
Abstract
p38 Mitogen-activated protein (MAP) kinase is involved in the apoptosis of nucleated cells. Although platelets are anucleated cells, apoptotic proteins have been shown to regulate platelet lifespan. However, the involvement of p38 MAP kinase in platelet apoptosis is not yet clearly defined. Therefore, we investigated the role of p38 MAP kinase in apoptosis induced by a mimetic of BH3-only proteins, ABT-737, and in apoptosis-like events induced by such strong platelet agonists as thrombin in combination with convulxin (Thr/Cvx), both of which result in p38 MAP kinase phosphorylation and activation. A p38 inhibitor (SB202190) inhibited the apoptotic events induced by ABT-737 but did not influence those induced by Thr/Cvx. The inhibitor also reduced the phosphorylation of cytosolic phospholipase A2 (cPLA2), an established p38 substrate, induced by ABT-737 or Thr/Cvx. ABT-737, but not Thr/Cvx, induced the caspase 3-dependent cleavage and inactivation of cPLA2. Thus, p38 MAPK promotes ABT-737-induced apoptosis by inhibiting the cPLA2/arachidonate pathway. We also show that arachidonic acid (AA) itself and in combination with Thr/Cvx or ABT-737 at low concentrations prevented apoptotic events, whereas at high concentrations it enhanced such events. Our data support the hypothesis that the p38 MAPK-triggered arachidonate pathway serves as a defense mechanism against apoptosis under physiological conditions.
Similar content being viewed by others

Main
Platelets play a key role in normal and pathological hemostasis through their ability to rapidly adhere to activated or injured endothelium and subendothelial matrix proteins (platelet adhesion), and to other activated platelets (platelet aggregation).1 Platelets are also considered as key mediators of thrombosis, vascular inflammation, and atherosclerosis.2, 3
Apoptosis play a significant role in platelet production from megakaryocytes, and circulating platelets contain many components of the apoptotic machinery.4 Indeed, cytochrome c, caspase 9, caspase 3, apoptotic protease-activating factor, and Bcl-2 family proteins (for example, BAK, BAX, Bcl-XL, Bcl-2, Bclw, Bim, Bid) are all expressed in platelets.5, 6, 7, 8
A variety of stimuli can induce apoptotic or apoptotic-like events in platelets, such as ABT-737, thrombin, collagen, and A23187. ABT-737, a potent mimetic of Bcl-2 homology BH3-only proteins (including Bim, Bid, and other proteins that can be important for binding and neutralizing antiapoptotic Bcl-2 family proteins), induces caspase-dependent platelet apoptosis that is associated with BAX translocation from the cytosol to mitochondria and homo-oligomerization.7 In highly activated platelets, several structural and functional changes, such as phosphatidylserine (PS) externalization, cell shrinkage, loss of mitochondrial membrane potential (ΔΨm), microparticle (MP) formation, and cleavage of gelsolin and protein kinase C-δ, are reminiscent of nucleated cell apoptosis.9, 10, 11, 12, 13 However, strong agonists either did not activate caspase 314 or did so very weakly.15, 16, 17 A total of 13 types of cell death have been characterized for nucleated cells, including caspase-dependent and caspase-independent intrinsic apoptosis, autophagic cell death, necroptosis and others.18 Although highly activated platelets exhibit the characteristic features of dying cells, there is no consensus in the literature clearly defining the type of strong activated platelet death. Some authors define them as necrotic cells19 whereas others11, 20, 21 consider them as apoptotic platelets.
Mitogen-activated protein (MAP) kinases represent a family of threonine/tyrosine-activated serine/threonine kinases that control many cellular responses, such as proliferation, migration, differentiation, and apoptosis. In platelets, p38 MAP kinase is phosphorylated and activated by different physiological agonists, including thrombin, collagen, and thromboxane A2 (TxA2).22, 23, 24, 25 However, the mechanisms of p38 activation and their downstream effects are controversial and have not been clearly defined. p38 has been suggested to regulate platelet adhesion to collagen26 and aggregation.24, 27 In contrast, using different p38 inhibitors, other authors did not find any significant effect of p38 on platelet activation.28, 29, 30, 31
One of the established p38 substrates in platelets is cytosolic phospholipase A2 (cPLA2), which is phosphorylated by p38 at serine505 in agonist-stimulated platelets.32, 33, 34 In platelets, cPLA2 activity is responsible for the release of arachidonic acid (AA) from membrane phospholipids.35 AA is then metabolized by cyclooxygenase to prostaglandins, which are further converted to TxA2 by thromboxane synthase (TxS). In addition, AA is involved in the regulation of reactive oxygen species (ROS) production. cPLA2-induced AA is a prominent requirement for the activation of NADPH oxidase and generation of ROS in phagocytes.36 ROS may also be generated as a by-product during the oxidation of AA by cyclooxygenase or lipoxygenase.37, 38 Therefore, platelet cPLA2 is involved in the regulation of two different functions, ROS generation and eicosanoid production. Excess ROS generation and AA release play essential roles in the initiation of apoptosis in platelets.21, 39 As p38/cPLA2 is involved in the regulation of nucleated cell apoptosis40, 41, 42, 43 and the possible involvement of p38/cPLA2 in the induction of platelet apoptotic events is not known, we compared the mechanism of p38/cPLA2-mediated signaling induced by platelet agonists and ABT-737.
