
Abstract
In this study, we have analysed the apoptotic effects of the ubiquitous environmental toxin benzo[a]pyrene (BP) in HaCaT cells and human keratinocytes. Although prolonged exposure to BP was not cytotoxic on its own, a strong enhancement of CD95 (Fas)-mediated apoptosis was observed with BP at concentrations activating the aryl hydrocarbon receptor (AhR). Importantly, the ultimately mutagenic BP-metabolite, that is, (+)-anti-BP-7,8-diol-9,10-epoxide (BPDE), failed to enhance CD95-mediated cell death, suggesting that the observed pro-apoptotic effect of BP is neither associated with DNA adducts nor DNA-damage related signalling. CD95-induced apoptosis was also enhanced by β-naphtoflavone, a well-known agonist of the AhR that does not induce DNA damage, thus suggesting a crucial role for AhR activation. Consistently, BP failed to sensitise for CD95L-induced apoptosis in AhR knockdown HaCaT cells. Furthermore, inhibition of CYP1A1 and/or 1B1 expression did not affect the pro-apoptotic crosstalk. Exposure to BP did not increase expression of CD95, but led to augmented activation of caspase-8. Enhancement of apoptosis was also observed with the TRAIL death receptors that activate caspase-8 and apoptosis by similar mechanisms as CD95. Together, these observations indicate an interference of AhR signalling with the activity of receptor-associated signalling intermediates that are shared by CD95 and TRAIL receptors. Our data thus suggest that AhR agonists can enhance cytokine-mediated adversity upon dermal exposure.
Similar content being viewed by others


Main
Benzo[a]pyrene (BP) is regarded as a carcinogenic lead compound among the group of polycyclic aromatic hydrocarbons (PAHs). BP is a strong activator of the aryl hydrocarbon receptor (AhR) and a powerful mutagen.1 Genotoxicity depends on its endogenous biotransformation into mutagenic intermediates, particularly the (+)-anti-BP-7,8-diol-9,10-epoxide (BPDE). The latter metabolite is regarded as the ultimate carcinogen that triggers formation of bulky DNA adducts and subsequent mutations. Metabolism of BP is initiated by cytochrome P450-dependent monooxygenases (CYPs), especially CYP1A1 (cytochrome P450 1A1) and CYP1B1 (cytochrome P450 1B1),2 resulting in the generation of a variety of metabolites. Besides BPDE, other metabolites are also of concern to human health. For example, oxidation of phenolic or catecholic intermediates can generate BP quinones.3 As H2O2 and superoxide anion radicals (O2˙−) are side products of the corresponding reactions, oxidative stress is generated and might be further enhanced by consecutive redox cycles, which depend on endogenous enzymes and reduction equivalents.
The biotransformation of BP is regulated via AhR. This receptor is also activated by other xenobiotics, notably 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), halogenated biphenyls and other mutagenic PAHs, such as dibenzo[a,l]pyrene or 3-methylcholanthrene. In response to agonist binding, AhR translocates into the nucleus, where it associates with the AhR nuclear translocator (ARNT) and triggers transcription of target genes, including CYP1A1 and CYP1B1, as well as several phase II enzymes (i.e., transferases). Thus, BP induces its own biotransformation via AhR. The relevance of endogenous ligands is still a matter of debate. However, there is growing evidence that the AhR protein affects additional physiological functions, besides its essential role in the biotransformation of xenobiotics.4 For example, AhR is proposed to link the adaptive immune response to environmental factors.5 It is also suggested to play an important role in autoimmunity.6 Notably, the structure of AhR is highly conserved and knockout mice show nonlethal developmental defects.7 Transcriptional activation of the xenobiotic response element (XRE) is not the only function of the AhR. It also interacts with NF-κB subunits as well as with the transcription factor E2F and was shown to affect several MAP kinases, including JNK and p38.8 AhR does further act as an ubiquitin ligase that triggers degradation of selected proteins,9 as, for instance, the oestrogen receptor.
Exposure of cells to BP can therefore induce multiple cellular effects that are either directly related to activation of the AhR or are due to the formation of toxic metabolites. Mutagenesis via BPDE is regarded as a carcinogenic risk factor to human health. Dependent on the magnitude of DNA lesions induced, genotoxicity can alternatively trigger apoptosis, as part of the DNA damage response. The apoptotic effects of BP have been observed in liver cells,10, 11 in mammary carcinoma MCF-7 cells12 as well as in lung cancer cells,13 but are not yet well defined. Intriguingly, BP has also been reported to enhance survival of injured cells. The overall capacity of BP to affect apoptosis thus remains enigmatic.
