
Abstract
Osteopontin (OPN) is a multifunctional protein involved in hepatic steatosis, inflammation, fibrosis and cancer progression. However, its role in hepatic injury induced by ischemia–reperfusion (I–R) has not yet been investigated. We show here that hepatic warm ischemia for 45 min followed by reperfusion for 4 h induced the upregulation of the hepatic and systemic level of OPN in mice. Plasma aspartate aminotransferase and alanine aminotransferase levels were strongly increased in Opn−/− mice compared with wild-type (Wt) mice after I–R, and histological analysis of the liver revealed a significantly higher incidence of necrosis of hepatocytes. In addition, the expression levels of inducible nitric oxide synthase (iNOS), tumor necrosis factor-α (TNFα), interleukin 6 (IL6) and interferon-γ were strongly upregulated in Opn−/− mice versus Wt mice after I–R. One explanation for these responses could be the vulnerability of the OPN-deficient hepatocyte. Indeed, the downregulation of OPN in primary and AML12 hepatocytes decreased cell viability in the basal state and sensitized AML12 hepatocytes to cell death induced by oxygen–glucose deprivation and TNFα. Further, the downregulation of OPN in AML12 hepatocytes caused a strong decrease in the expression of anti-apoptotic Bcl2 and in the ATP level. The hepatic expression of Bcl2 also decreased in Opn−/− mice versus Wt mice livers after I–R. Another explanation could be the regulation of the macrophage activity by OPN. In RAW macrophages, the downregulation of OPN enhanced iNOS expression in the basal state and sensitized macrophages to inflammatory signals, as evaluated by the upregulation of iNOS, TNFα and IL6 in response to lipopolysaccharide. In conclusion, OPN partially protects from hepatic injury and inflammation induced in this experimental model of liver I–R. This could be due to its ability to partially prevent death of hepatocytes and to limit the production of toxic iNOS-derived NO by macrophages.
Similar content being viewed by others

Main
Hepatic ischemia–reperfusion (I–R) injury is the main cause of liver damage that occurs during surgical procedures such as hepatic resection and transplantation. The molecular mechanisms leading to liver damage on I–R are complex, multi-factorial and affect all liver cellular components. In the ischemic phase, anoxic injury of hepatocytes (cells dependent on oxygen) is clearly the predominant process leading to injury, which is associated with an alteration in ATP homeostasis. In the reperfusion phase, an inflammatory response is initiated and enhances hepatocyte injury. Over-production and release of tumor necrosis factor-α (TNFα) from hepatic resident macrophages (Kupffer cells) may activate TNF receptors on hepatocytes to induce several pathways that promote hepatocyte death.1, 2, 3 The production of toxic nitric oxide (NO) by macrophage inducible NO synthase (iNOS) was also deleterious and the iNOS deficiency reduced liver injury after hepatic I–R.4, 5 However, the mechanisms and actors involved in hepatic I–R injury are still unknown or unclear.
Osteopontin (OPN) is expressed in a variety of liver cells including hepatic macrophages (resident Kupffer cells and infiltrated macrophages), stellate cells and hepatocytes6, 7, 8, 9 and is a versatile modulator of liver diseases. OPN has an important role in hepatic inflammation and fibrogenesis in alcoholic and nonalcoholic steatohepatitis.9, 10 OPN is also linked to progression and metastasis of hepatocellular carcinoma.9 In addition, we previously reported that hepatic expression of OPN correlated with hepatic steatosis, the level of alanine aminotransferase (ALT) and insulin resistance in morbidly obese patients.6 Hepatic OPN also increased with liver fibrosis in alcoholic patients and its circulating level was predictive of liver fibrosis in patients with alcoholic liver disease and chronic hepatitis C.10
The upregulation of OPN has been reported in rat retina that activated microglia on I–R,11 in cardiomyocytes in an ex-vivo hemoperfused working porcine heart model,12 in the brain during early cerebral I–R in rats13 but also in cultured rat aortic vascular smooth muscle cells in response to hypoxia.14 The role of OPN in I–R injury has largely been reported in the kidney and could have an unexpected protective and deleterious role. OPN may act as a ‘survival factor’ for the renal tubule, either through inhibition of iNOS15 or through inhibition of apoptosis.16, 17 The deficiency in OPN reduced tolerance to acute renal ischemia associated with increased iNOS, NO and I–R injury at 24 h after reperfusion.18 OPN also stimulated the development of renal fibrosis after acute ischemic insult.19 The overexpression of OPN via hyperactivation of Wnt (Wingless) signaling, as detected in Brown Norway rats, is also critical for the maintenance of their inherent ischemic resistance. OPN reduces mitochondrial cytochrome c release and caspase 3 activity after renal I–R.20 It has also been reported that OPN expressed in tubular epithelial cells regulates NK cell-mediated kidney I–R injury.21
Despite the fact that hepatic OPN is involved in a large number of liver diseases, its role in hepatic I–R injury has not yet been investigated. We focused our study on the expression of OPN in response to I–R and on its role in I–R-induced liver injury and inflammation using Opn−/− mice. We then examined hepatocytes and macrophages in vitro to better understand the potential roles of OPN.
Results
Liver I–R induced the upregulation of plasma and hepatic expression of OPN
We first estimated the circulating and hepatic level of OPN on I–R in wild-type (Wt) mice. The plasma level of OPN was evaluated before and after ischemia for 45 min followed by 4 h of reperfusion. As shown in Figure 1a, the circulating level of the OPN protein was strongly increased in response to I–R. The hepatic expression of OPN was also evaluated in the Wt I–R mice compared with SHAM mice. The expression of OPN in the liver markedly increased in response to I–R (Figure 1b). Hepatic I–R thus caused upregulation of OPN expression and abundance in the liver and in the systemic circulation.