In this study, we show that p38 plays a significant role in ABT-737-induced platelet apoptosis, whereas it has no effect on thrombin in combination with convulxin(Thr/Cvx)-induced apoptotic-like events. ABT-737, but not Thr/Cvx, induced the caspase-dependent cleavage and inactivation of cPLA2. We also assessed multiple influences of AA/ROS on apoptotic-like events in platelets and suggest that the p38-triggered arachidonate pathway serves as a defense mechanism under physiological conditions.
Results
Activation of p38 MAP kinase plays a significant role in ABT-737-induced platelet apoptosis
To clarify the function of p38 in platelet apoptosis induced by two independent pathways, the BH3 mimetic compound ABT-737 was used as an activator of the BAK/BAX-caspase pathway and a combination of Thr/Cvx as agonists of the second caspase-independent pathway.
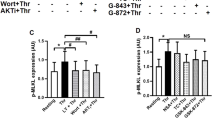
In our study, we used terms apoptosis-like events for platelets activated by strong agonists and apoptosis for ABT-737-induced platelet death. ABT-737 at concentrations ranging from 0.1 to 1 μM induced dose- and time-dependent increases in Annexin V binding (Figures 1a and c). Simultaneously, ABT-737 (0.1–1 μM) time dependently increased the phosphorylation of p38 (Figure 1b). In contrast to Thr/Cvx, ABT-737 did not activate αIIbβ3 integrins (Figure 1c) and did not induce MP formation (Figures 6a and b). The activation of p38 by thrombin and convulxin alone or in combination (Thr/Cvx) was transient, reaching a maximum after 5 min of incubation and then gradually decreasing to a level comparable with that of the control after 60 min (Figure 1d). Conversely, activation of p38 by ABT-373 was detectable only after 20 min of stimulation and remained constant even up to 90 min of stimulation (Figure 1a). A p38 inhibitor (SB202190, 1 μM) significantly decreased the amount of Annexin V-positive platelets (67±3% compared with ABT-737-stimulated platelets taken as 100%) and prevented decreases in ΔΨm (32±4%) stimulated by ABT-737 (Figures 2a and b). Similarly, SB202190 inhibited the cleavage of procaspase 3 by 31±3% (Figures 2 c and d), indicating that p38 is partly involved in ABT-737-induced caspase 3 activation and apoptosis.

BH3 mimetic (ABT-737) phosphorylates p38. (a) FACS analysis of Annexin V binding and (c) PAC-1 binding (integrin αIIbβ3 activation, mean fluorescence, AU) to WP (1 × 107/ml) incubated with ABT-737. Annexin V and PAC-1 binding are presented as the fold increase compared with the control taken as 1. Data are presented as means±S.E.M, n=5; +P<0.05 compared with the control. (b and d) Western blot analysis using WP (4 × 108/ml) of p38 phosphorylation after incubation with thrombin (5 mU/ml) and convulxin (5 ng/ml) or ABT-737 (0.1–1 μM) (data shown are representative of three independent experiments)

p38 MAP kinase inhibition prevents ABT-737-induced platelet apoptosis (a) FACS analysis of Annexin V-positive platelets (1 × 107/ml) and (b) mitochondrial membrane potential (ΔΨm) in WP incubated with ABT-737 (0.1 μM, 30 min) or pretreated (10 min, 1 μM) with p38 inhibitor (SB202190). (c) Western blot and (d) densitometry analyses of caspase 3 in human WP (4 × 108/ml) incubated with ABT-737 (0.1 μM) or pretreated (10 min, 1 μM) with p38 inhibitor (SB202190). In (a), ABT-737 was taken as 100%; in (b) and (d), the control was taken as 100%. Data are presented as means±S.E.M, n=4; +P<0.05 compared with the control, *P<0.05 compared with ABT-737
p38 MAP kinase is not involved in the generation of apoptotic-like events triggered by Thr/Cvx
We next investigated whether the activation of p38 is involved in Thr/Cvx-induced platelet death. The activation of platelets by Thr/Cvx significantly increased Annexin V binding 7.6±1.5 compared with control 1±0.14) and αIIbβ3 integrin activation (Figure 3a). The inhibition of p38 (SB202190, 1 μM), as assessed by the phosphorylation of its established substrate (HSP27) (Figures 3c and d), did not significantly inhibit Annexin V binding (Figure 3a) or decreases in ΔΨm (Figure 3b) and had no effect on αIIbβ3 integrin activation (Figure 3a), indicating that p38 is not involved in Thr/Cvx-induced platelet apoptotic-like events.

