided by H Mehmet, Merck Frosst, Quebec, QC, Canada), PARP-1 (clone C-2-10; Biomol, Plymouth, PA, USA), rat Abs to cIAP1 (cellular inhibitor of apoptosis protein 1)60 and cIAP2 (cellular inhibitor of apoptosis protein 2),61 β-tubulin (clone 2.1) and BIM (B7929) were from Sigma (St. Louis, MO, USA). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit, goat anti-rat IgG and goat anti-mouse IgG Abs and HRP-conjugated goat anti-mouse IgG1, IgG2a and IgG2b were obtained from Southern Biotechnology Associates (Birmingham, AL, USA).
Cell culture
Primary human keratinocytes were cultured in CnT-07 medium from Cell-N-Tech (Bern, Switzerland) for a total of 2–6 passages after isolation. The spontaneously transformed keratinocyte line HaCaT and the derived nonmetastatic clones A5 and A5-RT1, as well as the metastatic clone A5-RT3,26 were kindly provided by Dr. Petra Boukamp (DKFZ, Heidelberg, Germany). MET1, MET2 and MET4 cells62 were provided by I Leigh (Skin Tumor Laboratory, Cancer Research UK, London, UK). Cell lines were exactly cultured as described previously.63, 64 DJM and SCC25 cells were cultured in complete DMEM medium with 10% FCS, 1% HEPES and 1% sodium pyruvate.
Substrates
For cell death induction and the analysis of quality and quantity of cell death, the following substrates were used: ABT737 and the ABT737 Enantiomer were kindly provided by Abbott Laboratories (North Chicago, IL, USA) and Obatoclax was purchased from Adooq Bioscience (Irvine, CA, USA). His-FLAG-TRAIL (HF-TRAIL) was produced as previously described.65 For expression of Fc-CD95L, we used constructs published elsewhere,66 kindly provided by P Schneider (Epalinges, Switzerland). One unit of Fc-CD95L was determined as a 1 : 500 dilution of the stock Fc-CD95L supernatant, and 1 unit/ml of Fc-CD95L supernatant was sufficient to kill 50% (LD50) of A375 melanoma cells, as previously described.67 Propidium iodide was obtained from Sigma (St. Louis, MO, USA), crystal violet was from VWR International (Radnor, PA, USA) and TMRE (tetramethyl rhodamine ethyl ester perchlorate) was from Molecular Probes (Eugene, OR, USA).
Retroviral infection
To overexpress Bcl-2, we used the Pinco retroviral vector as described previously.68 The cDNA of Noxa was subcloned into pCFG5 retroviral vector. Sequence-confirmed vectors were used for transduction of HaCaT (Bcl-2) or MET1 and MET4 (Noxa) cells, respectively. For virus production, the amphotrophic producer cell line ΦNX was transfected with 10 μg of the retroviral vectors by calcium phosphate precipitation. Cell culture supernatants containing viral particles were generated by incubation of producer cells with HaCaT medium (DMEM containing 10% FCS) or MET medium (DMEM containing 10% FCS, sodium pyruvate and HEPES buffer) overnight. Following filtration (45 μm filter, Schleicher & Schuell, Dassel, Germany), culture supernatant was added to HaCaT cells seeded in 6-well plates 24 h earlier in the presence of 1 μg/ml polybrene. After centrifugation for 3 h at 30°C, viral particles containing supernatants were replaced by fresh medium. After 3 days of puromycin selection of bulk-infected HaCaT or 14 days of zeocine selection of bulk-infected MET cultures, FACS analysis for GFP expression (always >95%, data not shown) and western blot analysis was performed on polyclonal cells to confirm ectopic expression or successful knockdown of the respective molecules. The empty retroviral vectors served as controls. Aliquots of cells were used for the experiments between passages two and six after initial characterization for all subsequent studies.
Stable siRNA expression
For stable knockdown of XIAP, shRNA targeting XIAP sequence 241 (5′-GTGGTAGTCCTGTTTCAGC-3′) corresponding to mRNA sequence and as a control a hyper random sequence (HRS) with no corresponding part in the human genome (5′-GATCATGTAGATACGCTCA-3′) were cloned into pRETRO-SUPER using HindIII and BglII restriction sites as previously described.69 The resulting vectors were transfected into the amphotrophic producer cell line exactly as outlined above. The retrovirus-containing supernatant was used to infect MET1 and MET4 cells with HRS shRNA or XIAP shRNA, respectively, and selected with puromycin (1 μg/ml; Sigma, Taufkirchen, Germany) for 3 days to obtain puromycin-resistant bulk-infected cultures for further analysis. FACS analysis of GFP expression (always >95%, data not shown) and western blot analysis was performed on polyclonal cells to confirm successful knockdown. Aliquots of cells were used for cytotoxic assays and biochemical characterization between passages two and six following the antibiotic selection.