Here, we have analysed the pro-apoptotic effects of BP in human keratinocytes and HaCaT cells. These cells were used as model system to address the effects of dermal BP exposure that can occur via contact with PAH-contaminated synthetic materials. For example, tool handles, watch bands, plastic components of shoes and textile flockings have been recognised as potential sources for dermal exposure, besides the more established dietary and inhalation routes. Although BP does induce tumours in mouse skin, comparatively little is known about its biological effects in humans. Dermal expression and induction of the enzymes crucial for its metabolic conversion, including CYP1A1, CYP1B1 or CYP3A4, has been demonstrated. Recently, we have shown that BP is efficiently metabolised in human skin, keratinocytes as well as in in vitro 3D skin models.14 Here we show that, although direct apoptotic effects were only marginal, a strong enhancement of physiological inducers of apoptosis can be noticed. BP sensitised both keratinocytes and HaCaT cells toward CD95L (cluster of differentiation 95 (Fas) ligand)- and TRAIL (TNF-related apoptosis-inducing ligand)-induced apoptosis at concentrations that are likely to be reached in exposed human skin by contacts to contaminated materials, according to exposure models currently applied in risk assessment. CD95L and TRAIL are expressed by infiltrating immune cells and play an important role in the pathology of inflammatory skin diseases, especially in eczematous dermatitis.15 Unexpectedly, the BP-mediated enhancement of CD95L- and TRAIL-induced cell death was not related to genotoxic metabolites of BP, but dependent on activation of AhR. This became evident from experiments using the nongenotoxic AhR agonist β-naphtoflavone (β-NF) and variant HaCaT cell lines in which AhR expression was downregulated via small hairpin RNA (shRNA).
Results
Benzo[a]pyrene augments CD95-mediated apoptosis in keratinocytes
Dermal up-take of BP is a major route of exposure, but comparatively little is known about the BP-mediated adverse effects in human skin. We observed a strong activation of the AhR in human keratinocytes, leading to an increased expression of CYP1A1 at both RNA and protein levels (Figures 1a and b). Maximal transcriptional induction was observed at 3.5 μM BP. As exposure levels of >3 μM are feasible in human skin after contact with contaminated materials, we usually used a standard concentration of 3.5 μM BP for everything further.

Induction of CYP1A1 by BP in NHEKs and HaCaT cells. (a) Transriptional induction of the CYP1A1 gene by BP: transcripts of CYP1A1 were measured by real-time PCR in normal (primary) human epidermal keratinocytes (NHEKs) and HepG2 cells after exposure to BP for 6 h. Control cells were treated with solvent only (0.1% DMSO). Total RNA was isolated and real-time PCR analysis was performed as described in Materials and Methods. Data are expressed as fold induction of transcripts. Values represent means±S.D. of three independent experiments. (b) Induction of CYP1A1 protein levels by BP: cells were treated with 3.5 μM BP as indicated and analysed by western blotting. Molecular size markers are shown on the left in kDa
To address the capacity of BP to promote apoptosis in human skin, cultures of normal (primary) human epidermal keratinocytes (NHEKs) and HaCaT cells were treated with 3.5 and 10 μM BP (Figure 2). Cell viability was only moderately affected as determined by WST assay. As this method does not distinguish apoptotic and growth-arrested cells, induction of an apoptotic phenotype was directly assayed by annexin V staining. No indication for apoptosis was found in HaCaT cells. Treatment of NHEKs with 10 μM BP led to a 20% population of annexin V-positive cells after 48 h. However, only a marginal induction of annexin V-positive cells was observed after 24 h or in cells that had been treated with 3.5 μM BP. In contrast to cultured lung and liver cells,10, 11, 13 where BP-induced cell death was observed, in keratinocytes these effects are comparatively weak and not apparent under conditions where an optimal induction of AhR target genes is achieved. This led to the further question of whether BP can enhance physiological inducers of apoptosis, especially death receptors and their cytotoxic ligands that play a pivotal role in inflammatory skin diseases, such as CD95L.15, 16

BP does not induce apoptosis in HaCaT cells and NHEKs at exposure levels that trigger optimal induction of CYP1A1. (a) Cell viability and proliferation were determined by WST assays in the indicated cells after treatment with different concentrations of BP for 24 or 48 h. Values represent means±S.D. of three independent experiments. (b) Quantification of the apoptotic cell population by annexin V staining and flow cytometric analyses after BP treatment of cells for 24 or 48 h. Values represent means±S.D. of three independent experiments