p38 inhibitor does not prevent PS surface exposure, decreased ΔΨm, and integrin αIIbβ3 activation in platelets stimulated with Thr/Cvx (a) FACS analysis of Annexin V-positive cells and PAC-1 binding and (b) ΔΨm in WP (1 × 107/ml) stimulated by Thr/Cvx or pretreated (10 min, 1 μM) with p38 inhibitor (SB202190). In (a) and (d), Thr/Cvx was taken as 100%; in (b), control was taken as 100%. Data are presented as means±S.E.M, n=6; +P<0.05 compared with the control, *P<0.05 compared with platelets incubated with Thr/Cvx. (c) Western blot and (d) densitometry analyses of p38 and HSP27 phosphorylation in WP (3 × 108/ml) stimulated with Thr/Cvx or pretreated (10 min, 1 μM) with p38 inhibitor (SB202190). The data shown are representative of three independent experiments
Platelet cPLA2 cleavage by ABT-737 is caspase dependent
The stimulation of platelets by Cvx and Thr/Cvx (both 5 min) and by ABT-737 (30 min) induced p38 and cPLA2 phosphorylation, and the latter was partly (in the case of Cvx and Thr/Cvx) and fully (in the case of ABT-737) prevented by preincubation with SB202190 (Figures 4a and b). However, in contrast to Cvx and Thr/Cvx, ABT-737 induced the cleavage of cPLA2, which was prevented by a procaspase 3 inhibitor (z-DEVD-fmk) at concentrations of 50–100 μM (Figures 4c and d). We then tested whether the cleavage of cPLA2 would inhibit its activity. One of the main functions of cPLA2 in platelets is associated with the production of thromboxane (TxA2) TxS. ABT-737 (1 μM) induced only a weak activation of TxS (4±0.3-fold increase compared with the control taken as 1), Cvx and Thr/Cvx (Figure 4e) strongly activated TxS (20±1.3- and 52±1.3-fold, respectively). The inhibition of p38 (SB202190, 1 μM) decreased the TxS activity induced by Cvx or Thr/Cvx to 75±1.7% and 87±1.8%, respectively, compared with the agonist-induced activity taken as 100% (Figure 4f).

ABT-737 cleavage of platelet cPLA2 is caspase dependent. (a) Western blot and (b) densitometry analysis of p-cPLA2 in WP incubated with Cvx (5 ng/ml), Thr/Cvx (5 mU/5 ng/ml), or ABT-737 (1 μM) or pretreated (10 min, 1 μM) with p38 inhibitor (SB202190). (c) Western blot and (d) densitometry analysis of cPLA2 in WP after stimulation with ABT-737 or pretreated with the indicated concentrations of the caspase 3 inhibitor:z-DEVD. Western blot data are representative of three independent experiments of p38 and cPLA2 phosphorylation in WP (4 × 108/ml). (e and f) ELISA data for the TxS activity in WP (4 × 108/ml) activated by Cvx (5 ng/ml, 5 min), Thr/Cvx (5 mU/5 ng/ml, 5 min), or ABT-737 (1 μM, 30 min) and after preincubation with SB202190 (1 μM, 10 min)
Arachidonic acid prevents apoptosis in platelets at low concentrations and induces apoptosis in platelets at high concentrations
In nucleated cells, AA has been shown to contribute to mitochondrial dysfunction during apoptosis;44, 45 however, AA was used at very high concentrations (more than 10 μM) in these studies. The depletion of AA prevents apoptotic development in cold-stored platelets whereas incubation with extremely high concentrations of AA (50–100 μM) induces platelet apoptosis.39 In the present study, we tested whether AA would contribute to apoptosis in freshly isolated platelets. Washed platelets (WP) were incubated with different concentrations of AA (10−7 to 10−4 M), and apoptosis was assessed by decreases in ΔΨm, PS exposure, and procaspase 3 cleavage. At high concentrations (10−5 and 10−4 M), AA did indeed induce a decline in ΔΨm (Figure 5a) and an increase in Annexin V binding (Figure 5b) and cleavage of procaspase 3 after 24 h of incubation (Figure 5c). However, at low concentrations (10−6 and 10−7 M), AA appeared to have antiapoptotic effects: it did not affect Annexin V binding, slightly increased ΔΨm (Figure 5a), and decreased the procaspase 3 cleavage (Figures 5d and e) induced by ABT-737 and procaspase 3 cleavage in stored platelets after 24 h (Figure 5c). At low concentrations, AA also decreased the Annexin V binding induced by Thr/Cvx and ABT-737. In accordance with these data, AA induced MP formation only at high concentrations (Figures 6a and b).

Arachidonic acid at low concentrations prevents, and at high concentrations induces apoptosis in platelets. (a) FACS analysis of ΔΨm and (b) Annexin V binding in WP (1 × 107/ml) stimulated with AA for 5 min at the indicated concentrations. (f) Inhibition of Annexin V binding by AA (10−7–10−6 M) in platelets stimulated with Thr/Cvx (5 min) and ABT-737 (1 μM, 30 min). (c and d) Western blot analysis (data shown are representative of three independent experiments) of caspase 3 in WP (4 × 108/ml) after stimulation with AA at the indicated concentrations for 2 and 24 h (c) and after stimulation with ABT-737 alone and in combination with AA for 30 min (d) and (e) densitometry analysis of (d). In (e), the control was taken as 100%. Data are presented as means±S.E.M, n=6; +P<0.05 compared with the control, *P<0.05 compared with platelets incubated with ABT-737

Microparticle generation and ROS production from platelets activated by different stimulators. Human WP were stimulated with ABT-737 (1 μM, 60 min), Thr/Cvx (5 mU/5 ng/ml, 2 min), and AA at the indicated concentrations (5 min) and processed for FACS analysis of MP formation, as assessed by the SSC/FSC plot (a and b) and (c) ROS production (DCF-DA fluorescence). The MP counts and DCF-DA fluorescence are presented as a fold increase compared with the control taken as 1. Data are presented as means±S.E.M, n=4; *P<0.05 compared with the control. (a) Representative plots (SSC/FSC) of the original data
ROS generated by activated platelets are not directly correlated with apoptotic events
AA can be involved in the regulation of ROS production as a by-product during the oxidation of AA by cyclooxygenase or lipoxygenase.37 In addition, AA can directly activate NADPH oxidase, inducing ROS generation.46 As MP formation in platelets may be induced by ROS,47, 48 we compared MP formation and ROS production in platelets activated by ABT-737 (1 μM), AA (10−6 and 10−4 M), and Thr/Cvx (5 mU/5 ng/ml). AA at a concentration of 10−4 M induced the highest amount of MPs (2.7±0.34 fold compared with the control taken as 1), and Thr/Cvx induced 1.7±0.2; neither a low concentration of AA (10−6 M) nor 1 μM ABT-737 induced significant MP formation (Figures 6a and b).
