
Abstract
We examined the roles of Notch1 signaling and its cross-talk with other signaling pathways, including p53 and phosphatidylinositol-3-kinase (PI3K)/Akt, in cadmium-induced cellular damage in HK-2 human renal proximal tubular epithelial cells. Following exposure to cadmium chloride (CdCl2), the level of Notch intracellular domain (NICD), the cleaved form of the Notch1 receptor, was increased and accumulated in the nuclear fraction. Knockdown of Notch1 with siRNA or treatment with the γ-secretase inhibitor, DAPT (N-[N-(3,5-difluorophenacetyl-L-alanyl)]-S-phenylglycine t-butyl ester), prevented CdCl2-induced morphological change of HK-2 cells and reduction of cell viability. Knockdown of Jagged1 or Jagged2, the ligands of the Notch1 receptor, partially suppressed cadmium cytotoxicity. Inhibition of p53 activity with pifithrin-α or inhibition of PI3K with LY294002 suppressed CdCl2-induced cellular damage and elevation of Notch1-NICD. In addition, treatment with the epidermal growth factor receptor (EGFR) inhibitor, AG1478, and the insulin-like growth factor-1 receptor inhibitor, PPP, suppressed both Notch1-NICD accumulation and Akt phosphorylation in HK-2 cells exposed to CdCl2. However, knockdown of Notch1 did not affect CdCl2-induced p53 accumulation and phosphorylation but suppressed phosphorylation of EGFR, Akt, and p70 S6 kinase. Depletion of Notch1 suppressed CdCl2-induced reduction of E-cadherin expression and elevation of Snail expression. Furthermore, treatment with SB216763, an inhibitor of glycogen synthase kinase-3, suppressed the potency of LY294002 treatment to reduce Snail expression in HK-2 cells exposed to CdCl2. Knockdown of Snail with siRNA partially prevented HK-2 cells from CdCl2-induced reduction of E-cadherin expression and cellular damage. These results suggest that cadmium exposure induces the activation of Notch1 signaling in renal proximal tubular cells with cooperative activation by the p53 and PI3K/Akt signaling pathways; the resultant expression of Snail, a repressor of E-cadherin expression, might lead to cellular damage by decreasing cell–cell adhesion.
Similar content being viewed by others


Main
Cadmium is an occupational and environmental pollutant that damages various organs, especially renal proximal tubular cells.1 One of the primary actions of cadmium on epithelial cells is the disruption of cadherin-mediated cell–cell adhesion.2 Following cadmium exposure, E-cadherin and N-cadherin translocate from adhering junctions in the proximal tubule epithelium.3, 4, 5 In a rat renal proximal tubular cell model, cadmium induced a reduction of total cellular E-cadherin protein content,6 indicating that a loss of cadherin-mediated cell–cell adhesion might contribute to this cellular damage. Identification of the signaling molecules that regulate expression of E-cadherin in renal proximal tubular cells is important for the understanding of the molecular mechanisms responsible for cadmium-induced cellular damage.
The Notch pathway is an evolutionally conserved signaling pathway implicated in a wide variety of processes, including cell-fate determination, cell differentiation, proliferation, and cell death.7 In mammals, there are four Notch receptors (Notch1–4). Activation of Notch signaling requires the interaction of the Notch receptor with their ligands such as Jagged1 and 2 and Delta-like 1, 3, and 4 on neighboring cells. Ligand binding leads to sequential cleavages by ADAM (a-disinterring-and-metalloprotease) and the γ-secretase complex in Notch and results in the release of Notch intracellular domain (NICD) from the membrane. NICD translocates to the nucleus to activate specific target genes via the DNA-binding adaptor protein CBF1-Suppressor of Hairless.8, 9, 10, 11 The Notch signaling regulates gene expression in a highly context- and cell type-dependent manner.9, 12 In human normal kidney tubule epithelial cell lines, the levels of expression of Notch1 and Notch4 are high13 and the zinc finger transcription factor Snail, a repressor of E-cadherin expression,14, 15 has been shown to be positively regulated by Notch1 signaling in these cells.16 However, the effects of cadmium exposure on Notch1 signaling in renal proximal tubules and its possible involvement in Snail expression and cellular damage have not yet been examined.
In this study, we first examined whether cadmium chloride (CdCl2) exposure activates the Notch1 signaling pathway in HK-2 cells, a human renal proximal tubular epithelial cell line, by determining the accumulation of Notch1-NICD. We then used siRNAs against human Notch1, Jagged1, and Jagged2 or the γ-secretase inhibitor, DAPT (N-[N-(3,5-difluorophenacetyl-L-alanyl)]-S-phenylglycine t-butyl ester) to examine the role of Notch1 signaling on cellular damage in HK-2 cells after prolonged exposure to CdCl2. Evidence indicates that the Notch signaling pathway could interact with other signaling pathways, including p5317, 18, 19, 20 and phosphatidylinositol-3-kinase (PI3K)/Akt (also known as protein kinase B),17, 21, 22, 23, 24, 25, 26, 27, 28 both of which have been shown to be activated by cadmium exposure.29, 30 Therefore, we examined the possible interplay between Notch1 and the p53 or PI3K/Akt signaling pathways in HK-2 cells exposed to CdCl2. Finally, we examined the effects of the Notch1 and PI3K/Akt/glycogen synthase kinase-3 (GSK-3) signaling pathways on the expression of Snail, the upstream regulator of E-cadherin expression, following exposure to CdCl2.