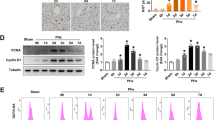
Plasma and hepatic OPN expression is increased in response to liver I–R and the OPN deficiency increased hepatic I–R injury. Wt (n=8–10) and Opn−/− (Opn−/−) (n=5) mice underwent ischemia for 45 min followed by 4 h of reperfusion. SHAM controls (n=4–6) underwent the same procedure but without vascular occlusion. (a) The plasma level of OPN was evaluated before and after I–R in Wt mice. (b) Hepatic expression of OPN was evaluated in I–R lobes from Wt mice (Wt I–R) versus SHAM mice. (c) The plasma level of ALT and AST was evaluated before and after I–R in Wt, Opn−/− and in SHAM mice. Results were expressed as means±S.E.M. Data were statistically analyzed using the Mann–Whitney test. *versus before I–R (a) or SHAM (b and c); $versus Wt I–R (c). P<0.05. (d) H&E staining of liver samples from Wt, Opn−/− after I–R and SHAM mice. Typical pictures are shown. Dotted lines and arrows limit areas of necrosis; PT indicates portal triad; CLV indicates centrolobular vein. (e) The TUNEL assay was performed on liver sections as described in the Materials and Methods section. The liver sections were then counterstained with hematoxylin. Typical pictures are shown. (f) The level of caspase 3 activity was evaluated from a total lysate of control and I–R lobes from Wt (Wt Ctr; Wt I–R) (n=7), Opn−/− (Opn−/− Ctr; Opn−/− I–R) (n=5) and SHAM mice (n=5). Results were expressed as means±S.E.M. Data were statistically analyzed using the Mann–Whitney test. ≠versus SHAM, #versus Wt Ctr and *versus Opn−/− Ctr. P<0.05
OPN deficiency increased the liver injury induced by hepatic I–R
The role of OPN in liver injury induced by I–R was then investigated using mice deficient for OPN. The plasma was collected before and after I–R of Wt and Opn−/− mice and in SHAM mice. As expected, post-I–R was associated with elevated aspartate aminotransferase (AST) and ALT levels in Wt mice (Figure 1c). The Opn−/− mice showed more liver damage on I–R as shown by the higher aminotransferase levels (AST, ALT). Further, histological analysis displayed extensive areas of necrosis (as indicated by arrows) in the liver of Opn−/− mice compared with livers of Wt mice after I–R (Figure 1d). The OPN deficiency seems to be more associated with necrosis than apoptosis in these experimental conditions. Indeed, apoptosis evaluated by TUNEL-positive cells (Figure 1e and data not shown) and the level of caspase 3 activity (Figure 1f) were not significantly aggravated with the deletion of OPN in response to I–R. These results indicated that the OPN deficiency resulted in aggravation of the liver injury induced by I–R.
OPN deficiency aggravated the liver inflammation induced by I–R
Immune and inflammatory cells including macrophages (Kupffer cells), neutrophils, CD4+ T lymphocytes are activated in response to reperfusion.2, 3 Several of the compounds released during the inflammatory response, such as high concentrations of NO and the cytokine TNFα, may produce cytotoxicity and induce additional cell injury from those induced by ischemia. We first analyzed the expression of iNOS in the liver of SHAM mice and in hepatic lobes that underwent ischemia and then reperfusion of Wt (Wt I–R) and Opn−/− (Opn−/− I–R) mice. As shown in Figure 2a, the gene expression of iNOS increased significantly in Opn−/− I–R compared with SHAM and Wt I–R mice. Furthermore, the iNOS expression relative to the vascularized lobe of Wt mice (Ctr: internal control lobe) was already upregulated in Opn−/− mice. Inflammation was also evaluated from the expressions of TNFα, interleukin 6 (IL6) and interferon-γ (IFNγ). The relative expression to the internal control lobe of Wt mice (Ctr), of TNFα, IL6 and IFNγ, was increased in response to I–R with a more marked rise in Opn−/− mice (Figures 2b–d). In the vascularized lobes, the OPN deficiency also mediated an increase in IFNγ expression. The OPN deficiency was thus associated with more pronounced inflammation in response to I–R.

OPN deficiency aggravated the liver inflammation induced by I–R. The gene expression of iNOS (a), TNFα (b), IL6 (c) and IFNγ (d) was evaluated for control and ischemic-reperfused lobes (ischemia for 45 min and then 4 h of reperfusion) from Wt (Wt Ctr; Wt I–R) (n=8–10), Opn−/− (Opn−/− Ctr; Opn−/− I–R) (n=5) and SHAM mice (n=4–6). The mRNA levels were normalized to RPLP0 mRNA levels and expressed as fold stimulation±S.E.M. versus SHAM or Wt Ctr. Data were statistically analyzed using the Mann–Whitney test. ¤versus SHAM, *versus Wt Ctr, $versus Wt I–R and #versus Opn−/− Ctr. P<0.05
OPN deficiency decreased hepatocyte viability and Bcl2 expression
We first evaluated the viability of hepatocytes freshly isolated from Wt and Opn−/− mice. As shown in Figure 3a, the Opn−/− versus Wt hepatocytes were more sensitive to cellular damage associated with the hepatocyte isolation procedure. Furthermore, the silencing of OPN by siRNA in AML12 hepatocytes caused reduced cell viability (Figure 3b) and increased cytotoxicity (lactate dehydrogenase (LDH) release) (Figure 3c). Interestingly, we found that the decrease in viability of AML12 hepatocytes after OPN silencing was associated with a substantial decrease in anti-apoptotic Bcl2 expression at the mRNA (Figure 3d) and protein level (Figure 3e). The knock out or silencing of OPN expression in primary hepatocytes also caused a decrease in gene expression of Bcl2 (Opn−/− versus Wt hepatocytes: Bcl2=0.32, n=1; si OPN versus si Ctr hepatocytes: Bcl2=0.57, n=1). Furthermore, the in vivo level of the Bcl2 protein decreased significantly in the Opn−/− I–R lobe compared with the Wt I–R lobe and SHAM liver in mice (Figures 3f and g).

OPN deficiency decreased primary and AML12 hepatocyte viability and caused decreased Bcl2 expression. (a) The viability of hepatocytes freshly isolated from Opn+/+ (n=10) and Wt livers (n=4) was evaluated by trypan blue exclusion. (b–e) After silencing of OPN with siRNA in AML12 cells, the cell viability (MTT assay) (b) and LDH release (c) were evaluated after 16 h in DMEM, 4.5 g/l glucose supplemented with 0.5% BSA (n=6). Expression of Bcl2 was also evaluated at the mRNA ((d), n=3) and protein levels ((e), n=2). (f–g) Hepatic expression of Bcl2 was evaluated in I–R lobes from Opn−/− and Wt mice (Wt I–R) versus SHAM mice by immunoblotting ((f), three mice per group). Results from (f) were analyzed by densitometry (g). (a–d and g) Results were expressed as means±S.E.M. Data were statistically analyzed using the Mann–Whitney or Student’s t-test. *P<0.05
OPN silencing sensitized AML12 hepatocytes to cell death induced by oxygen–glucose deprivation
To then explore the effect of the OPN deficiency on the sensitivity of hepatocytes to I–R, we used an in vitro model of oxygen–glucose deprivation (OGD).22 An initial short phase of OGD was performed by culturing the Ctr or OPN-silenced AML12 cells in a medium without glucose and supplemented with KCN (2.5 to 10 mM) for 1 h, followed by an overnight restoration phase. As OPN silencing affected basal cell viability, the following results were expressed as a percentage of basal state, as indicated. Compared with Ctr AML12 cells, the OPN-silenced AML12 cells displayed more sensitivity to OGD-induced cell death (significant at KCN at 10 mM) as evaluated by the reduced cell viability (MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay evaluated at 10 mM KCN) (Figure 4a) and the increased cytotoxicity (LDH release) (Figure 4b). To better evaluate the cell death mechanisms (apoptosis versus necrosis), less stringent conditions with KCN at 2.5 or 5 mM and only a 2-h restoration phase were performed. In these experimental conditions, the OPN-silenced AML12 cells exhibited more apoptosis, when evaluated from the levels of annexin V-PE-positive cells (Figure 4c) and activated (cleaved) caspase 3 (p17) (Figure 4d) in response to OGD versus Ctr AML12 hepatocytes.