ROS production was strongly induced by AA at a low but not at a high concentration (27±5.6 and 3±1.5-fold respectively, compared with the control taken as 1) (Figure 6c). Thr/Cvx stimulation also significantly increased ROS production (20±4-fold, compared with the control taken as 1), whereas ABT-737 had no effect on platelet ROS production (Figure 6c).
Discussion
In nucleated cells, stress-activated p38 MAPK is involved in apoptosis, cytokine production, cytoskeleton reorganization, and transcriptional regulation. p38 MAPK also appears to play a role in the pathogenesis of heart ischemia, sepsis, arthritis, human immunodeficiency virus infection, and Alzheimer’s disease.49 In platelets, p38 MAPK has been shown to be activated by almost all platelet agonists, including thrombin, collagen, ADP, and TxA2.22, 23, 24, 25 Nonetheless, the involvement of p38 in the mechanisms that trigger apoptosis in platelets has remained unclear. Therefore, we investigated the functions of p38 in the regulation of apoptosis induced by ABT-737 and the apoptosis-like events induced by strong agonists (Thr/Cvx). We found that ABT-737 dose- and time-dependently resulted in the phosphorylation of p38 (Figure 1). The inhibition of p38 significantly prevented platelet apoptosis, as assessed by PS-positive cells, a decrease in mitochondrial potential, and cleavage of caspase 3 (Figure 2). cPLA2 is an established p38 substrate in platelets,23, 32, 33, 34 and we show that the ABT-737-induced activation of p38 triggered cPLA2 phosphorylation. In nucleated cells undergoing apoptosis, cPLA2 is cleaved and inactivated by caspase 3 activation.50, 51 In platelets, cPLA2 was also cleaved upon ABT-737 activation and inactivated by a caspase 3-dependent mechanism (Figures 4a and b). In contrast, Thr/Cvx treatment induced the phosphorylation of cPLA2 but did not promote its degradation (Figure 4a). Consistent with these data, Thr/Cvx strongly enhanced cPLA2 activity compared with the effect of ABT-737 (Figure 4e). It is important to note that the inhibition of p38 significantly prevented the apoptosis induced by ABT-737 but had no effect on the apoptotic-like events triggered by Thr/Cvx stimulation (Figure 3). As ABT-737-induced platelet apoptosis was directly correlated with the cleavage and inactivation of cPLA2 and because Thr/Cvx stimulation strongly activated cPLA2, which was responsible for AA generation, we tested whether AA itself was involved in apoptosis. In nucleated cells, high concentrations of AA contribute to mitochondrial dysfunction and apoptotic cell death,44, 45, 52 and AA also promotes apoptosis in stored platelets.39 In addition, the concentration of AA can increase up to 10-fold during cerebral or heart ischemia compared with normal physiological conditions.53, 54 Therefore, we tested the effects of different AA concentrations on hallmarks of platelet apoptosis. AA at low concentrations (10−7–10−6 M) enhanced ΔΨm, but had no influence on PS externalization and did not cleave caspase 3. In contrast, AA at high concentrations (10−5–10−4 M) induced a decrease in ΔΨm, enhanced PS externalization, and activated caspase 3 (Figure 5). Thus, the amount of AA might be an essential factor for mediating pro or antiapoptotic-like effects in platelets. We next addressed whether different concentrations of AA could prevent or enhance the apoptotic events induced by Thr/Cvx and ABT-737. Accordingly, we found that AA at low concentrations (10−7 for Thr/Cvx and 10−6 for ABT-737) inhibited the apoptotic events induced by both stimuli (Figure 5). The higher concentrations of AA required for the inhibitory effects of ABT-737-induced apoptosis were most likely associated with the inactivation of cPLA2 and consequently a reduced basal AA concentration in this case.
ROS are involved in cellular responses to stress55 and may contribute to various cellular pathological events, including apoptosis56 and, in particular, MP formation.47, 48 We investigated whether MP formation induced by ABT-737, Thr/Cvx, and AA correlates with ROS production and found increases only in the Thr/Cvx- and 10−6 M AA-treated platelets (Figure 6c), whereas MPs were enhanced only in the Thr/Cvx- and 10−4 M AA-stimulated platelets (Figures 6a and b), indicating that ROS are most likely not directly linked to apoptotic events.
In summary, we show here that p38 MAP kinase plays a significant role in platelet apoptosis induced by ABT-737, but is not involved in the generation of the apoptotic-like events triggered by physiological stimuli (Thr/Cvx). Consistently, under physiological conditions, the p38-triggered activation of cPLA2, release of AA, and ROS generation may serve as mechanisms to protect cells from apoptosis. In contrast, a high concentration of AA (as often observed under pathological conditions) may promote proapoptotic events.