Lipofectamine-mediated transfection of Mcl-1- or Noxa-specific siRNA in HaCaT or A5RT3 cells
For transient knockdown of Mcl-1 or Noxa expression, Mcl-1-specific siRNA species ((HS Mcl-1 1–6 (#1); 1–2 (#2; 1–8 (#3); and 1–11 (#4) Flexi Tubes)) or NOXA-specific siRNA species ((HS PMAIP 1–1 (#1); 1–4 (#2); 1–5 (#3); and 1–6 (#4) Flexi Tubes), or a negative control siRNA (AllStar negative control siRNA) from Qiagen (Valencia, CA, USA) were used. In brief, 250 μl Opti-MEM (Life Technologies, Gibco, Darmstadt, Germany) containing 5 μl of respective siRNA duplex (20 μM) was mixed with 250 μl Opti-MEM containing 5 μl Lipofectamine (Life Technologies, Invitrogen, Darmstadt, Germany) for 25 min by room temperature. The formed siRNA containing liposomes were transferred onto adherent MET cells in a 6-well plate with previously changed medium and incubated for 24 h by 37°C. After 24 h, the medium was changed and cells were incubated for additional 24 h, harvested and analysed for Mcl-1 or Noxa knockdown. In parallel, the respective functional assays as depicted in the figures were performed.
Western blot analysis
Cell lysates were prepared as described previously70 and 5 μg of total cellular proteins were separated by SDS-PAGE on 4–12% gradient gels (Invitrogen, Karlsruhe, Germany) followed by transfer to nitrocellulose or PVDF membranes. Blocking of membranes and individual incubation with primary and appropriate secondary Abs was performed as recommended by the companies respectively. Bands were visualized with ECL detection kits (Amersham, Freiburg, Germany).
Cytotoxicity assays
Analysis of living attached cell by crystal violet staining
Crystal violet staining of attached, living cells was performed 18–24 h after stimulation with the indicated concentrations of ABT737, Obatoclax, TRAIL, CD95L, TNF alone or the respective combinations in 96-well plates. We routinely use triplicate wells per condition as described previously.67 The optical density (OD) of control cultures was normalized to 100% and compared with stimulated cells. In case of combined stimulation of ABT737 or DMSO or Enantiomer together with CD95L (Figure 4), TRAIL (Supplementary Figures 1a–d) or TNF (Supplementary Figure 1e), the spontaneous cytotoxic effect of single diluents and substrates was subtracted from each costimulation to solely show specific DL-induced cell death. For statistical analysis the S.E.M. was determined for three to five independent experiments of each cell line and stimulatory condition.
Hypodiploidy analysis
For quantify diploidic DNA content, cells were stimulated with the indicated concentrations of ABT737 for 24 or 48 h. Cells floating in the supernatant as well as attached cells were harvested, collected, washed with cold PBS and resuspended in buffer N (sodium citrate 0.1% (w/v), Triton X-100 0.1% (v/v) and propidium iodide 50 μg/ml). Cells were kept in the dark at 4°C for 36–48 h and then diploidy was measured by FACScan analysis and summarized with FCS Express version 3 program (De Novo software, Glendale, CA, USA).
MMP analysis
For determination of MMP, PK, MET1, MET4, HaCaT and A5RT3 cells were harvested by trypsinization upon stimulation with ABT737, Enantiomer or diluents after indicated time points. Cells were washed with cold PBS and resuspended with respective medium containing TMRE at a final concentration of 40 nM. Following a 10-min incubation period at 37°C in the dark, MMP was determined by FACScan analysis and summarized with FCS Express version 3 programs (De Novo software). Nonstimulated cells served as control for TMRE staining.
Analysis of colony formation
For colony formation assay, 5 × 104 cells of parental A5RT3 cells were seeded per well in a 12-well plate. After 24 h of incubation, adhering cells were either nonstimulated or separately prestimulated with zVAD-fmk (10 μM, for 1 h) or dimethyl sulphoxide (DMSO) or ABT737 or Enantiomer of ABT7373 (5 μM, for 30 min) alone or in respective indicated combination of the compounds for 6 h. At that time, medium was removed, cells were washed two times with sterile PBS and complete medium was added. Cells were cultured for 4 or 7 days, and subsequently colonies of viable cells were stained by crystal violet as indicated above.