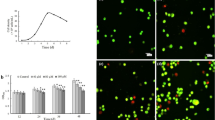
To analyse whether BP enhances the pro-apoptotic effects of CD95L, we first analysed CD95L-mediated apoptosis in BP-pretreated HaCaT cells revealing strong sensitisation over a wide range of concentrations (Figure 3a). No cell death was apparent in the presence of the pan-caspase inhibitor z-VAD (Figure 3a), indicating that cell death was due to the activation of caspases. Secondly, we demonstrated a synergistic pro-apoptotic effect under conditions where neither CD95L nor BP alone is capable of inducing apoptosis in HaCaT cells or NHEKs. When applying 2.5 ng/ml oligomerised CD95L for 24 h to these cells, up to 30% annexin-positive cells were observed after pretreatment with BP, but not in control experiments (Figures 3b and c). Similarly, an enhanced release of cytochrome c was observed in response to a combined application of BP and CD95L (Figure 3d). Consistent with these findings, a significant decrease in cell viability was observed in parallel (data not shown). These data demonstrate that BP can amplify effects of endogenous activators of apoptosis in human keratinocytes, although it failed to act as a direct inducer of apoptosis in human keratinocytes.

BP sensitises HaCaT cells and NHEKs for CD95L-induced apoptosis. (a) HaCaT cells were pretreated with 3.5 μM BP (white bars) or 0.1% DMSO (black bars) for 48 h and subsequently exposed to different concentrations of CD95L for further 24 h with or without 20 μM pan-caspase inhibitor z-VAD (right). Cell viability was determined by crystal violet staining. Values represent means±S.D. of three independent experiments. (b and c) Cells were pretreated with 3.5 μM BP or solvent (0.1% DMSO) for 48 h and subsequently exposed to 2.5 ng/ml CD95L for 24 h. The populations of apoptotic cells were determined by annexin V staining and flow cytometric analyses. Data from three independent experiments are summarised in (b), and images obtained in a typical experiment with HaCaT cells are shown in (c). (d) Induction of cytochrome c release in CD95L/BP-treated cells was determined by western blotting as described in Materials and Methods. The increase of the cytosolic fraction was quantified by densiometry and average based on three independent experiments in (left panel). The columns shown in the right panel correspond to the bar diagram shown in the lower panel. Molecular size markers are shown on the left in kDa
Enhancement of CD95-mediated apoptosis by AhR agonists do not depend on the formation of mutagenic metabolites
One possibility to explain how BP enhances CD95-mediated apoptosis refers to the mutagenic properties of its metabolites. According to this model, DNA adducts might trigger a DNA damage response, thus generating an intrinsic signal to cooperate with death receptor-induced apoptosis by p53-mediated upregulation of CD95. This hypothesis was however not supported in our investigation. When the ultimate DNA-damaging metabolite BPDE was directly applied to HaCaT cells, this compound failed to promote CD95L-induced apoptosis (Figure 4). We also investigated other metabolites of BP that had been previously identified in human skin by LC-MS analysis.14 Only BP, but none of the other tested metabolites, was capable of initiating a pro-apoptotic crosstalk with CD95L (Figure 4).

Pro-apoptotic crosstalk between CD95L and BP is not triggered by BP metabolites. (a and b) Cells were pretreated with 3.5 μM BP, 2 μM 3-OH-BP or 2 μM BP-1.6-dione (a), 6 μM BPDE (b) or solvent (0.1% DMSO) for 48 h and subsequently exposed to 2.5 ng/ml CD95L for further 24 h. The populations of apoptotic cells were determined by annexin V staining and flow cytometric analyses. Values represent means±S.D. of three independent experiments
This finding suggested that the observed pro-apoptotic effects of BP are not related to induction of DNA damage in cultured skin cells. Because activation of AhR is the initial cellular effect of BP, we next analysed whether this receptor plays a role in the enhancement of CD95-mediated apoptosis. We therefore tested whether alternate AhR agonists trigger similar effects as BP with respect to CD95L-induced apoptosis. We applied β-NF in these experiments, because this compound does not trigger DNA adducts or any other genotoxic effects in eukaryotic cells. The β-NF is a comparably strong activator of AhR signalling like BP, as indicated by transcriptional upregulation of the CYP1A1 gene (Supplementary Figure 1). Notably, β-NF revealed a comparable potency as BP to enhance CD95L-dependent apoptosis (Figure 5). Our data suggest that an augmented CD95-mediated apoptosis in BP-pretreated keratinocytes is based on the activation of the AhR and not related to the intrinsic toxicity of the various compounds or their metabolites.