Results
Involvement of Notch1 signaling in CdCl2-induced cellular damage in HK-2 cells
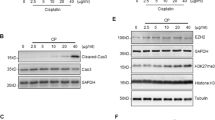
When HK-2 cells were incubated with 20 μM CdCl2, the levels of Notch1-NICD and Notch4-NICD increased after 6 or 9 h, without changing the level of actin (Figure 1a and Supplementary Figure S1), and Notch1-NICD accumulated in the nuclear fraction in a time-dependent manner (Figure 1b). Treatment of HK-2 cells with 1–50 μM CdCl2 for 12 h showed a dose-dependent increase in the level of Notch1-NICD (Supplementary Figure S2). However, the level of the transmembrane subunit including the intracellular region (Notch1-NTM) increased only slightly after 6 h and decreased after 12 h of exposure to 20 μM CdCl2 (Figure 1a and Supplementary Figure S2). The level of the hairy and enhancer of split-1 (HES1) transcription factor, a marker of Notch pathway activity,9, 31 increased at 3 h following exposure to CdCl2 (Supplementary Figure S3). To determine the pathological significance of Notch1 signaling, we knocked down Notch1 using siRNAs (Figure 1c) and then compared cellular damage in normal and Notch1-deficient HK-2 cells following exposure to CdCl2 (Figures 1d and e). Because cell viability of HK-2 cells exposed to 20 or 50 μM CdCl2 for 16 h was not reduced markedly,32 cellular damage was determined after incubation with 20 μM CdCl2 for >24 h. In comparison with cells transfected with negative control siRNA, transfection with siRNAs targeted against the human Notch1 gene (siRNA-1 and siRNA-2) almost completely abolished both Notch1-NICD and Notch1-NTM expression in HK-2 cells exposed to CdCl2 (Figure 1c, lanes 2 versus 4 or 6). Exposure to 20 μM CdCl2 for 24 h caused HK-2 cells to separate from each other and change from epithelioid to rounded shape (Figure 1d, top panel); knockdown of Notch1 with either siRNA-1 or siRNA-2 dramatically suppressed CdCl2-induced cellular damage (Figure 1d, middle and bottom panels). We aimed to quantify these cellular damage by performing trypan blue exclusion assay. Exposure of HK-2 cells to 20 μM CdCl2 for 30 h increased the ratio of dead cells relative to controls (Figure 1e), while knockdown of Notch1 with either siRNA-1 or siRNA-2 markedly suppressed CdCl2-induced cell death (Figure 1e, P<0.01 for siRNA-1, P<0.05 for siRNA-2). We also found that knockdown of Notch1 suppressed the CdCl2-induced increase in the number of propidium iodide-positive and annexin-V/propidium iodide double-positive cells, as well as the increase in the level of cytoplasmic nucleosomes in comparison with cells transfected with control siRNA (Supplementary Figure S4), suggesting the involvement of Notch1 signaling in apoptotic and non-apoptotic cell death of HK-2 cells.

Involvement of Notch1 signaling in CdCl2-induced cellular damage in HK-2 cells. (a and b) Cells were incubated with 20 μM CdCl2 (Cd) for the indicated time. The untreated control is labeled 0 h. Cell lysates were subjected to western blotting using antibodies against Notch1-NICD, Nohch1-NTM, and actin (a). Equal amounts of protein (20 μg) in nuclear and cytoplasmic extracts were subjected to western blotting using antibodies against Notch1-NICD, MEK1/2, lamin A/C, and actin. MEK1 and lamin A/C served as a loading control for cytoplasmic and nuclear extracts, respectively (b). (c–e) Cells transfected with control siRNA, Notch1 siRNA-1, or Notch1 siRNA-2 were incubated with or without 20 μM CdCl2 (Cd) for 12 h (c), 24 h (d), or 30 h (e). Cell lysates were subjected to western blotting using antibodies against Notch1-NICD, Notch1-NTM, and actin (c). Phase-contrast micrographs were taken (d). The viability of cells was determined by trypan blue exclusion assay. Each value is the percentage of trypan blue-positive cells and reflects the mean±S.D. of three experiments with duplicate assays in each experiment. *P<0.05, **P<0.01 versus CdCl2-treated cells transfected with control siRNA (e). (f–h) Cells were incubated with 0.1% DMSO or 40 μM DAPT for 1 h and then incubated with or without 20 μM CdCl2 (Cd) for 12 h (f) or 30 h (g and h). Cell lysates were subjected to western blotting using antibodies against Notch1-NICD, Notch1-NTM, and actin (f). Phase-contrast micrographs were taken (g). The viability of cells was determined by trypan blue exclusion assay. Each value is the percentage of trypan blue-positive cells and reflects the mean±S.D. of three experiments with duplicate assays in each experiment. **P<0.01 versus CdCl2-treated cells incubated with DMSO (h). Immunoblots shown are representative of at least three independent experiments
Next, we examined the role of γ-secretase in CdCl2-induced accumulation of Notch1-NICD and cellular damage in HK-2 cells. Treatment with DAPT, an inhibitor of γ-secretase, markedly suppressed the level of Notch1-NICD and slightly elevated Notch1-NTM level in HK-2 cells exposed to 20 μM CdCl2 for 12 h (Figure 1f, lanes 2 versus 4). Furthermore, DAPT suppressed both the CdCl2-induced morphological change (Figure 1g, lower panel) and the increase in the ratio of dead cells (Figure 1h, P<0.01). Consistent with these findings, hyperactivation of Notch1 signaling with the Notch1 extracellular-domain-deleted construct, Notch1 ΔEMV,33 resulted in the upregulation of HES1 expression and the reduction of cell viability in another type of kidney cells, human embryonic kidney 293T cells, following exposure to CdCl2 (Supplementary Figure S5). Collectively, these results indicate that cadmium exposure activates the Notch1 signaling pathway and that NICD cleaved by γ-secretase induces cellular damage in HK-2 cells.
Involvement of Jagged1 and Jagged2 in CdCl2-induced cellular damage in HK-2 cells
Expression of the Notch ligands Delta-like1, 3, and 4 was not definitively detected in HK-2 cells (data not shown). Therefore, we examined the role of Jagged1 and Jagged2 in Notch1 signaling and subsequent cellular damage in HK-2 cells exposed to CdCl2; exposure to CdCl2 reduced the levels of full-length Jagged1 and Jagged2 in a time-dependent manner (Figure 2a). Transfection of HK-2 cells with siRNAs targeted against human Jagged1 and Jagged2 almost completely abolished the expression of Jagged1 (Figure 2b, left, lanes 1 versus 3) and Jagged2 (right, lanes 1 versus 3), respectively. In addition, CdCl2-induced elevation of Notch1-NICD levels was markedly suppressed by silencing of either Jagged1 (Figure 2b, left, lanes 2 versus 4) or Jagged2 (right, lanes 2 versus 4). The morphological changes at 12 h (Figure 2c) and increase in the ratio of dead cells at 30 h after exposure to 20 μM CdCl2 (Figure 2d) were partially suppressed by knockdown of these Notch ligands (P>0.05 for Jagged1, P<0.05 for Jagged2). These results suggest that Jagged1 and Jagged2 are ligands that are responsible for the activation of Notch1 signaling leading to cellular damage in HK-2 cells exposed to CdCl2.