Cell fractionation to determine release of mitochondrial-derived proteins into cytoplasm
We used the selective lysis of digitonin-based buffers to identify the release of mitochondrial proteins such as cytochrome c, HtrA2/omi and smac/Diablo71 with modifications as published previously.25 Next, 1 × 106 A5RT3 cells were allowed to adhere overnight. Cells were either stimulated with ABT737 (5μM) or the respective Enantiomer, followed by Trypsin-mediated harvesting after the indicated time points. Cells were washed with cold PBS, centrifuged and resuspended in 60 μl PBS. Resuspended cells were lysed with the same volume of digitonin lysis buffer (150 μg Digitonin (Fluka 37006, Sigma Aldrich, Munich, Germany), 1 ml 500 mM Sucrose) for 10 min at room temperature followed by centrifugation (3000 r.p.m.) for 5 min at 4°C. The resulting supernatant was centrifuged again (14 000 r.p.m.) for 30 min at 4°C, and the same volume of the supernatants were transferred into another tube and used to isolate the cytoplasmic fraction. The remaining cell pellet that contains the mitochondrial fraction was resuspended and lysed with 100 μl DISC-Lysis buffer ((30 mM Tris-HCl (pH 7.0), 120 mM NaCl, 10% Glycerol, 1% Triton, complete protease inhibitor cocktail (Roche, Mannheim, Germany)) for 30 min on ice followed by centrifugation for 20 min by 14 000 r.p.m. Same volumes of supernatants were transferred into a fresh tube (and designated the mitochondrial fraction). Both cell fractions were analysed by western blot assay as described above for presence of mitochondria-based proteins (cytochrome c, smac and HtrA2). Purity of cytoplasmic fraction was assessed by confirming the absence of cytochrome oxidase (COXIV).
Abbreviations
- SCC:
-
squamous cell carcinoma
- Bcl-2:
-
B-cell lymphoma 2 protein
- BH:
-
Bcl-2 homology domains
- MMP:
-
mitochondrial membrane potential
- XIAP:
-
X-linked inhibitor of apoptosis protein
- smac/DIABLO:
-
second mitochondria-derived activator of caspases/direct IAP binding protein with low pI
- DR:
-
death receptor
- Mcl-1:
-
induced myeloid leukaemia cell differentiation protein
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- CD95:
-
cluster of differentiation 95
- DL:
-
death ligands
- FADD:
-
Fas-associated protein with death domain
- PARP-1:
-
poly-ADP-Ribose polymerase-1
- MOMP:
-
mitochondria outer membrane permeability
- Bax:
-
Bcl-2-associated X protein
- Bak:
-
Bcl-2 homologous antagonist/killer
- Bid:
-
BH3 interacting-domain death agonist
- Bim:
-
Bcl-2-like protein 11
- Bad:
-
Bcl-2-associated death promoter
- NOXA:
-
phorbol-12-myristate-13-acetate-induced protein 1
- Puma:
-
p53 upregulated modulator of apoptosis
- HNSCC:
-
head and neck SCC
- Bcl-XL:
-
B-cell lymphoma-extra large
- Bcl-w:
-
Bcl-2-like protein 2
- A1:
-
Bcl-2-related protein A1
- TNF:
-
tumour necrosis factor
- DISC:
-
death-inducing signalling complex
- cFLIP:
-
CASP8 and FADD-like apoptosis regulator
- PDX:
-
patient-derived xenograft
- cIAP1/2:
-
cellular inhibitor of apoptosis protein 1/2
- DMSO:
-
dimethyl sulphoxide
- TMRE:
-
tetramethylrhodamine ethyl ester
- HRS:
-
hyper random sequence
References
Kaufmann T, Strasser A, Jost PJ . Fas death receptor signalling: roles of Bid and XIAP. Cell Death Differ 2012; 19: 42–50.
Sprick MR, Weigand MA, Rieser E, Rauch CT, Juo P, Blenis J et al. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity 2000; 12: 599–609.
Kroemer G, Galluzzi L, Brenner C . Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007; 87: 99–163.
Deveraux QL, Takahashi R, Salvesen GS, Reed JC . X-linked IAP is a direct inhibitor of cell-death proteases. Nature 1997; 388: 300–304.
Czabotar PE, Lessene G, Strasser A, Adams JM . Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 2014; 15: 49–63.