Materials and Methods
Materials
Thrombin was obtained from Roche (Mannheim, Germany), and convulxin (Cvx) (ligand of glycoprotein VI from the snake venom Crotalus durissus terrificus) was purchased from LOXO (Dossenheim, Germany). SB202190 and z-DEVD-fmk were purchased from Calbiochem (Schwalbach, Germany), and ABT-737 was provided by Selleck Chemicals (Munich, Germany). Antibodies against p38, phospho-p38, and actin were obtained from Sigma (Munich, Germany). Antibodies against phospho-HSP27, phospho-cPLA2, cPLA2, and caspase 3 were from Cell Signaling (Frankfurt, Germany). Anti-rabbit and anti-mouse IgG conjugated with horseradish peroxidase were obtained from Amersham, Pharmacia Biotech (Freiburg, Germany). The fluorescent dyes Annexin-V-PE and PAC-1 were obtained from BD-Bioscience (Heidelberg, Germany). JC1 was obtained from Invitrogen (Eugen, Germany), and H2DCF-DA was purchased from Molecular Probes (Göttingen, Germany).
Platelet preparation
Human platelets were prepared and used as previously reported,57 with minor modifications. Blood was obtained from healthy volunteers according to our institutional guidelines and the Declaration of Helsinki. Our studies with human platelets were approved and recently (24 September, 2008) reconfirmed by the local ethics committee of the University of Würzburg (Studies Number 67/92 and 114/04).
Blood was collected into 1/7 volume of ACD solution (12 mM citric acid, 15 mM sodium citrate, 25 mM D-glucose, and 2 μM EGTA, final concentrations). Platelet rich plasma was obtained by a 5-min centrifugation at 330 × g. To reduce leukocyte contamination, Platelet rich plasma was diluted 1 : 1 with PBS and centrifuged at 240 × g for 10 min. The supernatant was centrifuged for 10 min at 430 × g, and the pelleted platelets were washed once in CGS buffer (120 mM sodium chloride, 12.9 mM trisodium citrate, and 30 mM D-glucose, pH 6.5) and resuspended in HEPES buffer (150 mM sodium chloride, 5 mM potassium chloride, 1 mM magnesium chloride, 10 mM D-glucose, and 10 mM HEPES, pH 7.4) without stirring; 1 mM CaCl2 was added directly before platelet stimulation.
WP were used at a concentration of 3 × 108/ml for western blot analysis and TxS activity, and a concentration of 1 × 107/ml was used for flow cytometry analysis.
Flow cytometry (FACS) analysis
FACS analysis was performed using a Becton Dickinson FACS Calibur with CELLQuest software, version 3.1f (Becton Dickinson, Heidelberg, Germany). For the detection of surface PS or activated αIIbβ3 integrins, WP (50 μl) were labeled with Annexin V-PE or PAC-1-FITC (both 1 : 10 dilution) for 10 min at RT after agonist stimulation. The platelets were then diluted 10 times with Annexin V-binding solution (140 mM NaCl, 10 mM HEPES, and 2.5 mM CaCl2) for Annexin V or PBS for PAC-1 and immediately analyzed by flow cytometry.
Analysis of mitochondrial membrane potential
The ΔΨm in human platelets was analyzed with JC1 dye (1.5 μM final concentration). JC1 was incubated with WP (50 μl) for 10 min at RT, and the samples were diluted (1 : 10) in PBS and analyzed by FACS. Green fluorescence (FL1) and red fluorescence (FL2) were measured in logarithmic scales using voltage settings of 710 and 588, respectively. Compensation settings (FL1-%FL2 and FL2-%FL1) were performed using FITC and PE beads (BD-Bioscience). JC1 is sensitive to ΔΨm, and the ratio of fluorescence in FL2 to FL1 corresponds to changes in ΔΨm.
Measurement of ROS production
Intracellular ROS production was analyzed by H2DCF-DA, as described previously.58 Briefly, WP were preloaded with 10 μM H2DCF-DA for 30 min at 37 °C in HEPES buffer. H2DCF-DA is a cell-permeable, nonfluorescent dye converted intracellularly to cell-impermeable H2DCF by intracellular esterases; the dye can then be oxidized by ROS to the fluorescent species (DCF) that is measured by flow cytometry. After incubation with agonists, the reaction was stopped by diluting the platelets in PBS for FACS analysis.
Measurement of thromboxane synthase activity
The TxS activity (TxA2 synthase) was measured as described59 using a method based on malondialdehyde formation by TxA2 synthase. For the experiments, 0.2-ml aliquots of WP (3 × 108/ml) were incubated with the reagents at 37 °C with gentle shaking; the reaction was stopped by the addition of 0.115 ml of ice-cold trichloroacetic acid (20% wt/vol). The samples were kept on ice for 10 min and then centrifuged for 10 min at 14 000 r.p.m. Equal volumes of thiobarbituric acid (0.53% wt/vol) and supernatant were combined and incubated for 30 min at 70 °C. The mixture was then incubated for 30 min at room temperature, and fluorescence was measured with a Wallac Victor2 1420 Multilabel counter (Perkin Elmer Wallac Life and Analytical Sciences, Boston, MA, USA) at excitation and emission wavelengths of 520 nM and 550 nM, respectively. Standards were prepared from the stable malondialdehyde derivate tetraethoxypropane with thiobarbituric acid in the presence of trichloroacetic acid.