Jagged1 and Jagged2 are involved in CdCl2-induced cellular damage in HK-2 cells. (a) Cells were incubated with or without 20 μM CdCl2 (Cd) for the indicated time. Cell lysates were subjected to western blotting using antibodies against Jagged1, Jagged2, and actin. (b–d) Cells transfected with control siRNA, Jagged1 siRNA, or Jagged2 siRNA were incubated with or without 20 μM CdCl2 (Cd) for 12 h (b), 12 h (c), or 30 h (d). Cell lysates were subjected to western blotting using antibodies against Notch1-NICD, Jagged1 (left), Jagged2 (right), and actin (b). Phase-contrast micrographs were taken (c). The viability of cells was determined by trypan blue exclusion assay. Each value is the percentage of trypan blue-positive cells and reflects the mean±S.D. of three experiments with duplicate assays in each experiment. *P<0.05 versus CdCl2-treated cells transfected with control siRNA (d). Immunoblots shown are representative of at least three independent experiments
Modulation of Notch1 signaling by p53 in HK-2 cells exposed to CdCl2
It has been reported that the p53 tumor suppressor interacts with the Notch1 signaling pathway via transcriptional activation of the Notch1 gene18 or members of the γ-secretase complex.19, 20 Therefore, we examined the possible interplay between p53 and the Notch1 signaling pathway in HK-2 cells exposed to CdCl2. Consistent with our previous findings,29 CdCl2 exposure resulted in both accumulation of p53 protein and its phosphorylation at Ser15, required for p53 stability,34 in HK-2 cells (Figure 3a). In addition, the level of p53 protein in the nuclear fraction increased following exposure to CdCl2 (Figure 3b). When HK-2 cells were treated with pifithrin-α, a specific inhibitor of p53 transcriptional activity, the morphological changes at 24 h (Figure 3c) and increase in the ratio of dead cells at 30 h after exposure to 20 μM CdCl2 (Figure 3d) were markedly suppressed compared with untreated cells (P<0.01), indicating that p53 has a role in CdCl2-induced cellular damage. Inhibition of p53 activity with pifithrin-α did not affect the levels of Notch1-NICD and Notch1-NTM in the absence of CdCl2 (Figure 3e, lanes 1 versus 3). However, CdCl2-induced elevation of Notch1-NICD and reduction of Notch1-NTM were apparently counteracted by pifithrin-α treatment (Figure 3e, lanes 2 versus 4). In contrast, knockdown of Notch1 had little effect on the expression and phosphorylation of p53 protein following exposure to CdCl2 (Figure 3f, lanes 2 versus 4). These findings suggest that p53 might positively regulate Notch1 signaling through the cleavage of Notch1 by γ-secretase resulting in cellular damage in HK-2 cells exposed to CdCl2.

Modulation of Notch1 signaling by p53 in HK-2 cells exposed to CdCl2. (a and b) Cells were incubated with 20 μM CdCl2 (Cd) for the indicated time. The untreated control is labeled 0 h. Cell lysates were subjected to western blotting using antibodies against p53, phopho-p53, and actin (a). Equal amounts of protein (20 μg) in nuclear and cytoplasmic extracts were subjected to western blotting using antibodies against p53, MEK1/2, lamin A/C, and actin. MEK1 and lamin A/C served as a loading control for cytoplasmic and nuclear extracts, respectively. Data are from the same experiment shown in Figure 1b (b). (c–e) Cells were incubated with 0.1% DMSO or 40 μM pifithrin-α for 1 h and then incubated with or without 20 μM CdCl2 (Cd) for 24 h (c), 30 h (d), or 12 h (e). Phase-contrast micrographs were taken (c). The viability of cells was determined by trypan blue exclusion assay. Each value is the percentage of trypan blue-positive cells and reflects the mean±S.D. of three experiments with duplicate assays in each experiment. **P<0.01 versus CdCl2-treated cells incubated with DMSO (d). Cell lysates were subjected to western blotting using antibodies against Notch1-NICD, Notch1-NTM, and actin (e). (f) Cells transfected with control siRNA or Notch1 siRNA-1 were incubated with or without 20 μM CdCl2 (Cd) for 12 h. Cell lysates were subjected to western blotting using antibodies against p53, phospho-p53, Notch1-NICD, and actin. Immunoblots shown are representative of at least three independent experiments
Cross-talk between the Notch1 and PI3K/Akt signaling pathways in HK-2 cells exposed to CdCl2
The PI3K/Akt pathway has been reported to be activated by Notch signaling in various cell types.21, 22, 23, 24 We examined the cross-talk between the Notch1 and PI3K/Akt signaling pathways in HK-2 cells exposed to CdCl2. Consistent with our previous findings,30 phosphorylation of the epidermal growth factor receptor (EGFR) at Tyr1106, Akt at Thr308, GSK-3α at Ser21, GSK-3β at Ser9, and p70 S6 kinase (S6K) at Thr389, a downstream effector molecule of mammalian target of rapamycin, was observed in HK-2 cells exposed to 20 μM CdCl2 for 3–15 h (Figure 4a). To examine the functional role of CdCl2-induced activation of the PI3K/Akt signaling pathway, HK-2 cells were treated with LY294002, a PI3K inhibitor, and MK2206, an Akt inhibitor, and cellular damage was determined. Treatment with either LY294002 (Figures 4b and d) or MK2206 (Figures 4c and e) suppressed both morphological change at 24 h (Figures 4b and c) and the increase in the ratio of dead cells at 30 h after exposure to 20 μM CdCl2 (Figures 4d and e, P<0.01), suggesting that activation of the PI3K/Akt pathway might result in cellular damage. To determine whether Notch1 signaling contributes to the detrimental effects observed from PI3K/Akt pathway activation, we examined the effects of Notch1 knockdown on CdCl2-induced PI3K/Akt activation in HK-2 cells. Treatment with Notch1 siRNA-1 suppressed CdCl2-induced phosphorylation of EGFR, Akt, and S6K (Figure 4f, lanes 2 versus 4). Furthermore, knockdown of Jagged1 or Jagged 2 reduced the phosphorylation of Akt in HK-2 cells exposed to CdCl2 but less markedly than depletion of Notch1 (Supplementary Figure S6). Treatment of HK-2 cells with AG1478, an EGFR inhibitor, PPP, an insulin-like growth factor-1 receptor (IGF-1R) inhibitor, and LY294002 also suppressed CdCl2-induced Notch1-NICD accumulation as well as Akt phosphorylation (Figure 4g, lanes 2 versus 4, 6, or 8). Collectively, our results indicate that there may be cross-talk between the Notch1 and PI3K/Akt signaling pathways in HK-2 cells that were exposed to CdCl2 for 12 h.