Pagliari LJ, Kuwana T, Bonzon C, Newmeyer DD, Tu S, Beere HM et al. The multidomain proapoptotic molecules Bax and Bak are directly activated by heat. Proc Natl Acad Sci USA 2005; 102: 17975–17980.
Nie C, Tian C, Zhao L, Petit PX, Mehrpour M, Chen Q . Cysteine 62 of Bax is critical for its conformational activation and its proapoptotic activity in response to H2O2-induced apoptosis. J Biol Chem 2008; 283: 15359–15369.
Westphal D, Dewson G, Czabotar PE, Kluck RM . Molecular biology of Bax and Bak activation and action. Biochim Biophys Acta 2011; 1813: 521–531.
Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD . The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 1997; 275: 1132–1136.
Wang C, Youle RJ . The role of mitochondria in apoptosis*. Annu Rev Genet 2009; 43: 95–118.
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997; 91: 479–489.
Zou H, Henzel WJ, Liu X, Lutschg A, Wang X . Apaf-1a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997; 90: 405–413.
Hardwick JM, Youle RJ . SnapShot: BCL-2 proteins. Cell 2009; 138: 404 404.e1.
Chipuk JE, Green DR . How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol 2008; 18: 157–164.
Bachmann PS, Piazza RG, Janes ME, Wong NC, Davies C, Mogavero A et al. Epigenetic silencing of BIM in glucocorticoid poor-responsive pediatric acute lymphoblastic leukemia, and its reversal by histone deacetylase inhibition. Blood 2010; 116: 3013–3022.
Drenning SD, Marcovitch AJ, Johnson DE, Melhem MF, Tweardy DJ, Grandis JR . Bcl-2 but not Bax expression is associated with apoptosis in normal and transformed squamous epithelium. Clin Cancer Res 1998; 4: 2913–2921.
Gallo O, Boddi V, Calzolari A, Simonetti L, Trovati M, Bianchi S . bcl-2 protein expression correlates with recurrence and survival in early stage head and neck cancer treated by radiotherapy. Clin Cancer Res 1996; 2: 261–267.
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435: 677–681.
Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 2008; 68: 3421–3428.
Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 2013; 19: 202–208.
Wang B, Ni Z, Dai X, Qin L, Li X, Xu L et al. The Bcl-2/xL inhibitor ABT-263 increases the stability of Mcl-1 mRNA and protein in hepatocellular carcinoma cells. Mol Cancer 2014; 13: 98.
Albershardt TC, Salerni BL, Soderquist RS, Bates DJ, Pletnev AA, Kisselev AF et al. Multiple BH3 mimetics antagonize antiapoptotic MCL1 protein by inducing the endoplasmic reticulum stress response and up-regulating BH3-only protein NOXA. J Biol Chem 2011; 286: 24882–24895.
Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci USA 2007; 104: 19512–19517.
Geserick P, Hupe M, Moulin M, Wong WW, Feoktistova M, Kellert B et al. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J Cell Biol 2009; 187: 1037–1054.
Leverkus M, Sprick MR, Wachter T, Mengling T, Baumann B, Serfling E et al. Proteasome inhibition results in TRAIL sensitization of primary keratinocytes by removing the resistance-mediating block of effector caspase maturation. Mol Cell Biol 2003; 23: 777–790.
Mueller MM, Peter W, Mappes M, Huelsen A, Steinbauer H, Boukamp P et al. Tumor progression of skin carcinoma cells in vivo promoted by clonal selection, mutagenesis, and autocrine growth regulation by granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor. Am J Pathol 2001; 159: 1567–1579.
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19: 107–120.
Fulda S, Galluzzi L, Kroemer G . Targeting mitochondria for cancer therapy. Nat Rev Drug Discov 2010; 9: 447–464.
Tait SW, Green DR . Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 2010; 11: 621–632.
Ricci JE, Munoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N et al. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 2004; 117: 773–786.
Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD et al. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol 1999; 144: 281–292.
Song JH, Kandasamy K, Kraft AS . ABT-737 induces expression of the death receptor 5 and sensitizes human cancer cells to TRAIL-induced apoptosis. J Biol Chem 2008; 283: 25003–25013.
Vogler M, Weber K, Dinsdale D, Schmitz I, Schulze-Osthoff K, Dyer MJ et al. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ 2009; 16: 1030–1039.
Certo M, Del GMV, Nishino M, Wei G, Korsmeyer S, Armstrong SA et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006; 9: 351–365.