Western blot analysis
For western blot analysis, WP were added directly to sodium dodecyl sulfate (SDS) gel-loading buffer and analyzed by SDS-polyacrylamide gel electrophoresis (PAGE), as described.57 The separated proteins were transferred to a nitrocellulose membrane and incubated with the primary antibody overnight at 4 °C. For visualization of the signal, goat anti-rabbit or anti-mouse IgG conjugated with horseradish peroxidase was used as a secondary antibody, followed by ECL detection (Amersham, Pharmacia Biotech).
Data analysis
All experiments were performed with at least n=4, and the combined data are expressed as means±S.E.M Differences between groups were analyzed by ANOVA, followed by Bonferroni's test; Student’s t-test was used when appropriate. A value of P<0.05 was considered statistically significant.
Abbreviations
- AA:
-
arachidonic acid
- cPLA2:
-
cytosolic phospholipase A2
- ΔΨm:
-
mitochondrial membrane potential
- MP:
-
microparticle
- ROS:
-
reactive oxygen species
- Thr/Cvx:
-
thrombin/convulxin
- TxA2:
-
thromboxane A2
- WP:
-
washed platelets
References
Versteeg HH, Heemskerk JW, Levi M, Reitsma PH . New fundamentals in hemostasis. Physiol Rev 2013; 93: 327–358.
Semple JW, Italiano JE Jr, Freedman J . Platelets and the immune continuum. Nat Rev Immunol 2011; 11: 264–274.
Projahn D, Koenen RR . Platelets: key players in vascular inflammation. J Leukoc Biol 2012; 92: 1167–1175.
Geddis AE . The regulation of proplatelet production. Haematologica 2009; 94: 756–759.
Li J, Xia Y, Bertino AM, Coburn JP, Kuter DJ . The mechanism of apoptosis in human platelets during storage. Transfusion 2000; 40: 1320–1329.
Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ 2007; 14: 943–951.
Kodama T, Takehara T, Hikita H, Shimizu S, Shigekawa M, Li W et al. BH3-only activator proteins Bid and Bim are dispensable for Bak/Bax-dependent thrombocyte apoptosis induced by Bcl-xL deficiency: molecular requisites for the mitochondrial pathway to apoptosis in platelets. J Biol Chem 2011; 286: 13905–13913.
Burkhart JM, Vaudel M, Gambaryan S, Radau S, Walter U, Martens L et al. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 2012; 120: e73–e82.
Pereira J, Soto M, Palomo I, Ocqueteau M, Coetzee LM, Astudillo S et al. Platelet aging in vivo is associated with activation of apoptotic pathways: studies in a model of suppressed thrombopoiesis in dogs. Thromb Haemost 2002; 87: 905–909.
Rand ML, Wang H, Bang KW, Poon KS, Packham MA, Freedman J . Procoagulant surface exposure and apoptosis in rabbit platelets: association with shortened survival and steady-state senescence. J Thromb Haemost 2004; 2: 651–659.
Remenyi G, Szasz R, Friese P, Dale GL . Role of mitochondrial permeability transition pore in coated-platelet formation. Arterioscler Thromb Vasc Biol 2005; 25: 467–471.
Rukoyatkina N, Begonja AJ, Geiger J, Eigenthaler M, Walter U, Gambaryan S . Phosphatidylserine surface expression and integrin alpha IIb beta 3 activity on thrombin/convulxin stimulated platelets/particles of different sizes. Br J Haematol 2009; 144: 591–602.
Tonon G, Luo X, Greco NJ, Chen W, Shi Y, Jamieson GA . Weak platelet agonists and U46619 induce apoptosis-like events in platelets, in the absence of phosphatidylserine exposure. Thromb Res 2002; 107: 345–350.
Wolf BB, Goldstein JC, Stennicke HR, Beere H, Amarante-Mendes GP, Salvesen GS et al. Calpain functions in a caspase-independent manner to promote apoptosis-like events during platelet activation. Blood 1999; 94: 1683–1692.
Leytin V, Allen DJ, Mykhaylov S, Lyubimov E, Freedman J . Thrombin-triggered platelet apoptosis. J Thromb Haemost 2006; 4: 2656–2663.
Lin KH, Chang HC, Lu WJ, Jayakumar T, Chou HC, Fong TH et al. Comparison of the relative activities of inducing platelet apoptosis stimulated by various platelet-activating agents. Platelets 2009; 20: 575–581.
Winkler J, Rand ML, Schmugge M, Speer O . Omi/HtrA2 and XIAP are components of platelet apoptosis signalling. Thromb Haemost 2013; 109: 532–539.
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19: 107–120.
Jackson SP, Schoenwaelder SM . Procoagulant platelets: are they necrotic? Blood 2010; 116: 2011–2018.
Leung R, Gwozdz AM, Wang H, Bang KW, Packham MA, Freedman J et al. Persistence of procoagulant surface expression on activated human platelets: involvement of apoptosis and aminophospholipid translocase activity. J Thromb Haemost 2007; 5: 560–570.
Lopez JJ, Salido GM, Gomez-Arteta E, Rosado JA, Pariente JA . Thrombin induces apoptotic events through the generation of reactive oxygen species in human platelets. J Thromb Haemost 2007; 5: 1283–1291.