Sensitisation of HaCaT cells and NHEKs for CD95L-induced apoptosis by the non-genotoxic AhR agonist β-naphtoflavone (β-NF). (a) Cell viability in HaCaT cells was determined by crystal violet staining after treatment with β-NF for 24 and 48 h. Values represent means±S.D. of three independent experiments. (b) HaCaT cells were pretreated with 10 μM β-NF (white bars) or 0.1% DMSO (black bars) for 48 h and subsequently exposed to CD95L as indicated. Cell viability was then determined by crystal violet staining. Values represent means±S.D. of three independent experiments. (c) β-NF sensitises HaCaT cells and NHEKs for CD95L-induced apoptosis. Cells were pretreated with β-NF as indicated or with solvent (0.1% DMSO) for 48 h and subsequently exposed to 2.5 ng/ml CD95L for further 24 h. The populations of apoptotic cells were determined by annexin V staining and flow cytometric analyses. Values represent means±S.D. of three independent experiments
A crucial role of the AhR for BP-mediated enhancement of CD95-induced apoptosis was further demonstrated in HaCaT cells, in which AhR expression was permanently downregulated by RNA interference (Figure 6).17 In contrast to control cell, which carry the parental (empty) lentiviral expression vector (e.v.), no AhR protein was detectable in the knockdown (k.d.) cells, indicating an efficient downregulation (Figure 6a). Preincubation of cells with BP or β-NF failed to enhance the apoptotic capacity of CD95L in AhR-deficient cells (Figure 6b). As expected, synergistic apoptotic effects between AhR agonists and CD95L could be confirmed in simultaneous experiments using AhR-proficient (e.v.) control cells. The magnitude of this crosstalk was comparable to the effects seen before in keratinocytes and wild-type HaCaT cells (cf., above). From this we conclude that the pro-apoptotic crosstalk between CD95L and AhR agonists depends on AhR activation. As AhR acts as a transcription factor, an important question relates to putative target genes that might affect apoptosis. Therefore, we have analysed whether expression of CD95 is affected in NHEK and in wild-type or AhR-deficient HaCaT cells. Our data show that expression levels of CD95 are not affected during the course of 48 h treatment, although a minute transcriptional induction (1.3–1.5-fold) was observed (Supplementary Figure 2). These results were also confirmed by FACS analysis (Supplementary Figure 3). Furthermore, exposure of NHEK to BP did not lead to a detectable p53 expression (data not shown), which might otherwise account for an indirect upregulation of CD95.18 Taken together, our data do not support transcriptional upregulation of CD95 as underlying mechanism for the pro-apoptotic crosstalk with AhR agonists.