Barnhart BC, Alappat EC, Peter ME . The CD95 type I/type II model. Semin Immunol 2003; 15: 185–193.
Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 2009; 460: 1035–1039.
He L, Torres-Lockhart K, Forster N, Ramakrishnan S, Greninger P, Garnett MJ et al. Mcl-1 and FBW7 control a dominant survival pathway underlying HDAC and Bcl-2 inhibitor synergy in squamous cell carcinoma. Cancer Discov 2013; 3: 324–337.
Nakajima W, Hicks MA, Tanaka N, Krystal GW, Harada H . Noxa determines localization and stability of MCL-1 and consequently ABT-737 sensitivity in small cell lung cancer. Cell Death Dis 2014; 5: e1052.
Billard C . BH3 mimetics: status of the field and new developments. Mol Cancer Ther 2013; 12: 1691–1700.
Merino D, Khaw SL, Glaser SP, Anderson DJ, Belmont LD, Wong C et al. Bcl-2, Bcl-x(L), and Bcl-w are not equivalent targets of ABT-737 and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood 2012; 119: 5807–5816.
Witham J, Valenti MR, De-Haven-Brandon AK, Vidot S, Eccles SA, Kaye SB et al. The Bcl-2/Bcl-XL family inhibitor ABT-737 sensitizes ovarian cancer cells to carboplatin. Clin Cancer Res 2007; 13: 7191–7198.
Tagscherer KE, Fassl A, Sinkovic T, Combs SE, Roth W . p53-dependent regulation of Mcl-1 contributes to synergistic cell death by ionizing radiation and the Bcl-2/Bcl-XL inhibitor ABT-737. Apoptosis 2012; 17: 187–199.
Huang S, Sinicrope FA . BH3 mimetic ABT-737 potentiates TRAIL-mediated apoptotic signaling by unsequestering Bim and Bak in human pancreatic cancer cells. Cancer Res 2008; 68: 2944–2951.
Leverkus M, Diessenbacher P, Geserick P . FLIP ing the coin? Death receptor-mediated signals during skin tumorigenesis. Exp Dermatol 2008; 17: 614–622.
Boisvert-Adamo K, Longmate W, Abel EV, Aplin AE . Mcl-1 is required for melanoma cell resistance to anoikis. Mol Cancer Res 2009; 7: 549–556.
Reuland SN, Goldstein NB, Partyka KA, Smith S, Luo Y, Fujita M et al. ABT-737 synergizes with Bortezomib to kill melanoma cells. Biol Open 2012; 1: 92–100.
Meynet O, Zunino B, Happo L, Pradelli LA, Chiche J, Jacquin MA et al. Caloric restriction modulates Mcl-1 expression and sensitizes lymphomas to BH3 mimetic in mice. Blood 2013; 122: 2402–2411.
Soderquist R, Pletnev AA, Danilov AV, Eastman A . The putative BH3 mimetic S1 sensitizes leukemia to ABT-737 by increasing reactive oxygen species, inducing endoplasmic reticulum stress, and upregulating the BH3-only protein NOXA. Apoptosis 2014; 19: 201–209.
Okumura K, Huang S, Sinicrope FA . Induction of Noxa sensitizes human colorectal cancer cells expressing Mcl-1 to the small-molecule Bcl-2/Bcl-xL inhibitor, ABT-737. Clin Cancer Res 2008; 14: 8132–8142.
Labi V, Grespi F, Baumgartner F, Villunger A . Targeting the Bcl-2-regulated apoptosis pathway by BH3 mimetics: a breakthrough in anticancer therapy? Cell Death Differ 2008; 15: 977–987.
Miller LA, Goldstein NB, Johannes WU, Walton CH, Fujita M, Norris DA et al. BH3 mimetic ABT-737 and a proteasome inhibitor synergistically kill melanomas through Noxa-dependent apoptosis. J Invest Dermatol 2009; 129: 964–971.
Lucas KM, Mohana-Kumaran N, Lau D, Zhang XD, Hersey P, Huang DC et al. Modulation of NOXA and MCL-1 as a strategy for sensitizing melanoma cells to the BH3-mimetic ABT-737. Clin Cancer Res 2012; 18: 783–795.
Bauer JA, Trask DK, Kumar B, Los G, Castro J, Lee JS et al. Reversal of cisplatin resistance with a BH3 mimetic, (-)-gossypol, in head and neck cancer cells: role of wild-type p53 and Bcl-xL. Mol. Cancer Ther 2005l; 4: 1096–1104.