Cross-talk between the Notch1 and PI3K/Akt signaling pathways in HK-2 cells exposed to CdCl2. (a) Cells were incubated with 20 μM CdCl2 (Cd) for the indicated time. The untreated control is labeled 0 h. Cell lysates were subjected to western blotting using antibodies against phospho-EGFR, total EGFR, phospho-Akt, total Akt, phospho-GSK-3α, phospho-GSK-3β, total GSK-3α/β, phospho-S6K, and actin. (b–e) Cells were incubated with 0.1% DMSO, 25 μM LY294002 (b and d), or 10 μM MK2206 (c and e) for 1 h and then incubated with or without 20 μM CdCl2 (Cd) for 24 h (b and c) or 30 h (d and e). Phase-contrast micrographs were taken (b and c). The viability of cells was determined by trypan blue exclusion assay. Each value is the percentage of trypan blue-positive cells and reflects the mean±S.D. of three experiments with duplicate assays in each experiment. **P<0.01 versus CdCl2-treated cells incubated with DMSO (d and e). (f) Cells transfected with control siRNA or Notch1 siRNA-1 were incubated with or without 20 μM CdCl2 (Cd) for 12 h. Cell lysates were subjected to western blotting using antibodies against phospho-EGFR, total EGFR, phospho-Akt, total Akt, phospho-S6K, Notch1-NICD, and actin. Data are from the same experiment shown in Figure 3f. (g) Cells were incubated with 0.1% DMSO, 2.5 μM AG1478, 5 μM PPP, or 25 μM LY294002 for 1 h and then incubated with or without 20 μM CdCl2 (Cd) for 12 h. Cell lysates were subjected to western blotting using antibodies against Notch1-NICD, phospho-Akt, total Akt, and actin. Immunoblots shown are representative of at least three independent experiments
Involvement of the Notch1 and PI3K/Akt signaling pathways in the regulation of Snail and E-cadherin expression in HK-2 cells exposed to CdCl2
Notch1 signaling can induce the expression of Snail in vitro and in vivo.16, 35, 36, 37 Inhibition of GSK-3α and GSK-3β increases Snail expression in epithelial cells,38 and the activity of GSK-3 is inhibited by Akt-mediated phosphorylation at Ser21 of GSK-3α and at Ser9 of GSK-3β.39 Therefore, we examined the roles of Notch1 and PI3K/Akt signaling in the regulation of Snail and E-cadherin expression in HK-2 cells exposed to CdCl2. HK-2 cells exposed to 20 μM CdCl2 showed reduced E-cadherin expression associated with increased Snail expression in a time-dependent manner (Figure 5a), and depletion of Notch1 suppressed the CdCl2-induced reduction of E-cadherin expression and elevation of Snail expression (Figure 5b, lanes 2 versus 4). Treatment with LY294002 weakened CdCl2-induced reduction of E-cadherin expression and elevation of Snail expression (Figure 5c, lanes 3 versus 4) in comparison with cells without LY294002 (lanes 1 versus 2). Treatment of HK-2 cells with SB216763, an inhibitor of GSK-3, did not affect CdCl2-induced elevation of Snail and Notch1-NICD (Figure 5d, lanes 2 versus 4) but could suppress the potency of LY294002 treatment to reduce Snail expression without elevating the level of Notch1-NICD in the presence of CdCl2 (lanes 6 versus 8). These results suggest that not only the activation of the Notch1 signaling pathway but also the inhibition of GSK-3 activity by PI3K/Akt might result in the upregulation of Snail in HK-2 cells exposed to CdCl2.

The Notch1 and PI3K/Akt signaling pathways are involved in the regulation of Snail and E-cadherin expression in HK-2 cells exposed to CdCl2. (a) Cells were incubated with 20 μM CdCl2 (Cd) for the indicated time. The untreated control is labeled 0 h. Cell lysates were subjected to western blotting using antibodies against E-cadherin, Snail, and actin. Data are from the same experiment shown in Figure 4a. (b) Cells transfected with control siRNA or Notch1 siRNA-1 were incubated with or without 20 μM CdCl2 (Cd) for 12 h. Cell lysates were subjected to western blotting using antibodies against E-cadherin, Snail, Notch1-NICD, and actin. Data are from the same experiment shown in Figure 3f. (c) Cells were incubated with 0.1% DMSO or 25 μM LY294002 for 1 h and then incubated with or without 20 μM CdCl2 (Cd) for 12 h. Cell lysates were subjected to western blotting using antibodies against E-cadherin, Snail, phospho-Akt, total Akt, and actin. (d) Cells were incubated with 0.2% DMSO, 20 μM SB216763 (SB), 25 μM LY294002 (LY), or 20 μM SB216763 and 25 μM LY294002 (SB+LY) for 1 h and then incubated with or without 20 μM CdCl2 (Cd) for 12 h. Cell lysates were subjected to western blotting using antibodies against Snail, Notch1-NICD, phospho-Akt, total Akt, and actin. Immunoblots shown are representative of at least three independent experiments
Effects of Snail knockdown on CdCl2-induced cellular damage in HK-2 cells
Transfection with siRNAs targeted against the human SNAI1 gene (siRNA-1 and siRNA-2) suppressed CdCl2-induced elevation of Snail expression and partially prevented HK-2 cells from CdCl2-induced reduction of E-cadherin expression (Figure 6a, lanes 2 versus 4 or 6). Knockdown of Snail with either siRNA-1 or siRNA-2 suppressed the morphological changes at 12 h (Figure 6b, middle and bottom panels) and increase in the ratio of dead cells at 30 h after exposure to 20 μM CdCl2 (Figures 6c, P<0.01 for siRNA-1, P<0.01 for siRNA-2). These results suggest that Snail expression has an important role in CdCl2-induced cellular damage in HK-2 cells.

Effects of Snail knockdown on CdCl2-induced cellular damage in HK-2 cells. Cells transfected with control siRNA, SNAI1 siRNA-1, or SNAI1 siRNA-2 were incubated with or without 20 μM CdCl2 (Cd) for 12 h (a and b) or 30 h (c). Cell lysates were subjected to western blotting using antibodies against E-cadherin, Snail, and actin. Immunoblots shown are representative of at least three independent experiments (a). Phase-contrast micrographs were taken (b). The viability of cells was determined by trypan blue exclusion assay. Each value is the percentage of trypan blue-positive cells and reflects the mean±S.D. of three experiments with duplicate assays in each experiment. **P<0.01 versus CdCl2-treated cells transfected with control siRNA (c)
Discussion
We found that exposure of HK-2 cells to CdCl2 increased the level of Notch1-NICD, the cleaved form of the Notch1 receptor, in a time-dependent manner. Furthermore, Notch1-NICD accumulated in the nuclear fraction and was accompanied by the upregulation of the Notch target gene HES1. Knockdown of Notch1 with siRNAs prevented HK-2 cells from undergoing CdCl2-induced morphological change, including cell rounding and cell separation, as well as apoptotic and non-apoptotic cell death. To clarify the role of NICD accumulation in the cytotoxicity of cadmium, HK-2 cells were treated with the γ-secretase inhibitor DAPT; reduction in Notch1-NICD levels by DAPT treatment also suppressed CdCl2-induced cellular damage. In addition, hyperactivation of Notch1 signaling by the introduction of Notch1 ΔEMV resulted in the enhancement of cellular damage caused by CdCl2 exposure. These findings demonstrate, to our knowledge for the first time, that cadmium activates the Notch1 signaling pathway and that NICD cleaved by γ-secretase is responsible for cellular damage in renal proximal tubular cells. Our data are consistent with recent findings that Notch signaling promotes the apoptotic cell death induced by various stress stimuli, including reactive oxygen species (ROS),40 hyperthermia,41 high glucose,42 and resveratrol.43 In addition, treatment with DAPT has been reported to block cadmium cytotoxicity in T47D human breast cancer cells.44 Inhibition of Notch1 signaling suppressed CdCl2-induced cellular damage less significantly in human hepatoma HepG2 cells and human neuroblastoma SH-SY5Y cells than in HK-2 cells (Supplementary Figure S7), indicating that the pathological significance of cadmium-induced activation of Notch1 signaling is different among cell types and might be significant in renal proximal tubular cells.