AhR agonists fail to promote apoptosis in AhR-deficient cells. (a) Expression of AhR in AhR-deficient (knockdown (k.d.)) HaCaT cells compared with wild-type (w.t.) and empty vector (e.v.) HaCaT cells, analysed by western blotting. Molecular size markers are shown on the left in kDa. (b) Sensitisation of HaCaT cells for CD95L-induced apoptosis depends on the presence of a functional AhR protein. AhR e.v., parental (empty) control vector (black bars); AhR k.d., shRNA AhR k.d. (white bars). Cells were pretreated with either 3.5 μM BP or 10 μM β-NF for 48 h and subsequently exposed to 2.5 ng/ml CD95L for further 24 h. The populations of apoptotic cells were determined by annexin V staining and flow cytometric analyses. Values represent means±S.D. of three independent experiments. (c) HaCaT cultures were transfected using HiPerfect (Qiagen, Hilden, Germany) and predesigned siRNAs against AhR (Applied Biosystems), CYP1A1 (Qiagen), CYP1B1 (Applied Biosystems) and a mixture of the CYP1A1 and CYP1B1 siRNAs. Cells were subsequently exposed to 3.5 mM BP for 48 h, followed by an exposure to 200 ng/ml TRAIL until significant cell death was observed (∼2–3 h). Cell viability was determined using crystal violet and compared with transfected cells not exposed to TRAIL. A control siRNA from Applied Biosystems was included as experimental control (right). Inhibition of EROD (CYP1A) activities after AhR, CYP1A1 and CYP1B1 k.d. was demonstrated for transfected HaCaT populations in parallel
AhR agonists cooperate with both CD95L and TRAIL to promote apoptosis
To characterise the putative mechanism that leads to enhanced apoptosis in response to AhR activation, we analysed next whether this effect is limited to CD95. TRAIL was used in further experiments, because it was previously demonstrated to induce apoptosis in HaCaT cells19 via two receptors (TRAILR1 and TRAILR2). These receptors belong to the death receptor subgroup of the TNF receptor superfamily and thus induce cell death by mechanisms closely related to CD95. Pretreatment of wild-type and AhR-proficient HaCaT variants with BP did strongly enhance TRAIL-induced apoptosis, indicating that the cytotoxic capacity of AhR activation is not specifically associated with CD95 (Figure 7a). These data suggest an involvement of downstream signalling intermediates that are shared by CD95 and the TRAIL death receptors in the pro-apoptotic crosstalk with AhR agonists. These effects were again completely inhibited by the pan-caspase inhibitor z-VAD, which was applied in control experiments. In contrast to AhR-proficient cells, the corresponding AhR-deficient HaCaT variants were protected from TRAIL-induced apoptosis after sensitising with BP (Figures 7b and c). Consistent with these data, simultaneous application of the inhibitory AhR antagonist α-NF to AhR-proficient HaCaT cells did also abolish the capacity of BP to enhance death receptor-induced apoptosis (Supplementary Figure 4).

AhR-dependent enhancement of death receptor-mediated apoptosis. (a) HaCaT cells were pretreated with 3.5 μM BP (white bars) or 0.1% DMSO (black bars) for 48 h and subsequently exposed to different concentrations of TRAIL for further 24 h with or without 20 μM pan-caspase inhibitor z-VAD (right). Cell viability was then determined by crystal violet staining. Values represent means±S.D. of three independent experiments. (b) HaCaT AhR parental (empty) control vector cells were pretreated with 3.5 μM BP (white bars) or 0.1% DMSO (black bars) for 48 h and subsequently exposed to different concentrations of CD95L or TRAIL for further 24 h with or without 20 μM pan-caspase inhibitor z-VAD (right). Cell viability was then determined by crystal violet staining. Values represent means±S.D. of three independent experiments. (c) HaCaT shRNA AhR k.d. cells were pretreated with 3.5 μM BP (white bars) or 0.1% DMSO (black bars) for 48 h and subsequently exposed to different concentrations of CD95L or TRAIL for further 24 h with or without 20 μM pan-caspase inhibitor z-VAD (right). Cell viability was then determined by crystal violet staining. Values represent means±S.D. of three independent experiments. (d) Sensitisation of undifferentiated (KBM) and differentiating (KBM−) NHEKs for TRAIL-induced apoptosis: to trigger differentiation, keratinocytes were switched to growth supplement deprived KBM− medium or maintained in KBM medium for 24 h. Expression of distinct differentiation markers was confirmed by real-time PCR (Supplementary Figure 5). Both populations were pretreated with 3.5 μM BP for 48 h and subsequently exposed to 250 ng/ml (left) or 100 ng/ml (right) TRAIL until signs of apoptosis were observed by light microscopy. Proportions of apoptotic cells were determined by annexin V staining and flow cytometric analyses. Values represent means±S.D. of three experiments
Pro-apoptotic effects of AhR are independent from transcriptional induction of CYP1 enzymes and occur in both undifferentiated and differentiating keratinocytes
We have analysed next whether the pro-apoptotic crosstalk between death receptors and AhR agonists depends on classical AhR target genes, especially CYP1A1 and 1B1, which play important roles in oxidative biotransformation of xenobiotics. Transient downregulation of AhR using independent siRNAs confirmed its crucial importance for enhanced death receptor-mediated apoptosis, consistent with our previous observations in stable k.d. cells. Inhibition of both CYP enzymes was ascertained at a functional level in all transfected cell populations by EROD assays (Figure 6c). In contrast to AhR, inhibition of both CYP1A1 and 1B1 did not affect the capacity of BP to enhance TRAIL-induced apoptosis (Figure 6c). We conclude that this effect neither depends on oxidative metabolism of endogenous or xenobiotic CYP1 substrates nor on secondary genotoxic injuries. Again, this observation confirms our finding that BP metabolites do not sensitise cells for apoptosis.