Song JH, Kandasamy K, Zemskova M, Lin YW, Kraft AS . The BH3 mimetic ABT-737 induces cancer cell senescence. Cancer Res 2011; 71: 506–515.
Malik SA, Orhon I, Morselli E, Criollo A, Shen S, Marino G et al. BH3 mimetics activate multiple pro-autophagic pathways. Oncogene 2011; 30: 3918–3929.
Cohen NA, Stewart ML, Gavathiotis E, Tepper JL, Bruekner SR, Koss B et al. A competitive stapled peptide screen identifies a selective small molecule that overcomes MCL-1-dependent leukemia cell survival. Chem Biol 2012; 19: 1175–1186.
Doi K, Li R, Sung SS, Wu H, Liu Y, Manieri W et al. Discovery of marinopyrrole A (maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. J Biol Chem 2012; 287: 10224–10235.
Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M . Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J Med Chem 2003; 46: 4259–4264.
Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R . A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell 2001; 8: 613–621.
Silke J, Kratina T, Chu D, Ekert PG, Day CL, Pakusch M et al. Determination of cell survival by RING-mediated regulation of inhibitor of apoptosis (IAP) protein abundance. Proc Natl Acad Sci USA 2005; 102: 16182–16187.
Vince JE, Pantaki D, Feltham R, Mace PD, Cordier SM, Schmukle AC et al. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (tnf) to efficiently activate nf-{kappa}b and to prevent tnf-induced apoptosis. J Biol Chem 2009; 284: 35906–35915.
Popp S, Waltering S, Holtgreve-Grez H, Jauch A, Proby C, Leigh IM et al. Genetic characterization of a human skin carcinoma progression model: from primary tumor to metastasis. J Invest Dermatol 2000; 115: 1095–1103.
Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE . Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol 1988; 106: 761–771.
Proby CM, Purdie KJ, Sexton CJ, Purkis P, Navsaria HA, Stables JN et al. Spontaneous keratinocyte cell lines representing early and advanced stages of malignant transformation of the epidermis. Exp Dermatol 2000; 9: 104–117.
Diessenbacher P, Hupe M, Sprick MR, Kerstan A, Geserick P, Haas TL et al. NF-kappaB inhibition reveals differential mechanisms of TNF versus TRAIL-induced apoptosis upstream or at the level of caspase-8 activation independent of cIAP2. J Invest Dermatol 2008; 128: 1134–1147.
Bossen C, Ingold K, Tardivel A, Bodmer JL, Gaide O, Hertig S et al. Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human. J Biol Chem 2006; 281: 13964–13971.
Geserick P, Drewniok C, Hupe M, Haas TL, Diessenbacher P, Sprick MR et al. Suppression of cFLIP is sufficient to sensitize human melanoma cells to TRAIL- and CD95L-mediated apoptosis. Oncogene 2008; 27: 3211–3220.
Stassi G, Di LD, Todaro M, Zeuner A, Ricci-Vitiani L, Stoppacciaro A et al. Control of target cell survival in thyroid autoimmunity by T helper cytokines via regulation of apoptotic proteins. Nat Immunol 2000; 1: 483–488.
Vogler M, Walczak H, Stadel D, Haas TL, Genze F, Jovanovic M et al. Targeting XIAP bypasses Bcl-2-mediated resistance to TRAIL and cooperates with TRAIL to suppress pancreatic cancer growth in vitro and in vivo. Cancer Res 2008; 68: 7956–7965.
Kavuri SM, Geserick P, Berg D, Dimitrova DP, Feoktistova M, Siegmund D et al. Cellular FLICE-inhibitory protein (cFLIP) isoforms block. J Biol Chem 2011; 286: 16631–16646.
Single B, Leist M, Nicotera P . Simultaneous release of adenylate kinase and cytochrome c in cell death. Cell Death Differ 1998; 5: 1001–1003.
Acknowledgements
We thank P Boukamp for providing HaCaT and HaCaT-derived cell lines. We are indebted to PH Krammer, H Mehmet, M MacFarlane, G Stassi, and S Fulda for providing antibodies or constructs. ML is supported by grants from the DFG (Le 953/6-1 and Le 953/8-1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by T Brunner
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Geserick, P., Wang, J., Feoktistova, M. et al. The ratio of Mcl-1 and Noxa determines ABT737 resistance in squamous cell carcinoma of the skin. Cell Death Dis 5, e1412 (2014). https://doi.org/10.1038/cddis.2014.379
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.379