OPN silencing sensitized AML12 hepatocytes to cell death induced by OGD and caused increased NQO1 expression and decreased ATP levels. After silencing of OPN with siRNA in AML12 cells, cell viability (MTT assay) (a) (n=6), LDH release (b) (n=6), cell apoptosis (c) (n=4), cleaved caspase 3 level (d) (n=2), the ATP level (e) (n=4) and gene expression of NQO1 (f) (n=3) were evaluated in the basal state and in response to KCN (2.5, 5 or 10 mM as indicated) after 1 h in DMEM without glucose, followed by a 16- (a, b and f) or 2-h (c, d and e) restoration period in DMEM, 4.5 g/l glucose supplemented with 0.5% BSA. Results relative to either its own control (si Ctr or si OPN) (a and b) or si Ctr (c, d and f) were expressed as means±S.E.M. Data were statistically analyzed using Student’s t-test. *P<0.05
OPN silencing in AML12 hepatocytes altered the ATP level and could enhance oxidative stress
In addition to the low expression level of Bcl2 (Figures 3d and e), the drop-off in cell viability of OPN-silenced AML12 hepatocytes in response to OGD could not only be due to an alteration in the recovery of ATP stores but also due to defense response against oxidative stress. We thus evaluated the cellular level of ATP and the NRF2-dependent antioxidant response including nicotinamide adenine dinucleotide phosphate (NADPH) quinine oxidoreductase 1 (NQO1) as an indirect indicator of oxidative stress. In the basal state, OPN silencing mediated a decrease in the ATP level (at 2 h-restoration period: Figure 4e) and an increase in NQO1 expression (at 16 h-restoration period: Figure 4f; at 2 h-restoration period: +3.28±0.64, P=0.01). These responses were further amplified by the OGD treatment (Figures 4e and f). The regulation of NQO1 by OPN silencing and OGD treatment was abolished by the silencing of NRF2 with siRNA (data not shown). This indicated that OPN silencing in AML12 hepatocytes altered the ATP level and could enhance oxidative stress.
The OPN deficiency sensitized hepatocytes to cell death induced by TNFα
As TNFα was strongly upregulated in the liver of Opn−/− mice on I–R (Figure 2b) and could mediate hepatocyte death, we then evaluated the effect of the OPN deficiency on the sensitivity to cell death induced by TNFα. As OPN silencing affected basal cell viability (Figure 3b), the following results were expressed as a percentage of basal state, as indicated. Although treatment of cells with TNFα alone was without an effect on cell viability and cytotoxicity, the downregulation of OPN sensitized AML12 hepatocytes to cell death as evaluated by the MTT assay (Figure 5a) and LDH release induced by TNFα (Figure 5b). Taken together, these results indicated that OPN had a hepatoprotective role under basal conditions and in response to OGD and TNFα.

OPN silencing sensitized AML12 hepatocytes to cell death induced by TNFα. After silencing of OPN with siRNA in AML12 cells, the cell viability (MTT assay) (a) and LDH release (b) were evaluated in response to TNFα (20 ng/ml) for 16 h in DMEM, 4.5 g/l glucose supplemented with 0.5% BSA (n=3). As OPN silencing affected the basal cell viability, the results were expressed as a function of the corresponding basal state (si Ctr or si OPN, respectively) and as means±S.E.M. Data were statistically analyzed using Student’s t-test. *P<0.05
OPN silencing increased iNOS expression in the basal state and sensitized RAW macrophages to inflammatory signals
Liver macrophages are activated in response to I–R, which leads to cell injury that is mainly due to the high production of NO and the cytokine TNFα.3 As we reported that OPN was strongly upregulated on I–R (Figures 1a and b) and that OPN regulates motility, NO production and cytokine expression in macrophages,9, 23, 24 we first investigated the effect of the OPN silencing in RAW macrophages on expression of integrins and inflammatory markers. As shown in Figure 6a, OPN silencing with siRNA caused substantial modifications to integrin and CD44 expression. Under basal conditions, the expression of αV, β1 and CD44 decreased and the expression of β3 increased after silencing of OPN. OPN silencing of RAW cells also displayed upregulation of iNOS, IL1β and IL6 compared with control cells in the basal state (Figure 6b). This response was also strongly amplified by inflammatory signals such as endotoxin (lipopolysaccharide (LPS)). A higher expression of iNOS ( × 2.3±0.3), TNFα ( × 1.7±0.3), IL1β ( × 2.3±0.3) and particularly IL6 ( × 52.4±13.6) was detected in OPN-silenced versus Ctr RAW cells in response to LPS (Figure 6c). Although validation in isolated hepatic macrophages (Kupffer cells) has to be assessed, these results could indicate that the deficiency in OPN modified the properties of macrophages with higher production of NO as evaluated by iNOS expression and higher responsiveness to inflammatory signals.