Kramer RM, Roberts EF, Strifler BA, Johnstone EM . Thrombin induces activation of p38 MAP kinase in human platelets. J Biol Chem 1995; 270: 27395–27398.
Kramer RM, Roberts EF, Um SL, Borsch-Haubold AG, Watson SP, Fisher MJ et al. p38 mitogen-activated protein kinase phosphorylates cytosolic phospholipase A2 (cPLA2) in thrombin-stimulated platelets. Evidence that proline-directed phosphorylation is not required for mobilization of arachidonic acid by cPLA2. J Biol Chem 1996; 271: 27723–27729.
Saklatvala J, Rawlinson L, Waller RJ, Sarsfield S, Lee JC, Morton LF et al. Role for p38 mitogen-activated protein kinase in platelet aggregation caused by collagen or a thromboxane analogue. J Biol Chem 1996; 271: 6586–6589.
Adam F, Kauskot A, Rosa JP, Bryckaert M . Mitogen-activated protein kinases in hemostasis and thrombosis. J Thromb Haemost 2008; 6: 2007–2016.
Mazharian A, Roger S, Maurice P, Berrou E, Popoff MR, Hoylaerts MF et al. Differential Involvement of ERK2 and p38 in platelet adhesion to collagen. J Biol Chem 2005; 280: 26002–26010.
Canobbio I, Reineri S, Sinigaglia F, Balduini C, Torti M . A role for p38 MAP kinase in platelet activation by von Willebrand factor. Thromb Haemost 2004; 91: 102–110.
Begonja AJ, Geiger J, Rukoyatkina N, Rauchfuss S, Gambaryan S, Walter U . Thrombin stimulation of p38 MAP kinase in human platelets is mediated by ADP and thromboxane A2 and inhibited by cGMP/cGMP-dependent protein kinase. Blood 2007; 109: 616–618.
Borsch-Haubold AG, Pasquet S, Watson SP . Direct inhibition of cyclooxygenase-1 and -2 by the kinase inhibitors SB 203580 and PD 98059. SB 203580 also inhibits thromboxane synthase. J Biol Chem 1998; 273: 28766–28772.
Kato H, Takai S, Matsushima-Nishiwaki R, Adachi S, Minamitani C, Otsuka T et al. HSP27 phosphorylation is correlated with ADP-induced platelet granule secretion. Arch Biochem Biophys 2008; 475: 80–86.
Kuliopulos A, Mohanlal R, Covic L . Effect of selective inhibition of the p38 MAP kinase pathway on platelet aggregation. Thromb Haemost 2004; 92: 1387–1393.
Lin LL, Wartmann M, Lin AY, Knopf JL, Seth A, Davis RJ . cPLA2 is phosphorylated and activated by MAP kinase. Cell 1993; 72: 269–278.
Borsch-Haubold AG, Kramer RM, Watson SP . Phosphorylation and activation of cytosolic phospholipase A2 by 38-kDa mitogen-activated protein kinase in collagen-stimulated human platelets. Eur J Biochem 1997; 245: 751–759.
Waterman WH, Molski TF, Huang CK, Adams JL, Sha'afi RI . Tumour necrosis factor-alpha-induced phosphorylation and activation of cytosolic phospholipase A2 are abrogated by an inhibitor of the p38 mitogen-activated protein kinase cascade in human neutrophils. Biochem J 1996; 319 Pt 1 17–20.
Lin LL, Lin AY, Knopf JL . Cytosolic phospholipase A2 is coupled to hormonally regulated release of arachidonic acid. Proc Natl Acad Sci USA 1992; 89: 6147–6151.
Shmelzer Z, Haddad N, Admon E, Pessach I, Leto TL, Eitan-Hazan Z et al. Unique targeting of cytosolic phospholipase A2 to plasma membranes mediated by the NADPH oxidase in phagocytes. J Cell Biol 2003; 162: 683–692.
Kukreja RC, Kontos HA, Hess ML, Ellis EF . PGH synthase and lipoxygenase generate superoxide in the presence of NADH or NADPH. Circ Res 1986; 59: 612–619.
Edderkaoui M, Hong P, Vaquero EC, Lee JK, Fischer L, Friess H et al. Extracellular matrix stimulates reactive oxygen species production and increases pancreatic cancer cell survival through 5-lipoxygenase and NADPH oxidase. Am J Physiol Gastrointest Liver Physiol 2005; 289: G1137–G1147.
van der Wal DE, Gitz E, Du VX, Lo KS, Koekman CA, Versteeg S et al. Arachidonic acid depletion extends survival of cold-stored platelets by interfering with the (glycoprotein Ibalpha-14-3-3zeta) association. Haematologica 2012; 97: 1514–1522.
De Genaro P, Simon MV, Rotstein NP, Politi LE . Retinoic acid promotes apoptosis and differentiation in photoreceptors by activating the P38 MAP kinase pathway. Invest Ophthalmol Vis Sci 2013; 54: 3143–3156.
Mayr M, Hu Y, Hainaut H, Xu Q . Mechanical stress-induced DNA damage and rac-p38MAPK signal pathways mediate p53-dependent apoptosis in vascular smooth muscle cells. Faseb J 2002; 16: 1423–1425.