Interaction of ligands with Notch receptors is required for the activation of Notch signaling.8, 9, 10 In renal injury, the upregulation of Jagged1 in mice with ureteral obstruction45 and Delta-1 in rats using the ischemia–reperfusion model46 have been reported. In the present study, exposure to CdCl2 reduced the levels of Jagged1 and Jagged2 probably through the degradation by the lysosome,47 and knockdown of Jagged1 or Jagged2 partially suppressed CdCl2-induced Notch1-NICD accumulation and cellular damage in HK-2 cells. These results suggest that the upregulation of Notch ligands is an unlikely mechanism for activation of Notch1 signaling in HK-2 cells exposed to CdCl2. In addition, our data indicate that the interaction of Jagged1 or Jagged2 at the Notch1 receptor might be necessary initially, but insufficient, for the full cellular stress responses induced by cadmium exposure. The contribution of ROS, including superoxide anion, hydrogen peroxide, and hydroxyl radicals, production of which is enhanced by cadmium exposure,48 to the activation and subsequent modification of the Notch1 signaling pathway remains to be investigated.
We also examined the interaction between the p53 and Notch1 signaling pathways in HK-2 cells exposed to CdCl2. In response to CdCl2 exposure, p53 protein was accumulated in the nuclear fraction and was also phosphorylated at Ser15, indicating the activation of p53 signaling as has been reported previously.29 Furthermore, inhibition of p53 activity with pifithrin-α suppressed CdCl2-induced cellular damage, elevation of Notch1-NICD levels, and reduction in the level of Notch1-NTM in HK-2 cells. Neither the level of Jagged1 or Jagged2 was affected in HK-2 cells treated with pifithrin-α (data not shown). On the other hand, silencing of Notch1 expression failed to change the accumulation and phosphorylation of p53 in HK-2 cells exposed to CdCl2. These findings suggest that p53 signaling, which is responsible for cadmium cytotoxicity in renal tubular epithelial cells,49 promotes the production of Notch1-NICD without increasing the levels of Notch1 and its ligands. It has been demonstrated that p53 directly or indirectly regulates the expression and transcription of members of the γ-secretase complex, including presenilin 1, presenilin 2, and Pen-2.19, 20 We also found that treatment of HK-2 cells with pifithrin-α decreased the expression of presenilin 2 (Supplementary Figure S8). Although it is not known whether cadmium exposure affects the expression and function of γ-secretase, treatment of HK-2 cells with DAPT suppressed cadmium-induced elevation of Notch1-NICD. These findings raise a possibility that p53 affects γ-secretase activity in HK-2 cells, leading to enhance Notch1 signaling. Further investigations are required to elucidate the involvement of Notch1 signaling in the p53-mediated apoptotic cell death in response to cadmium exposure.
Although Akt is a well-known cellular survival signal, it may facilitate rather than inhibit cell death under certain conditions.50, 51 It has been reported that neuronal cell death induced by cadmium exposure was suppressed by wortmannin, a PI3K inhibitor, and overexpression of PTEN (phosphatase and tensin homolog deleted on chromosome 10), a negative regulator of PI3K/Akt pathway.52 Our results are consistent with a pro-cell death function of PI3K/Akt in cadmium-exposed HK-2 cells; that is, the PI3K/Akt signaling pathway was activated in response to CdCl2 exposure and treatment with either LY294002 or MK2206 suppressed CdCl2-induced cellular damage. Furthermore, knockdown of Notch1 or its ligands suppressed CdCl2-induced phosphorylation of EGFR, Akt, and S6K. Conversely, treatment with AG1478, PPP, or LY294002 reduced the level of Notch1-NICD in HK-2 cells exposed to CdCl2. These findings indicate that there might be a reciprocal regulatory loop between the Notch1 and PI3K/Akt signaling pathways leading to cadmium-induced cellular damage. The mechanism of this cooperation of two signaling pathways appears to be complex and multifactorial, including the Notch-mediated EGFR activation,21 IGF-1R activation,23, 53 and PTEN downregulation,23, 54 and the Akt-mediated nuclear factor κB activation, hypoxia-inducible factor-1α stabilization,25 and β-catenin activation,27 as well as the association of Notch1 with PI3K.22 Although the functional role of EGFR in cellular damage via Notch1 signaling has not been assessed, it is noted that knockdown of Notch1 reduced CdCl2-induced auto-phosphorylation of EGFR at Tyr1106 in HK-2 cells.
Involvement of the Notch1 and PI3K/Akt signaling pathways in the expression of Snail in HK-2 cells exposed to CdCl2 was also examined. Cadmium-induced reduction of E-cadherin level was associated with the elevation of Snail expression, compatible with the fact that Snail is a repressor of E-cadherin expression.14, 15 Furthermore, these changes were attenuated by Notch1 knockdown. Treatment of HK-2 cells with the γ-secretase inhibitor, Compound E, has been shown to inhibit transforming growth factor β1 (TGFβ1)-induced decrease in the level of E-cadherin mRNA.55 Overexpression of Notch1-NICD or inhibition of Snail degradation by lithium chloride led to a further decrease in E-cadherin protein levels in primary renal tubular epithelial cells concurrently exposed to TGFβ1 and angiotensin II, while silencing of Snail blocked this effect.16 Transfection of microRNA-34a into HK-2 cells abolished hypoxia-induced expression of Notch1 and Snail.56 These results and our present results indicate that Notch1 signaling promotes the reduction of E-cadherin levels through the expression of Snail in renal tubular epithelial cells treated with different stimuli, including cadmium. In addition, inhibition of GSK-3 activity with SB216763 diminished the potency of LY294002 treatment to reduce Snail expression without changing the level of Notch1-NICD in HK-2 cells exposed to CdCl2, suggesting that PI3K/Akt/GSK-3 signaling also has a role in Snail expression. Although the possibility of cleavage of E-cadherin by activated γ-secretase44 remains to be excluded, our data suppose that the cooperative modulation of Snail expression by Notch1 and PI3K/Akt signaling leads to the reduction of E-cadherin level in renal proximal tubular cells exposed to cadmium. Overexpression of full-length E-cadherin in WKPT-0293 Cl.2 rat kidney proximal tubule cells is protective against cadmium-induced disruption of the cellular junctions, detachment, and cell death.57 We also found that knockdown of Snail partially prevented HK-2 cells from CdCl2-induced reduction of E-cadherin expression and cellular damage. Therefore, Notch1 activation and its cross-talk with the PI3K/Akt axis might be responsible for the detrimental effects of cadmium in renal proximal tubules.