TRAIL has previously been shown to induce cell death in dividing NHEKs. Differentiating NHEKs, however, displayed decreased expression of TRAIL receptors and TRAIL-induced apoptosis.20 We have therefore analysed whether the apoptotic crosstalk between TRAIL and AhR agonists was affected by keratinocyte differentiation. In our experiments, differentiation was induced by growth factor withdrawal and confirmed by increased expression of keratins 1 and 10 and involucrin (Supplementary Figure 5). Annexin V staining demonstrated sensitising effects of BP on TRAIL-induced apoptosis in both undifferentiated and differentiating cells, but this effect was less apparent in the second population (Figure 7d). Furthermore, differentiating keratinocytes displayed decreased membrane expression of TRAIL receptors 1 and 2. However, AhR agonists did not affect expression of these receptors in both HaCaT cells and dividing keratinocytes, thus excluding altered receptor levels as putative mechanism (Supplementary Figure 3).
We have further investigated the expression of cytokines in the HaCaT model, including endogenous CD95L, TRAIL, interferon-γ (INF-γ) and tumour necrosis factor-α (TNF-α) (Supplementary Figure 6). Although some analysed factors displayed altered expression levels upon BP or β-NF exposure, none of these patterns was consistent for explaining the comparable sensitisation of HaCaT (e.v.) cells by both agonists and the lack of sensitisation in AhR k.d. cells. Exposure to BP and subsequently TRAIL did heavily induce (∼700-fold) expression of several pro-apoptotic and inflammatory molecules, including CD95L and INF-γ, which was only observed in cultures showing markedly increased cell death (data not shown). This finding putatively reflects a secondary effect in the process of apoptosis itself.
Apoptotic crosstalk between BP and death receptors involves enhanced processing of caspase-8
Activation of death receptors triggers recruitment of FADD and subsequent processing of caspase-8. In contrast to caspase-3, which integrates multiple endogenous and extracellular stimuli, caspase-8 is specifically activated within the death receptor-induced signalling complex (DISC). To test whether AhR agonists promote apoptosis at the level of receptor complexes, we have investigated processing of pro-caspase-8 by western blotting (Figure 8). In these experiments, varying concentrations of CD95L and TRAIL were applied and adopted to cell viability tests to assure an optimal enhancement of apoptosis by BP. A strongly enhanced formation of the pro-caspase-8 cleavage products (p43/41 and p18) was observed after 8 h in HaCaT cells that had been pretreated with BP (Figure 8a). As expected, an equivalent processing pattern was found for the downstream effector caspase-3. Processing of either pro-caspase-8 or pro-caspase-3 in response to TRAIL was only observed in cells that had been pretreated with BP (Figure 8a). As the pro-apoptotic effect of BP involves enhanced processing of pro-caspase-8, the observed crosstalk between BP and CD95L or TRAIL is expected to occur at the receptor complex level.

BP promotes activation of caspase-8 and caspase-3 in AhR-proficient but not in AhR-deficient cells. (a) HaCaT cells were pretreated with 3.5 μM BP or 0.1% DMSO for 48 h, subsequently exposed to indicated concentrations of CD95L (upper panel) or TRAIL (lower panel) and analysed by western blotting. Caspase cleavage products, FLIP long and actin are indicated. In addition, PARP cleavage was analysed in the TRAIL experiment. (b) HaCaT parental (empty) control vector cells (e.v.) and HaCaT shRNA AhR knockdown cells (k.d.) were pretreated with 3.5 μM BP or 0.1% DMSO for 48 h, subsequently exposed for different time intervals to 20 ng/ml of CD95L-Fc (upper panel) or 100 ng/ml TRAIL (lower panel) and analysed by western blotting. Caspase cleavage products, FLIP long and actin are indicated
Next, we analysed whether enhanced processing of pro-caspase-8 and -3 was dependent on AhR, using again the variant HaCaT k.d. cells and the corresponding e.v. control cells (Figure 8b). Consistent with the functional data on apoptosis induction and our findings in wild-type HaCaT cells, an enhanced processing of both pro-caspases was exclusively observed in the control (AhR-proficient cells) cell line and only after preincubation with BP (Figure 8b). In contrast, the AhR-deficient cells were again protected, indicating that AhR signalling is required for the processing of both caspase precursors.