OPN deficiency increased iNOS expression in the basal state and sensitized macrophages to inflammatory signals. After silencing of OPN with siRNA in RAW macrophages, the gene expression of OPN, its receptors (CD44, integrins β1, αV and β3) (a) and inflammation (iNOS and the cytokines IL1β, TNFα and IL6) (b) were evaluated in the basal state (n=3). (c) After silencing of OPN with siRNA, RAW macrophages were incubated or not with LPS (100 ng/ml) for 6 h. The gene expression of iNOS, IL1β, TNFα and IL6 was then evaluated (n=3). The mRNA levels were normalized to RPLP0 mRNA levels and expressed as fold stimulation±S.E.M. versus si Ctr or si Ctr plus LPS. Data were statistically analyzed using the Mann–Whitney or Student’s t-test. *versus si Ctr and $versus si Ctr plus LPS. P<0.05
Discussion
We first described that hepatic I–R promoted the upregulation of hepatic and plasma OPN. Interestingly, it has been reported that OPN could be regulated by I–R in other organs. For example, OPN was elevated in renal I–R injury,21 in the brain during early cerebral I–R in rats13 and in the porcine heart during hemoperfusion.12 In the liver, the cellular origin of increased OPN expression in response to I–R has not been investigated. A large number of liver cells could express OPN and its upregulation was first reported in activated macrophages and stellate cells after intoxication with carbon tetrachloride.8 We and others have shown that hepatocytes could also be a source of OPN6, 7, 25 and TNFα enhanced its expression in vitro in hepatocytes.7 Future investigation is necessary to identify the source and the molecular mechanism responsible for this response. However, we report here that induction of OPN is associated with hepatic I–R injury (elevated AST/ALT, hepatocyte death) and this seems to be a conserved response to liver injury. Indeed, the upregulation of hepatic OPN has also been reported in the liver of patients with nonalcoholic fatty liver, alcoholic and nonalcoholic liver cirrhosis, primary biliary cirrhosis, autoimmune hepatitis and primary sclerosing cholangitis.6, 26 Recent reports further suggested that systemic OPN levels were predictive of liver fibrosis in patients with chronic hepatitis B, chronic hepatitis C, alcoholic and nonalcoholic liver disease.9, 10
We then reported that OPN provides endogenous protection of the liver from I–R injury. The OPN deficiency aggravated liver injury induced by I–R as evaluated by the elevated AST and ALT levels and extensive areas of necrosis in the liver. Under these experimental conditions, the protective effect of OPN seems to be more associated with prevention of necrosis than apoptosis. Indeed, apoptosis evaluated with the TUNEL assay and from the level of caspases 3 activity were not aggravated after ablation of OPN. However, we cannot rule out the protective effect of OPN on cell apoptosis. This protective effect could occur in early stages and necrosis, in some circumstances, can also be viewed as aborted apoptosis, due to insufficient ATP as induced by I–R to drive the apoptotic program.27 In accordance with this, our in vitro studies have shown that an OPN deficiency caused more apoptosis in low stringent conditions of OGD. The OPN deficiency also caused a strong decrease in the ATP level, which was amplified in response to OGD. Interestingly, the protective effect of OPN on liver injury has also been reported with carbon tetrachloride intoxication. OPN-deficient mice were more susceptible to carbon tetrachloride treatment, displaying more necrosis and higher ALT levels during the initial steps.28 In contrast, liver necrosis and ALT levels were lower in the transgenic mice overexpressing OPN in hepatocytes when treated with carbon tetrachloride.29
A protective effect of OPN against cardiac and renal ischemic injury has also been reported. Wang et al. reported that patients undergoing mitral valve replacement and with high plasma OPN levels had more activated transcription factors, nuclear factor kappa B and signal transducer and activator of transcription 3, higher expression of effector proteins and better cardioprotective effects. Further, OPN treatment displayed cardioprotective effects on neonatal cardiomyocytes 24 h after anoxia–reoxygenation injury, when assessed from cell viability, LDH activity, MDA content and SOD activity.30 OPN has the potential to modulate different phases of injury, healing and myocardial remodeling. Genetically engineered mouse studies provide evidence that increased expression of OPN may have a protective role against left ventricular dilation after myocardial infarction. However, in the infarct remodeling stage, OPN may exacerbate unfavorable fibrosis.31 In renal ischemic injury, a protective role for OPN is supported by the finding that increased OPN expression occurs in the distal tubules, which contain cells that are resistant to ischemic injury. In contrast, proximal tubules, which are less tolerant of ischemic injury, show very little OPN expression after ischemia.32 The deficiency in OPN reduced the tolerance to acute renal ischemia associated with increased I–R injury at 24 h after reperfusion.18 The overexpression of OPN via the hyperactivation of Wnt signaling, as detected in Brown Norway rats, is also critical for the maintenance of their inherent ischemic resistance.20 However, OPN again stimulated the development of renal fibrosis after acute ischemic insult.19
In the current study, this protective role of OPN from I–R injury was first associated with the protection of hepatocytes from cell death. The OPN deficiency decreased hepatocyte viability in vitro and further sensitized hepatocytes to cell death induced by OGD and TNFα. The OPN deficiency in AML12 hepatocytes enhanced apoptosis and the level of activated caspase 3. Interestingly, we found that the decrease in viability of primary and AML12 hepatocytes after OPN silencing was associated with a substantial decline in Bcl2 expression (Figure 3). In addition, a deficiency in OPN aggravated hepatic Bcl2 downregulation on I–R (Figure 3). It is well documented that Bcl2 is important for liver development by preventing apoptosis33 and its hepatic overexpression prevented the liver cell necrosis induced by hypoxia.34 Hepatic I–R in our mice was also associated with substantial upregulation of TNFα (Figure 2b), which is also involved in cell injury. As Bcl2 protects hepatocytes from TNFα-induced apoptosis,35 its downregulation induced by silencing of OPN could also sensitize hepatocytes to cell death induced by TNFα. Importantly, the regulation of Bcl2 by OPN seemed to be specific to hepatocytes, as silencing of OPN in RAW macrophages was without any effect on Bcl2 expression (data not shown). This association between OPN and Bcl2 expression was recently reported in liver carcinoma cells.36 Indeed, OPN silencing resulted in strong downregulation of the anti-apoptotic Bcl2 family members, including Bcl2. This response has been associated with a blockade of NF-κB activation and induction of mitochondria-mediated apoptosis.36
The level of OPN induction is thus determinant and its high expression conferred resistance of cells to hypoxia–reoxygenation injury. The cell survival role of OPN in hypoxia–reoxygenation-induced cell death has recently been reported for cancer cells. The upregulation of OPN in response to hypoxia–reoxygenation mediated the protective function of OPN via sustained AKT activation. However, OPN could be inactivated via proteolytic cleavage by caspase 8 and accumulation of caspase-generated OPN fragments could induce cell death via p53.37 In Brown Norway rats with inherent ischemic resistance, the overexpression of OPN reduced mitochondrial cytochrome c release and caspase 3 activity after renal I–R.20
The aggravation of liver injury in OPN null mice could also be due to the more pronounced inflammatory response induced by I–R. In the current study, we report that the deficiency in OPN was associated with a higher hepatic expression of TNFα, IL6, IFNγ and iNOS compared with the littermate mice on I–R. This massive release of TNFα and of iNOS-derived NO could induce hepatocyte damage. It is well established that TNFα is a crucial mediator in hepatic reperfusion injury and the inhibition of TNFα signaling by TNFα antiserum or genetic inactivation of TNF-receptor 1 ameliorated hepatic reperfusion injury and prolonged survival.1, 38, 39 iNOS-derived NO was also deleterious and the iNOS deficiency reduced liver injury (the AST/ALT level and hepatic areas of necrosis) after hepatic I–R.4 Ablation of iNOS or treatment with a specific iNOS inhibitor also resulted in complete protection against hypoxia-reperfusion-induced ALT release.5
iNOS and iNOS-derived NO were enhanced by inflammatory signals (DAMPs, dead cell-induced macrophage activation) but could also be negatively regulated by OPN. In Opn−/− mice, iNOS was already upregulated in the internal control lobe compared with Wt mice and strongly increased on I–R. The silencing of OPN in RAW macrophages also resulted in an increase in iNOS in the basal state and in response to an inflammatory signal (LPS). This role of OPN in the negative regulation of iNOS was recently described in macrophages. In response to inflammation, the iNOS-derived NO-increased OPN expression leads to the degradation of STAT1 and consequently inhibition in STAT1-dependent iNOS expression.24 OPN thus mediated negative feedback of iNOS expression and limited iNOS-derived NO synthesis in response to inflammation in macrophages. We also report that silencing of the OPN deficiency in macrophages resulted in an increase in pro-inflammatory cytokine synthesis in the basal state and in response to LPS.