In summary, our study shows that cadmium exposure induces the accumulation of Notch1-NICD following the possible interaction of Notch1 and its ligands, Jagged1 and Jagged2, in HK-2 cells. Other signaling pathways, including p53 and PI3K/Akt, further modulate the activation of Notch1 signaling leading to the cellular damage caused by cadmium. The transcription factor Snail, a repressor of E-cadherin expression, is upregulated cooperatively by the activation of Notch1 signaling and the inhibition of GSK-3, a downstream molecule of PI3K/Akt. In addition to the cellular adhesion experiments, the dysregulation of activity and spatiotemporal expression of canonical or non-canonical Notch ligands in renal proximal tubules exposed to cadmium will need to be investigated.
Materials and Methods
Chemicals
CdCl2 was obtained from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). LY294002 and SB216763 were obtained from Cayman Chemical Company (Ann Arbor, MI, USA). AG1478 and PPP were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). DAPT and pifithrin-α were obtained from Wako. MK-2206 was obtained from Active Biochem (Maplewood, NJ, USA). Antibodies against phospho-EGFR (Tyr1068), total EGFR (D38B1) XP, phospho-Akt (Thr308) (C31E5E), total Akt (pan) (C67E7), phospho-p70 S6 kinase (Thr389) (108D2), phospho-GSK-3α/β (Ser21/9) (37F11), total GSK3-α/β (D75D3) XP, cleaved Notch1 (Val1744) (D3B8), Notch1 (D1E11) XP, Jagged1 (28H8), Jagged2 (C23D2), Snail (C15D3), E-cadherin (24E10), phospho-p53 (Ser15), and MEK1/2 were obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA). p53 (DO-1), Notch1 (C-20), actin (I-19), and lamin A/C (14/LaminAC) antibodies were obtained from Santa Cruz Biotechnology. The siRNAs targeted against the human Notch1 (siRNA-1: Hs_NOTCH1_3 FlexiTube siRNA, SI00119028, siRNA-2: Hs_NOTCH1_4 FlexiTube siRNA, SI00119035), Jagged1 (Hs_JAG1_5 FlexiTube siRNA, SI02780134), Jagged2 (Hs_JAG2_2 FlexiTube siRNA, SI03095764), SNAI1 (siRNA-1: Hs_SNAI1_1 FlexiTube siRNA, SI00083398, siRNA-2: Hs_SNAI1_5 FlexiTube siRNA, SI02636424), and non-target siRNA (AllStars Negative Control siRNA) were purchased from Qiagen (Hilden, Germany).
Cell culture and treatments
HK-2 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and grown in Dulbecco’s modified Eagle’s medium/Nutrient Mixture F-12 supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (GIBCO, Invitrogen Corp., Carlsbad, CA, USA) in a humidified atmosphere of 5% CO2 and 95% air at 37 °C. Exponentially growing HK-2 cells were seeded at 4 × 105 cells/well in six-well culture plates and cultured for 1 day before each experiment. CdCl2 was dissolved in water and sterilized by filtration. Cells were incubated in serum-free medium containing the appropriate concentration of CdCl2 for 3–30 h at 37 °C. AG1478, DAPT, LY294002, MK-2206, pifithrin-α, PPP, and SB216763 were dissolved in dimethyl sulfoxide (DMSO). After incubating cells in serum-free medium with DMSO (0.1 or 0.2%) or one of the inhibitors for 1 h, HK-2 cells were treated with 20 μM CdCl2 for the indicated time.
Preparation of whole cell lysates
After incubation, cells were washed with phosphate-buffered saline and lysed with sodium dodecyl sulfate-polyacrylamide gel Laemmli sample buffer. Cell lysates were collected, sonicated, and boiled for 5 min. Protein concentrations were determined using the RC DC Protein Assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Preparation of cytoplasmic and nuclear fractions
Cytoplasmic and nuclear fractions were prepared using the Nuclear Extract Kit (Active Motif, Carlsbad, CA, USA) following the manufacturer’s protocol.
Western blotting
Western blotting was carried out as described previously.32 Equal amounts of protein (20 μg) were subjected to sodium dodecyl sulfate-10% polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane (Hybond-ECL, Amersham Pharmacia Biotech, Buckinghamshire, England). The membrane was blocked with 5% non-fat milk in Tris-buffered saline containing 0.1% Tween 20 for 1 h at room temperature. The membrane was then incubated overnight at 4 °C with the primary antibody, and protein was detected with a Phototope-HRP Western blot detection kit (Cell Signaling Technology, Inc.).
Gene knockdown of Notch1, Jagged1 or Jagged2 by siRNA
Transfection of siRNA against human Notch1, Jagged1, Jagged2, and non-target siRNA into HK-2 cells was done using Lipofectamine RNAiMAX (Invitrogen Corp.) according to the manufacturer’s instructions with some adjustments. The siRNAs were dissolved in nuclease-free water and diluted to 0.2 μM with 250 μl Opti-MEM (Invitrogen Corp.). Five microliters of Lipofectamine RNAiMAX was also diluted 50-fold with Opti-MEM. Equal volumes of these two solutions were mixed (500 μl total) and immediately added to 2 ml culture medium at the time of cell plating. After incubation for 24 h, cells were washed with medium and used for experiments.
Trypan blue exclusion assay
Culture medium was aspirated and reserved. After trypsinization, cells were suspended in Dulbecco’s modified Eagle’s medium/Nutrient Mixture F-12 medium, and the culture medium was returned. The mixture was centrifuged to pellet the cells. Cellular suspension and 0.4% trypan blue in Hank’s Balanced Salt Solution were mixed, and the number of viable cells was counted using a TC10 Automated Cell Counter (Bio-Rad Laboratories, Inc.).
Statistical analysis
Results are expressed as the mean±S.D. Statistical significance was determined by the Student’s t-test or Welch’s t-test. A value of P<0.05 was considered to be statistically significant.