Shou Y, Li L, Prabhakaran K, Borowitz JL, Isom GE . p38 mitogen-activated protein kinase regulates Bax translocation in cyanide-induced apoptosis. Toxicol Sci 2003; 75: 99–107.
Tripathi T, Smith AD, Abdi M, Alizadeh H . Acanthamoeba-cytopathic protein induces apoptosis and proinflammatory cytokines in human corneal epithelial cells by cPLA2 alpha activation. Invest Ophthalmol Vis Sci 2012; 53: 7973–7982.
Penzo D, Tagliapietra C, Colonna R, Petronilli V, Bernardi P . Effects of fatty acids on mitochondria: implications for cell death. Biochim Biophys Acta 2002; 1555: 160–165.
Scorrano L, Penzo D, Petronilli V, Pagano F, Bernardi P . Arachidonic acid causes cell death through the mitochondrial permeability transition. Implications for tumor necrosis factor-alpha aopototic signaling. J Biol Chem 2001; 276: 12035–12040.
Shiose A, Sumimoto H . Arachidonic acid and phosphorylation synergistically induce a conformational change of p47phox to activate the phagocyte NADPH oxidase. J Biol Chem 2000; 275: 13793–13801.
Brill A, Chauhan AK, Canault M, Walsh MT, Bergmeier W, Wagner DD . Oxidative stress activates ADAM17/TACE and induces its target receptor shedding in platelets in a p38-dependent fashion. Cardiovasc Res 2009; 84: 137–144.
Nardi MA, Gor Y, Feinmark SJ, Xu F, Karpatkin S . Platelet particle formation by anti GPIIIa49-66 Ab, Ca2+ ionophore A23187, and phorbol myristate acetate is induced by reactive oxygen species and inhibited by dexamethasone blockade of platelet phospholipase A2, 12-lipoxygenase, and NADPH oxidase. Blood 2007; 110: 1989–1996.
Obata T, Brown GE, Yaffe MB . MAP kinase pathways activated by stress: the p38 MAPK pathway. Crit Care Med 2000; 28: N67–N77.
Adam-Klages S, Schwandner R, Luschen S, Ussat S, Kreder D, Kronke M . Caspase-mediated inhibition of human cytosolic phospholipase A2 during apoptosis. J Immunol 1998; 161: 5687–5694.
Atsumi G, Tajima M, Hadano A, Nakatani Y, Murakami M, Kudo I . Fas-induced arachidonic acid release is mediated by Ca2+-independent phospholipase A2 but not cytosolic phospholipase A2, which undergoes proteolytic inactivation. J Biol Chem 1998; 273: 13870–13877.
Kriem B, Sponne I, Fifre A, Malaplate-Armand C, Lozac'h-Pillot K, Koziel V et al. Cytosolic phospholipase A2 mediates neuronal apoptosis induced by soluble oligomers of the amyloid-beta peptide. Faseb J 2005; 19: 85–87.
Prinzen FW, Van der Vusse GJ, Arts T, Roemen TH, Coumans WA, Reneman RS . Accumulation of nonesterified fatty acids in ischemic canine myocardium. Am J Physiol 1984; 247: H264–H272.
Westerberg E, Deshpande JK, Wieloch T . Regional differences in arachidonic acid release in rat hippocampal CA1 and CA3 regions during cerebral ischemia. J Cereb Blood Flow Metab 1987; 7: 189–192.
Jiang F, Zhang Y, Dusting GJ . NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance and tissue repair. Pharmacol Rev 2011; 63: 218–242.
Lambeth JD . NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 2004; 4: 181–189.
Gambaryan S, Geiger J, Schwarz UR, Butt E, Begonja A, Obergfell A et al. Potent inhibition of human platelets by cGMP analogs independent of cGMP-dependent protein kinase. Blood 2004; 103: 2593–2600.
Begonja AJ, Gambaryan S, Geiger J, Aktas B, Pozgajova M, Nieswandt B et al. Platelet NAD(P)H-oxidase-generated ROS production regulates alphaIIbbeta3-integrin activation independent of the NO/cGMP pathway. Blood 2005; 106: 2757–2760.
Aktas B, Utz A, Hoenig-Liedl P, Walter U, Geiger J . Dipyridamole enhances NO/cGMP-mediated vasodilator-stimulated phosphoprotein phosphorylation and signaling in human platelets: in vitro and in vivo/ex vivo studies. Stroke 2003; 34: 764–769.
Acknowledgements
This study was supported by the DFG (SFB688, TP A2) and BMBF (01EO1003).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Stephanou
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Rukoyatkina, N., Mindukshev, I., Walter, U. et al. Dual role of the p38 MAPK/cPLA 2 pathway in the regulation of platelet apoptosis induced by ABT-737 and strong platelet agonists . Cell Death Dis 4, e931 (2013). https://doi.org/10.1038/cddis.2013.459
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.459
Keywords
This article is cited by
-
Down-regulation of platelet adhesion receptors is a controlling mechanism of thrombosis, while also affecting post-transfusion efficacy of stored platelets
Thrombosis Journal (2019)
-
Unconjugated Bilirubin exerts Pro-Apoptotic Effect on Platelets via p38-MAPK activation
Scientific Reports (2015)
-
Chronic hepatitis C virus infection triggers spontaneous differential expression of biosignatures associated with T cell exhaustion and apoptosis signaling in peripheral blood mononucleocytes
Apoptosis (2015)