In primary mouse kidney proximal tubule epithelial cells, OPN also suppressed iNOS-derived NO synthesis induced by the inflammatory mediators IFNγ and LPS. The inflammatory mediators increased iNOS and recombinant human OPN inhibited this response after neutralization of OPN. The inhibition of NO synthesis by OPN could be mediated via alpha v beta 3 integrin, which is known to be an OPN receptor.15 Further, the deficiency in OPN reduced tolerance to acute renal ischemia associated with increased iNOS, NO and I–R injury at 24 h after reperfusion.18
In conclusion, the hepatic I–R with 45 min of ischemia followed by 4 h of reperfusion induced upregulation of OPN and revealed its protective role in I–R injury (Figure 7). Endogenous OPN in hepatocytes conferred partial resistance to the cell death induced by OGD and TNFα. This could be mediated by the regulation of Bcl2 and the ATP level by OPN. This decreased rate of death of hepatocytes could consequently decrease the activation of liver macrophages and thus macrophage-mediated NO and TNFα release. Finally, lowering TNFα and NO levels could prevent additional hepatocyte injury induced by inflammation. Interestingly, this vicious cycle in I–R injury has been reported in mice with complete absence of NF-κB activation in hepatocytes (inactivation of NEMO).40 Endogenous OPN in macrophages also mediated negative feedback in iNOS-derived NO production and partially limited macrophage activation in response to an inflammatory signal. Therefore, OPN is an important factor in I–R-induced injury and additional studies need to carefully delineate the role of OPN in parenchymal and non-parenchymal cells as a function of the experimental model of liver I–R used (long lasting versus short period of ischemia and/or reperfusion). As OPN has an important role in immune responses, in tissue remodeling and in hepatic fibrosis, the role of OPN in late adverse post-ischemic effects has to be investigated.

Schematic representation showing the potential roles of OPN in hepatic I–R injury. The hepatic I–R with 45 min of ischemia followed by 4 h of reperfusion induced upregulation of OPN and revealed its protective role in I–R injury. Endogenous OPN in hepatocytes conferred partial resistance to cell death induced by OGD and TNFα. This could be mediated by the regulation of the Bcl2 and ATP levels by OPN. Endogenous OPN in macrophages also mediated negative feedback on iNOS-derived NO production and partially limited liver macrophage activation and TNFα production in response to an inflammatory signal. The decreased rate of death of hepatocytes could consequently decrease the activation of liver macrophages and thus macrophage-mediated NO and TNFα release. Finally, lowering TNFα and NO levels could prevent additional hepatocyte injury induced by inflammation
Materials and Methods
Animals and surgical protocol
Wt and OPN-deficient mice (Opn−/−) (B6.129S6(cg)-spp1 tm1Blh/J from Jackson Laboratory, Bar Harbor, ME, USA) C57BL/6 male mice (10–12 weeks of age) had free access to water and were fed a normal diet ad libitum. Mice were anesthetized with intraperitoneal injection of pentobarbital (60 mg/kg) and blood was collected from the tail (200 μl). After laparotomy, an atraumatic clip (FD562; Aesculap, Tuttlingen, Germany) was used to interrupt the arterial and portal venous blood supply to the left and middle liver lobes. The right lobe of the liver continued to be vascularized and was used as internal control lobe (Ctr lobe). The abdominal wall was closed with a running suture. After 45 min of partial warm ischemia, the clamp was removed to initiate hepatic reperfusion. Mice were killed 4 h after reperfusion. Blood was collected from the inferior vena cava. Left and middle liver lobes (I–R lobes) and the right liver lobe (Ctr lobe) were differentiated, and for each, one part was fixed in 4% formaldehyde and the other one was frozen in liquid nitrogen and stored at −80 °C. SHAM controls underwent the same procedure but without vascular occlusion. No differences have been noted between SHAM Wt and SHAM Opn−/− mice (data not shown). The partial warm I–R is referred to as I–R throughout the manuscript. The guidelines of laboratory animal care were followed, and the local ethical committee approved the animal experiments.
Circulating levels of transaminases and OPN
Determination of plasma transaminases (AST/ALT) was performed using in vitro test with pyridoxal phosphate activation on Roche/Hitachi cobas c systems (ASTPM, ALTPM, cobas, Meylan, France). Roche/Hitachi cobas c systems automatically calculate the analyte concentration of each sample. Plasma OPN levels were evaluated with the Quantikine ELISA Mouse/Rat OPN Immunoassay (R&D Systems), as per manufacturer’s instructions.
Light microscopy
For conventional light microscopy, liver lobes were fixed in 4% neutral-buffered formaldehyde solution and embedded in paraffin. Sections (4 μm thick) were stained with hematoxylin eosin safran. Histopathological features were observed: localization and the extent of hepatocyte injury were evaluated.
TUNEL assay
Liver biopsies were incubated in formol, paraffin embedded and sectioned. The TUNEL assay was performed as per manufacturer’s instructions of ApopTag Plus Peroxidase In Situ Apoptosis Detection kit (Millipore, Meylan, France). The liver sections were then counterstained with Mayer's hematoxylin (Carl Roth, Karlsruhe, Germany). Specimens were evaluated by light microscopy.
Cellular models and treatments
Mouse hepatocytes were isolated with a two-step collagenase procedure. Briefly, mouse livers were perfused with HEPES buffer containing 8 g/l NaCl, 33 mg/l Na2HPO4, 200 mg/l KCl and 2.38 g/l HEPES, pH 7.5, supplemented with 0.5 mM EGTA for 3 min at 3 ml/min, then with HEPES buffer for 3 min at 3 ml/min and finally with HEPES buffer supplemented with 1.5 g/l CaCl2 and 0.026% collagenase type IV (Sigma-Aldrich; C5138, Saint-Quentin-Fallavier, France) for 7 min at 3 ml/min. Livers were then carefully removed and minced in Williams’ E medium (Life Technologies, St Aubin, France) supplemented with 10% fetal bovine serum (PAA Laboratories, Vélizy-Villacoublay, France), 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine and 0.02 UI/ml insulin (Umulin, Lilly France, Neuilly-sur-Seine, France). The cell suspension was then filtered (250 μm) and hepatocytes were collected by centrifugation at 50 × g for 5 min. Viability was evaluated by trypan blue exclusion (Sigma-Aldrich). Mouse AML12 hepatocytes (CRL-2254, ATCC, Manassas, VA, USA) and RAW 264.7 macrophages (TIB-71, ATCC) were cultured in ‘cell medium’ (DMEM, 4.5 g/l glucose, 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine) supplemented with 10% fetal bovine serum (PAA Laboratories), under 5% CO2 at 37 °C.