Abbreviations
- CdCl2:
-
cadmium chloride
- DAPT:
-
N-[N-(3,5-difluorophenacetyl-L-alanyl)]-S-phenylglycine t-butyl ester
- DMSO:
-
dimethyl sulfoxide
- EGFR:
-
epidermal growth factor receptor
- GSK-3:
-
glycogen synthase kinase-3
- HES1:
-
hairy and enhancer of split-1
- NICD:
-
Notch intracellular domain
- PI3K:
-
phosphatidylinositol-3-kinase
- S6K:
-
p70 S6 kinase
- TGFβ1:
-
transforming growth factor β1
References
Nordberg GF, Nogawa K, Nordberg M, Friberg LT . Cadmium In: Nordberg GF, Fowler BA, Nordberg M, Friberg LT, (eds). Handbook on the Toxicology of Metals. Academic Press: Burlington, MA, USA, 2007; pp 445–486.
Prozialeck WC, Edwards JR . Mechanisms of cadmium-induced proximal tubule injury: new insights with implications for biomonitoring and therapeutic interventions. J Pharmacol Exp Ther 2012; 343: 2–12.
Prozialeck WC, Lamar PC, Lynch SM . Cadmium alters the localization of N-cadherin, E-cadherin, and β-catenin in the proximal tubule epithelium. Toxicol Appl Pharmacol 2003; 189: 180–195.
Prozialeck WC, Niewenhuis RJ . Cadmium (Cd2+) disrupts Ca2+-dependent cell-cell junctions and alters the pattern of E-cadherin immunofluorescence in LLC-PK1 cells. Biochem Biophys Res Commun 1991; 181: 1118–1124.
Edwards JR, Kolman K, Lamar PC, Chandar N, Fay MJ, Prozialeck WC . Effects of cadmium on the sub-cellular localization of β-catenin and β-catenin-regulated gene expression in NRK-52E cells. Biometals 2013; 26: 33–42.
Thévenod F, Wolff NA, Bork U, Lee W-K, Abouhamed M . Cadmium induces nuclear translocation of β-catenin and increases expression of c-myc and Abcb1a in kidney proximal tubule cells. Biometals 2007; 20: 807–820.
Yin L, Velazquez OC, Liu Z-J . Notch signaling: emerging molecular targets for cancer therapy. Biochem Pharmacol 2010; 80: 690–701.
Kopan R . Notch signaling. Cold Spring Harb Perspect Biol 2012; 4: a011213.
Capaccione KM, Pine SR . The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis 2013; 34: 1420–1430.
D’Souza B, Meloty-Kapella L, Weinmaster G . Canonical and non-canonical Notch ligands. Curr Top Dev Biol 2010; 92: 73–129.
Bray S, Bernard F . Notch targets and their regulation. Curr Top Dev Biol 2010; 92: 253–275.
Schwanbeck R, Martini S, Bernoth K, Just U . The Notch signaling pathway: molecular basis of cell context dependency. Eur J Cell Biol 2011; 90: 572–581.
Sun S, Du R, Gao J, Ning X, Xie H, Lin X et al. Expression and clinical significance of Notch receptors in human renal cell carcinoma. Pathology 2009; 41: 335–341.
Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG et al. The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2000; 2: 76–83.
Batlle E, Sancho E, Franci C, Domínguez D, Monfar M, Baulida J et al. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2000; 2: 84–89.
Saad S, Stanners SR, Yong R, Tang O, Pollock CA . Notch mediated epithelial to mesenchymal transformation is associated with increased expression of the Snail transcription factor. Int J Biochem Cell Biol 2010; 42: 1115–1122.
Dang TP . Notch, apoptosis and cancer. Adv Exp Med Biol 2012; 727: 199–209.
Yugawa T, Handa K, Narisawa-Saito M, Ohno S, Fujita M, Kiyono T . Regulation of Notch1 gene expression by p53 in epithelial cells. Mol Cell Biol 2007; 27: 3732–3742.
Dunys J, Sevalle J, Giaime E, Pardossi-Piquard R, Vitek MP, Renbaum P et al. p53-dependent control of transactivation of the Pen2 promoter by presenilins. J Cell Sci 2009; 122: 4003–4008.
Checler F, Dunys J, Pardossi-Piquard R, Alves da Costa C . p53 is regulated by and regulates members of the γ-secretase complex. Neurodegener Dis 2010; 7: 50–55.
Díaz B, Yuen A, Iizuka S, Higashiyama S, Courtneidge SA . Notch increases the shedding of HB-EGF by ADAM12 to potentiate invadopodia formation in hypoxia. J Cell Biol 2013; 201: 279–292.
Sade H, Krishna S, Sarin A . The anti-apoptotic effect of Notch-1 requires p56lck-dependent, Akt/PKB-mediated signaling in T cells. J Biol Chem 2004; 279: 2937–2944.
Hales EC, Taub JW, Matherly LH . New insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling axis: targeted therapy of γ-secretase inhibitor resistant T-cell acute lymphoblastic leukemia. Cell Signal 2014; 26: 149–161.
Chang L, Wong F, Niessen K, Karsan A . Notch activation promotes endothelial survival through a PI3K-Slug axis. Microvasc Res 2013; 89: 80–85.
Bedogni B, Warneke JA, Nickoloff BJ, Giaccia AJ, Powell MB . Notch1 is an effector of Akt and hypoxia in melanoma development. J Clin Invest 2008; 118: 3660–3670.
Calzavara E, Chiaramonte R, Cesana D, Basile A, Sherbet GV, Comi P . Reciprocal regulation of Notch and PI3K/Akt signalling in T-ALL cells in vitro. J Cell Biochem 2008; 103: 1405–1412.
Zhang J, Fukuhara S, Sako K, Takenouchi T, Kitani H, Kume T et al. Angiopoietin-1/Tie2 signal augments basal Notch signal controlling vascular quiescence by inducing delta-like 4 expression through AKT-mediated activation of β-catenin. J Biol Chem 2011; 286: 8055–8066.
Xu L, Zhu Y, Xu J, Wu K, Li J, Xu W et al. Notch1 activation promotes renal cell carcinoma growth via PI3K/Akt signaling. Cancer Sci 2012; 103: 1253–1258.
Matsuoka M, Igisu H . Cadmium induces phosphorylation of p53 at serine 15 in MCF-7 cells. Biochem Biophys Res Commun 2001; 282: 1120–1125.
Fujiki K, Inamura H, Matsuoka M . PI3K signaling mediates diverse regulation of ATF4 expression for the survival of HK-2 cells exposed to cadmium. Arch Toxicol 2014; 88: 403–414.
Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li Y-M et al. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res 2006; 66: 7445–7452.
Iwatsuki M, Inageda K, Matsuoka M . Cadmium induces phosphorylation and stabilization of c-Fos in HK-2 renal proximal tubular cells. Toxicol Appl Pharmacol 2011; 251: 209–216.