Discussion
Transcriptional induction of metabolising enzymes remains a central function of AhR function, despite a much wider spectrum of activities that had been recognised in recent years. However, oxidative biotransformation is always linked to generation of reactive intermediates that might lead to cellular injuries. Several AhR agonists, including BP, trigger adverse effects, including formation of DNA adducts or induction of oxidative stress. A tight regulation is therefore required to limit such adverse effects at the cellular level. Importantly, prolonged activation and nuclear translocation of AhR induces ubiquitination and subsequent degradation of the receptor itself,21 thus retaining the capacity of AhR to induce CYP1A monooxygenases and trigger oxidative biotransformation of toxicological relevant xenobiotics. Intriguingly, although AhR can act as ubiquitin E3 ligase, no auto-ubiquitination has yet been described in the literature. AhR forms an atypical E3 complex that mediates proteasome-dependent degradation of defined substrates, notably steroid receptors, including oestrogen receptors α and β, as well as the androgen receptor.9 Besides these specific E3 ligase activities, activated AhR might also induce ubiquitination of alternate target proteins. For example, the long isoform of FLIP, an inactive homologue of caspase-822, 23 was found to be decreased in BP-treated cells (Figure 8). On the other hand, chemical inhibition of the proteasomal activity did not exert any effect on the pro-apoptotic functions of BP-activated AhR (Supplementary Figure 7). Therefore, the pro-apoptotic function of AhR reveals unrelated to its E3 ubiquitin ligase activity. Alternatively, AhR activation might inhibit expression of FLIP, which is partly controlled by NF-κB,24 or regulate other target genes that remain to be characterised.
A further mechanism to limit putative mutagenic effects as consequence of excessive CYP activation might involve regulation of cell cycle progression and apoptosis. In fact, prolonged activation of AhR led to G1 phase cell cycle arrest in serum released mouse liver.25 It was proposed that CYP1A1 removes putative endogenous ligands, leading again to an inactivation of this receptor. In contrast, cell cycle arrest is maintained in the presence of stable xenobiotic ligands, such as TCDD.25 On the other hand, there is only limited evidence that activated AhR is sufficient to enhance elimination of potentially injured cells via apoptosis. Sustained AhR activity via a constitutively active mutant led to inflammation,26 as well as tumour promotion.27 This is consistent with our data, showing no significant apoptosis induction by BP in keratinocytes. However, the physiological context is changed when cells are exposed to apoptotic stimuli, as it can occur during inflammatory skin diseases. In human skin, death receptors, especially CD95, play a pivotal role in inflammatory diseases,28 such as eczematous dermatitis. Initially, apoptosis is only triggered in few keratinocytes by CD95L that is expressed by infiltrating T cells.29 Enhanced apoptosis by chemical exposures could thus disproportionately increase the overall tissue damage, thereby modulating the pathology of inflammatory diseases. Notably, CD95-induced apoptosis plays a pivotal role in toxic epidermal necrolysis (TEN), a rare skin disease that is also affected by exposures to drugs and chemicals.30 The pathology of TEN is marked by excessive apoptosis and can be blocked by antagonising CD95-specific antibodies.31
Our data indicate that AhR agonists can sensitise keratinocytes for death receptor-induced apoptosis, independently from putative genotoxic metabolites that can be formed from subsequent biotransformation. This finding is in agreement with other data,32 showing that hepatic mouse AhR knockout cells as well as fibroblasts are less susceptible to CD95-mediated apoptosis when compared with the corresponding reconstituted cells. Our data argue that apoptosis is a mechanism to prevent prolonged AhR signalling especially in cells that had been exposed to cytotoxic cytokines. The precise molecular mediators of AhR agonists to enhance receptor-induced apoptosis remains to be identified, but could involve transcriptional control of putative AhR target genes. Apoptosis-inducing ligands, especially CD95L, have been shown to induce proinflammatory cytokines and signalling in keratinocytes34, 35, 36, 37 and could potentially be affected by AhR signalling. In our study, however, we did not find any evidence for altered cytokine expression levels contributing to the pro-apoptotic effects of AhR described here. However, we provide evidence that DISC, and especially caspase-8, is a primary target for the enhancement of apoptosis by AhR agonists. Altogether, our results suggest that receptor-induced apoptosis can be modulated by environmentally relevant AhR agonists such as BP, the carcinogenic lead compound from the class of PAHs.