Cells were transfected with OPN siRNA (MSS209393, Life Technologies) (referred to as si OPN) or control siRNA (Life Technologies) (referred to as si Ctr) at 30 nM using Lipofectamine RNAiMAX (Life Technologies), according to the manufacturer’s instructions. After 48 h, cells were treated as indicated: (a) with TNFα (20 ng/ml) (Pepro Tech, Rocky Hill, NJ, USA) for 16 h in ‘cell medium’ supplemented with 0.5% bovine serum albumin; (b) with KCN (60179, Fluka chemika, Buchs, Switzerland) (2.5, 5 or 10 mM) for 1 h in DMEM without glucose and supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine and 0.5% bovine serum albumin, followed by a 2- or 16-h restoration period (with oxygen and glucose) as indicated, in ‘cell medium’ supplemented with 0.5% bovine serum albumin; (c) with LPS (L3024, Sigma-Aldrich) (100 ng/ml) for 6 h in ‘cell medium’ supplemented with 0.5% bovine serum albumin.
The si Ctr corresponds to a RNAi duplex designed with a comparable GC content (%GC=68) as the OPN RNAi duplex (si OPN) (%GC=52). The si Ctr is used as a negative control in the RNAi experiment as suggested by the manufacturer. To validate it, we evaluated the expression of gene including OPN, iNOS, TNFα and IL6 after transfection with si Ctr versus without the RNAi duplex (mock). The si Ctr did not modify the expression of these genes in RAW cells (OPN: mock: 1, si Ctr: 0.95±0.06, P=0.229; iNOS: mock: 1, si Ctr: 1.05±0.06, P=0.435; TNFα: mock: 1, si Ctr: 1.04±0.06, P=0.275; IL6: mock: 1, si Ctr: 1.23±0.46, P=0.320; n=3) and the expression of OPN in AML12 cells (OPN: mock: 1, si Ctr: 0.96, n=1).
LDH release assay
The cytotoxicity induced by TNFα and KCN was assessed by LDH release into the ‘cell medium’. Following treatment as indicated, the cell medium was collected and centrifuged at 50 × g for 5 min to obtain a cell-free supernatant. The activity of LDH in the medium was determined using the cytotoxicity detection kit (Roche Diagnostics, Meylan, France) according to the manufacturer’s instructions. Absorbance was recorded at 490 nm using a microplate spectrophotometer system (ELX800, Bio-TEK instruments, Colmar, France) and the results are expressed as a percentage of cytotoxicity ((treated cells DO−control cells DO)/(maximum LDH release DO−control cells DO) × 100). Reagents did not interfere with the determination of LDH.
MTT assay
The assay is dependent on the ability of viable cells to metabolize a water-soluble tetrazolium salt into a water-insoluble formazan product. Following treatment as indicated, cells were incubated for 2 h with 0.5 mg/ml MTT in serum-free medium (DMEM). After removing the supernatant, DMSO was added to completely dissolve the formazan product. Aliquots of the resulting solutions were transferred to 96-well plates and the absorbance was recorded at 550 nm using the microplate spectrophotometer system (ELX800, Bio-TEK instruments). Results are presented as a percentage of the control values.
Cell apoptosis
Cell apoptosis was evaluated by flow cytometry following double staining with annexin V-PE and 7-AAD according the manufacturer’s instructions (Annexin V-PE apoptosis detection kit I, BD Biosciences, Pont de claix, France).
ATP measurement
Cells were lysed in lysis buffer (see ‘Immunoblotting’). Lysates were diluted in the dilution buffer according to the manufacturer’s instructions (ATP Bioluminescence Assay Kit HS II, Roche Diagnostics) and then loaded in duplicate on a dark 96-well plate (Greiner Bio-One, Courtaboeuf, France). Luciferase was added and the luminescence measured immediately using a luminometer (FLUOstar OPTIMA, BMG LABTECH, Ortenberg, Germany). The ATP content was evaluated for each sample, which was standardized to the protein content.
DEVDase activity measurement
After the indicated treatments, cells were lysed in lysis buffer (see ‘Immunoblotting’). Lysates were standardized to the protein content and loaded onto a black 96-well plate (Greiner Bio-One) in the presence of 0.2 mmol/l caspase 3 substrate Ac-DEVD-AMC diluted in the following buffer: 50 mmol/l HEPES (pH 7.5), 150 mmol/l NaCl, 20 mmol/l EDTA and 10 mmol/l DTT. The caspase activity was determined on a fluoroscan at 460 nm with or without 1 μmol/l Ac-DEVD-CHO, and the specific activity was expressed as the change in absorbance per minute per milligram of protein.
Immunoblotting
Cells or frozen tissues were solubilized in lysis buffer (20 mM Tris, pH 7.4, 150 mM NaCl, 10 mM EDTA, 150 mM NaF, 2 mM sodium orthovanadate, 10 mM pyrophosphate, proteases inhibitors cocktail, and 1% Triton X-100) for 45 min at 4 °C. Lysates were cleared (14 000 r.p.m., 15 min). Proteins were quantified (BCA Protein assay kit, 23225, Thermo Fisher Scientific Inc., Waltham, MA, USA) and separated by SDS-PAGE and immunoblotted as described.41 The proteins were probed with anti-bcl2 ((50E3) 2870, Cell signaling, Danvers, MA, USA), anti-β actin ((C4) sc-47778, Santa Cruz Biotechnologies Inc., Dallas, TX, USA) and anti-caspase 3 (9662, Cell signaling) antibodies (1 μg/ml).