Kopan R, Schroeter EH, Weintraub H, Nye JS . Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc Natl Acad Sci USA 1996; 93: 1683–1688.
Shieh S-Y, Ikeda M, Taya Y, Prives C . DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997; 91: 325–334.
Matsuno Y, Coelho AL, Jarai G, Westwick J, Hogaboam CM . Notch signaling mediates TGF-β1-induced epithelial-mesenchymal transition through the induction of Snai1. Int J Biochem Cell Biol 2012; 44: 776–789.
Timmerman LA, Grego-Bessa J, Raya A, Bertrán E, Pérez-Pomares JM, Díez J et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev 2004; 18: 99–115.
Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U . Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci USA 2008; 105: 6392–6397.
Bachelder RE, Yoon S-O, Franci C, de Herreros AG, Mercurio AM . Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: implications for the epithelial-mesenchymal transition. J Cell Biol 2005; 168: 29–33.
Cross DAE, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA . Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995; 378: 785–789.
Ding X, Zhu F, Li T, Zhou Q, Hou FF, Nie J . Numb protects renal proximal tubular cells from puromycin aminonucleoside-induced apoptosis through inhibiting Notch signaling pathway. Int J Biol Sci 2011; 7: 269–278.
Basile A, Biziato D, Sherbet GV, Comi P, Cajone F . Hyperthermia inhibits cell proliferation and induces apoptosis: relative signaling status of P53, S100A4, and Notch in heat sensitive and resistant cell lines. J Cell Biochem 2008; 103: 212–220.
Gao F, Yao M, Shi Y, Hao J, Ren Y, Liu Q et al. Notch pathway is involved in high glucose-induced apoptosis in podocytes via Bcl-2 and p53 pathways. J Cell Biochem 2013; 114: 1029–1038.
Lin H, Xiong W, Zhang X, Liu B, Zhang W, Zhang Y et al. Notch-1 activation-dependent p53 restoration contributes to resveratrol-induced apoptosis in glioblastoma cells. Oncol Rep 2011; 26: 925–930.
Park CS, Kim OS, Yun S-M, Jo SA, Jo I, Koh YH . Presenilin 1/ γ-secretase is associated with cadmium-induced E-cadherin cleavage and COX-2 gene expression in T47D breast cancer cells. Toxicol Sci 2008; 106: 413–422.
Morrissey J, Guo G, Moridaira K, Fitzgerald M, McCracken R, Tolley T et al. Transforming growth factor-β induces renal epithelial jagged-1 expression in fibrotic disease. J Am Soc Nephrol 2002; 13: 1499–1508.
Kobayashi T, Terada Y, Kuwana H, Tanaka H, Okado T, Kuwahara M et al. Expression and function of the Delta-1/Notch-2/Hes-1 pathway during experimental acute kidney injury. Kidney Int 2008; 73: 1240–1250.
Koutelou E, Sato S, Tomomori-Sato C, Florens L, Swanson SK, Washburn MP et al. Neuralized-like 1 (Neurl1) targeted to the plasma membrane by N-myristoylation regulates the Notch ligand Jagged1. J Biol Chem 2008; 283: 3846–3853.
Liu J, Qu W, Kadiiska MB . Role of oxidative stress in cadmium toxicity and carcinogenesis. Toxicol Appl Pharmacol 2009; 238: 209–214.
Tokumoto M, Fujiwara Y, Shimada A, Hasegawa T, Seko Y, Nagase H et al. Cadmium toxicity is caused by accumulation of p53 through the down-regulation of Ube2d family genes in vitro and in vivo. J Toxicol Sci 2011; 36: 191–200.
Los M, Maddika S, Erb B, Schulze-Osthoff K . Switching Akt: from survival signaling to deadly response. Bioessays 2009; 31: 492–495.
Kerr BA, Ma L, West XZ, Ding L, Malinin NL, Weber ME et al. Interference with Akt signaling protects against myocardial infarction and death by limiting the consequences of oxidative stress. Sci Signal 2013; 6: ra67.
Chen L, Xu B, Liu L, Luo Y, Zhou H, Chen W et al. Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death. Free Radic Biol Med 2011; 50: 624–632.
Eliasz S, Liang S, Chen Y, De Marco MA, Machek O, Skucha S et al. Notch-1 stimulates survival of lung adenocarcinoma cells during hypoxia by activating the IGF-1R pathway. Oncogene 2010; 29: 2488–2498.
Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med 2007; 13: 1203–1210.
Nyhan KC, Faherty N, Murray G, Cooey LB, Godson C, Crean JK et al. Jagged/Notch signalling is required for a subset of TGFβ1 responses in human kidney epithelial cells. Biochim Biophys Acta 2010; 1803: 1386–1395.
Du R, Sun W, Xia L, Zhao A, Yu Y, Zhao L et al. Hypoxia-induced down-regulation of microRNA-34a promotes EMT by targeting the Notch signaling pathway in tubular epithelial cells. PLoS One 2012; 7: e30771.
Chakraborty PK, Lee W-K, Molitor M, Wolff NA, Thévenod F . Cadmium induces Wnt signaling to upregulate proliferation and survival genes in sub-confluent kidney proximal tubule cells. Mol Cancer 2010; 9: 102.
Acknowledgements
We thank Dr Raphael Kopan and Dr Jeffrey Nye for providing pCS2 Notch1 ΔEMV-6MT (Addgene plasmid 41737). We thank Dr Motoyuki Itoh for helpful discussion. This study was supported in part by Grants-in-Aid for Scientific Research (24870025 and 26460175) from the Japan Society for the Promotion of Science (JSPS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by J Chipuk
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Fujiki, K., Inamura, H. & Matsuoka, M. Detrimental effects of Notch1 signaling activated by cadmium in renal proximal tubular epithelial cells. Cell Death Dis 5, e1378 (2014). https://doi.org/10.1038/cddis.2014.339
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.339
This article is cited by
-
Blockade of ALK4/5 signaling suppresses cadmium- and erastin-induced cell death in renal proximal tubular epithelial cells via distinct signaling mechanisms
Cell Death & Differentiation (2019)
-
Identification of ARNT-regulated BIRC3 as the target factor in cadmium renal toxicity
Scientific Reports (2017)
-
Probing the PI3K/Akt/mTor pathway using 31P-NMR spectroscopy: routes to glycogen synthase kinase 3
Scientific Reports (2016)
-
Endoplasmic reticulum stress eIF2α–ATF4 pathway-mediated cyclooxygenase-2 induction regulates cadmium-induced autophagy in kidney
Cell Death & Disease (2016)
-
An RNA interference screen identifies new avenues for nephroprotection
Cell Death & Differentiation (2016)