Real-time quantitative PCR analysis
Cell or total liver RNA was extracted using the RNeasy Mini Kit (74104, Qiagen, Hilden, Germany) and treated with Turbo DNA-free (AM 1907, Thermo Fisher scientific Inc.) following the manufacturer’s protocol. The quantity and quality of the RNA were determined using the Agilent 2100 Bioanalyzer with RNA 6000 Nano Kit (5067-1511, Agilent Technologies, Santa Clara, CA, USA). Total RNA (1 μg) was reverse transcribed with a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher scientific Inc.). Real-time quantitative PCR was performed in duplicate for each sample using the StepOne Plus Real-Time PCR System (Thermo Fisher scientific Inc.) as previously described.41, 42 TaqMan gene expression assays were purchased from Thermo Fisher scientific Inc.: RPLP0 (ribosomal protein, large, P0) (Mm99999223_gH); TNFα (Mm00443258_m1); IL1β (Mm00434228_m2); OPN (Mm00436767_m1); iNOS (Mm01309897_m1); IL6 (Mm00446190_m1); IFNγ (Mm01168134_m1); CD44 (Mm01277163_m1); β1 integrin (Mm01253227_m1); αv integrin (Mm00434506_m1); β3 integrin (Mm00443980_m1); NQO1 (NAD(P)H dehydrogenase, quinone 1) (Mm00500821_m1) and Bcl2 (Mm00477631_m1). Gene expression was normalized to the mouse housekeeping gene RPLP0 and calculated based on the comparative cycle threshold Ct method (2–ΔΔCt).
Statistical analysis
Statistical significance of differential gene expression between two study groups was determined using the nonparametric Mann–Whitney test with the ΔCt of each group. Other data from mice and cell preparations were statistically analyzed using the Mann–Whitney test or the Student t-test. Data from cell lines were statistically analyzed using the Student t-test. P<0.05 was considered as significant.
Abbreviations
- I–R:
-
ischemia–reperfusion
- OPN:
-
osteopontin
- ALT:
-
alanine aminotransferase
- AST:
-
aspartate aminotransferase
- LPS:
-
lipopolysaccharide
- TNF:
-
tumor necrosis factor
- IL:
-
interleukin
- iNOS:
-
inducible nitric oxide synthase
- IFN:
-
interferon
References
Schwabe RF, Brenner DA . Mechanisms of liver injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol 2006; 290: G583–G589.
de Groot H, Rauen U . Ischemia-reperfusion injury: processes in pathogenetic networks: a review. Transplant Proc 2007; 39: 481–484.
Peralta C, Jimenez-Castro MB, Gracia-Sancho J . Hepatic ischemia and reperfusion injury: effects on the liver sinusoidal milieu. J Hepatol 2013; 59: 1094–1106.
Hamada T, Duarte S, Tsuchihashi S, Busuttil RW, Coito AJ . Inducible nitric oxide synthase deficiency impairs matrix metalloproteinase-9 activity and disrupts leukocyte migration in hepatic ischemia/reperfusion injury. Am J Pathol 2009; 174: 2265–2277.
Taniai H, Hines IN, Bharwani S, Maloney RE, Nimura Y, Gao B et al. Susceptibility of murine periportal hepatocytes to hypoxia-reoxygenation: role for NO and Kupffer cell-derived oxidants. Hepatology 2004; 39: 1544–1552.
Bertola A, Deveaux V, Bonnafous S, Rousseau D, Anty R, Wakkach A et al. Elevated expression of osteopontin may be related to adipose tissue macrophage accumulation and liver steatosis in morbid obesity. Diabetes 2009; 58: 125–133.
Sahai A, Malladi P, Melin-Aldana H, Green RM, Whitington PF . Upregulation of osteopontin expression is involved in the development of nonalcoholic steatohepatitis in a dietary murine model. Am J Physiol Gastrointest Liver Physiol 2004; 287: G264–G273.
Kawashima R, Mochida S, Matsui A, YouLuTu ZY, Ishikawa K, Toshima K et al. Expression of osteopontin in Kupffer cells and hepatic macrophages and Stellate cells in rat liver after carbon tetrachloride intoxication: a possible factor for macrophage migration into hepatic necrotic areas. Biochem Biophys Res Commun 1999; 256: 527–531.
Nagoshi S . Osteopontin: versatile modulator of liver diseases. Hepatol Res 2014; 44: 22–30.
Patouraux S, Bonnafous S, Voican CS, Anty R, Saint-Paul MC, Rosenthal-Allieri MA et al. The osteopontin level in liver, adipose tissue and serum is correlated with fibrosis in patients with alcoholic liver disease. PLoS One 2012; 7: e35612.
Chidlow G, Wood JP, Manavis J, Osborne NN, Casson RJ . Expression of osteopontin in the rat retina: effects of excitotoxic and ischemic injuries. Invest Ophthalmol Vis Sci 2008; 49: 762–771.
Kossmehl P, Schonberger J, Shakibaei M, Faramarzi S, Kurth E, Habighorst B et al. Increase of fibronectin and osteopontin in porcine hearts following ischemia and reperfusion. J Mol Med (Berl) 2005; 83: 626–637.
Baliga SS, Merrill GF, Shinohara ML, Denhardt DT . Osteopontin expression during early cerebral ischemia-reperfusion in rats: enhanced expression in the right cortex is suppressed by acetaminophen. PLoS One 2011; 6: e14568.
Sodhi CP, Phadke SA, Batlle D, Sahai A . Hypoxia stimulates osteopontin expression and proliferation of cultured vascular smooth muscle cells: potentiation by high glucose. Diabetes 2001; 50: 1482–1490.
Hwang SM, Lopez CA, Heck DE, Gardner CR, Laskin DL, Laskin JD et al. Osteopontin inhibits induction of nitric oxide synthase gene expression by inflammatory mediators in mouse kidney epithelial cells. J Biol Chem 1994; 269: 711–715.
Truong LD, Sheikh-Hamad D, Chakraborty S, Suki WN . Cell apoptosis and proliferation in obstructive uropathy. Semin Nephrol 1998; 18: 641–651.
Scatena M, Almeida M, Chaisson ML, Fausto N, Nicosia RF, Giachelli CM . NF-kappaB mediates alphavbeta3 integrin-induced endothelial cell survival. J Cell Biol 1998; 141: 1083–1093.
Noiri E, Dickman K, Miller F, Romanov G, Romanov VI, Shaw R et al. Reduced tolerance to acute renal ischemia in mice with a targeted disruption of the osteopontin gene. Kidney Int 1999; 56: 74–82.
Persy VP, Verhulst A, Ysebaert DK, De Greef KE, De Broe ME . Reduced postischemic macrophage infiltration and interstitial fibrosis in osteopontin knockout mice. Kidney Int 2003; 63: 543–553.
Vinas JL, Sola A, Jung M, Mastora C, Vinuesa E, Pi F et al. Inhibitory action of Wnt target gene osteopontin on mitochondrial cytochrome c release determines renal ischemic resistance. Am J Physiol Renal Physiol 2010; 299: F234–F242.
Zhang ZX, Shek K, Wang S, Huang X, Lau A, Yin Z et al. Osteopontin expressed in tubular epithelial cells regulates NK cell-mediated kidney ischemia reperfusion injury. J Immunol 2010; 185: 967–973.
Bailly-Maitre B, Fondevila C, Kaldas F, Droin N, Luciano F, Ricci JE et al. Cytoprotective gene bi-1 is required for intrinsic protection from endoplasmic reticulum stress and ischemia-reperfusion injury. Proc Natl Acad Sci USA 2006; 103: 2809–2814.
Guo H, Cai CQ, Schroeder RA, Kuo PC . Osteopontin is a negative feedback regulator of nitric oxide synthesis in murine macrophages. J Immunol 2001; 166: 1079–1086.
Gao C, Guo H, Mi Z, Grusby MJ, Kuo PC . Osteopontin induces ubiquitin-dependent degradation of STAT1 in RAW264.7 murine macrophages. J Immunol 2007; 178: 1870–1881.
Sahai A, Pan X, Paul R, Malladi P, Kohli R, Whitington PF . Roles of phosphatidylinositol 3-kinase and osteopontin in steatosis and aminotransferase release by hepatocytes treated with methionine-choline-deficient medium. Am J Physiol Gastrointest Liver Physiol 2006; 291: G55–G62.
Syn WK, Choi SS, Liaskou E, Karaca GF, Agboola KM, Oo YH et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 2011; 53: 106–115.
Malhi H, Gores GJ, Lemasters JJ . Apoptosis and necrosis in the liver: a tale of two deaths? Hepatology 2006; 43 (2 Suppl 1): S31–S44.
Lorena D, Darby IA, Gadeau AP, Leen LL, Rittling S, Porto LC et al. Osteopontin expression in normal and fibrotic liver. altered liver healing in osteopontin-deficient mice. J Hepatol 2006; 44: 383–390.
Koh HS, Matsui A, Mimura S, Inao M, Saitoh E, Ohno A et al. Increased cytoprotective function in the liver of transgenic mice expressing osteopontin in hepatocytes. Hepatol Res 2005; 32: 46–51.
Wang Y, Chen B, Shen D, Xue S . Osteopontin protects against cardiac ischemia-reperfusion injury through late preconditioning. Heart Vessels 2009; 24: 116–123.
Okamoto H, Imanaka-Yoshida K . Matricellular proteins: new molecular targets to prevent heart failure. Cardiovasc Ther 2012; 30: e198–e209.
Persy VP, Verstrepen WA, Ysebaert DK, De Greef KE, De Broe ME . Differences in osteopontin up-regulation between proximal and distal tubules after renal ischemia/reperfusion. Kidney Int 1999; 56: 601–611.
Lacronique V, Mignon A, Fabre M, Viollet B, Rouquet N, Molina T et al. Bcl-2 protects from lethal hepatic apoptosis induced by an anti-Fas antibody in mice. Nat Med 1996; 2: 80–86.
Yamabe K, Shimizu S, Kamiike W, Waguri S, Eguchi Y, Hasegawa J et al. Prevention of hypoxic liver cell necrosis by in vivo human bcl-2 gene transfection. Biochem Biophys Res Commun 1998; 243: 217–223.
Neuman MG . Apoptosis in diseases of the liver. Crit Rev Clin Lab Sci 2001; 38: 109–166.
Zhao J, Dong L, Lu B, Wu G, Xu D, Chen J et al. Down-regulation of osteopontin suppresses growth and metastasis of hepatocellular carcinoma via induction of apoptosis. Gastroenterology 2008; 135: 956–968.
Kim HJ, Lee HJ, Jun JI, Oh Y, Choi SG, Kim H et al. Intracellular cleavage of osteopontin by caspase-8 modulates hypoxia/reoxygenation cell death through p53. Proc Natl Acad Sci USA 2009; 106: 15326–15331.
Colletti LM, Remick DG, Burtch GD, Kunkel SL, Strieter RM, Campbell DA Jr. . Role of tumor necrosis factor-alpha in the pathophysiologic alterations after hepatic ischemia/reperfusion injury in the rat. J Clin Invest 1990; 85: 1936–1943.
Rudiger HA, Clavien PA . Tumor necrosis factor alpha, but not Fas, mediates hepatocellular apoptosis in the murine ischemic liver. Gastroenterology 2002; 122: 202–210.
Beraza N, Ludde T, Assmus U, Roskams T, Vander Borght S, Trautwein C . Hepatocyte-specific IKK gamma/NEMO expression determines the degree of liver injury. Gastroenterology 2007; 132: 2504–2517.
Anty R, Bekri S, Luciani N, Saint-Paul MC, Dahman M, Iannelli A et al. The inflammatory C-reactive protein is increased in both liver and adipose tissue in severely obese patients independently from metabolic syndrome, Type 2 diabetes, and NASH. Am J Gastroenterol 2006; 101: 1824–1833.
Bekri S, Gual P, Anty R, Luciani N, Dahman M, Ramesh B et al. Increased adipose tissue expression of hepcidin in severe obesity is independent from diabetes and NASH. Gastroenterology 2006; 131: 788–796.
Acknowledgements
We thank Dr MC Brahimi-Horn for editorial correction. We thank Véronique Corcelle and the INSERM U1065 animal facility staff for their excellent care of mice; E Gouze and Y Le Marchand-Brustel are thanked for critical reading of the manuscript. This work was supported by grants from the INSERM (France), the University of Nice, the Programme Hospitalier de Recherche Clinique (Centre Hospitalier Universitaire of Nice) and charities (Association Française pour l’Etude du Foie (AFEF)/LFB and European Foundation for the study of Diabetes/Lilly European Diabetes Research Programme to PG). This work was also funded by the French Government (National Research Agency, ANR) through the ‘Investments for the Future’ LABEX SIGNALIFE: program reference #ANR-11-LABX-0028-01. VJL was supported by the Programme Hospitalier de Recherche Clinique (Centre Hospitalier Universitaire of Nice) and the Association pour la Recherche sur le Cancer (France). SP was supported by the Fondation Recherche Médicale.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by C Munoz-Pinedo
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Patouraux, S., Rousseau, D., Rubio, A. et al. Osteopontin deficiency aggravates hepatic injury induced by ischemia–reperfusion in mice. Cell Death Dis 5, e1208 (2014). https://doi.org/10.1038/cddis.2014.174
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.174
Keywords
This article is cited by
-
Spatial transcriptomics analysis of zone-dependent hepatic ischemia-reperfusion injury murine model
Communications Biology (2023)
-
L-Cysteine attenuates osteopontin-mediated neuroinflammation following hypoxia-ischemia insult in neonatal mice by inducing S-sulfhydration of Stat3
Acta Pharmacologica Sinica (2022)
-
MCD diet-induced steatohepatitis generates a diurnal rhythm of associated biomarkers and worsens liver injury in Klf10 deficient mice
Scientific Reports (2020)
-
Osteopontin protects against lung injury caused by extracellular histones
Mucosal Immunology (2019)
-
Metabolic modulation of acetaminophen-induced hepatotoxicity by osteopontin
Cellular & Molecular Immunology (2019)