Materials and Methods
Reagents
BP and β-NF were purchased from Sigma-Aldrich (Sigma-Aldrich Chemie GmbH, Munich, Germany). BPDE, BP-3-phenol and BP-1,6-dione were obtained from NCI Chemical Carcinogen Reference Standards Repository, Midwest Research Institute, Kansas City, MO, USA. Liquor Carbonis Detergens was obtained from Caesar & Loretz (Caesar & Loretz GmbH, Hilden, Germany). Monoclonal ANTI-FLAG M2 antibody was purchased from Sigma-Aldrich (F3165).
Cell culture
The human keratinocyte cell line HaCaT was cultured in DMEM containing L-glutamine (2 mM), 10% fetal calf serum (v/v) and penicillin/streptomycin (100 U/ml). Only cells before passage no. 55 were used in this study. Lentiviral transduced HaCaT cells, containing either shRNA sequences for AhR silencing or nonsilencing, are described elsewhere.17 Primary human keratinocytes (i.e., NHEKs) were isolated from foreskin tissue samples by overnight trypsin digestion. The culture medium was a 3 : 1 mixture of Ham’s F-12 medium (L-glutamine (2 mM) and penicillin/streptomycin (100 U/ml)) and DMEM (low glucose (1 g/l), L-glutamine (8 mM) and penicillin/streptomycin (100 U/ml)), supplemented with adenine, insulin, fetal calf serum, hydrocortisone, epidermal growth factor and forskoline. Cell culture dishes were coated with collagen (2 mg/ml) diluted to 10% (v/v) in sterile water for 30 min at 37°C and then washed twice with PBS before cell culture. Alternatively, NHEKs were cultured to confluence in Keratinocyte Growth Medium 2 (KBM, PromoCell, Heidelberg Germany) on noncoated cell culture plastic. Differentiation was triggered by withdrawal of all supplements, except bovine pituitary extract and CaCl2, 24 h before the start of experiments.
Cytotoxicity assay
BP and β-NF stock solutions were prepared in DMSO at a final concentration of 10 mM. Working concentration (3.5–10 μM) of BP, as well as of applied metabolites, had been verified by LC-MS/MS (data not shown). Compound-dependent cytotoxicity in cell lines and NHEKs was evaluated with WST assays and crystal violet staining according to Scaffidi et al.22 Cells were plated at a density of 2 × 104 cells per cm2 and grown overnight. Culture medium was replaced before treatment with BP or other indicated reagents. The final concentration of the solvent did not exceed 0.1% (v/v) in the culture medium. The water-soluble dye WST-1 was added and absorbance was measured in a Tecan (Männedorf, Switzerland) microplate reader at 450 nm.
RNA analysis
RNA was isolated using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) and cDNA was prepared using either the High Capacity RNA-to-cDNA Kit (Applied Biosystems, Foster City, CA, USA) or AMV reverse transriptase (Promega, Madison, WI, USA). Real-time PCR was carried out using the pre-designed TaqMan assays Hs00153120_m1, Hs00236330_m1 and Hs99999903_m1, or using a SYBR green assay (5Prime, Hamburg, Germany) with the primers listed in Supplementary Table 1. Actin or HPRT were used as reference genes.
Western blot analysis
Cells were lysed on ice in RIPA buffer containing 50 mM Tris/HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Igepal and 0.25% sodium deoxycholate, with protease inhibitor cocktail (Calbiochem, San Diego, CA, USA). Equal amounts of proteins were applied to SDS-PAGE, transferred onto nitrocellulose membranes and immunoblotted according to the manufacturer’s instructions. Antibodies against the following proteins were used: CYP1A1/1B1 1 : 200 (sc-20772, sc-32882; Santa Cruz Biotechnology, Santa Cruz, CA, USA), cytochrome c 1 : 200 (556433; BD Biosciences, Franklin Lakes, NJ, USA), AhR 1 : 200 (sc-8087; Santa Cruz Biotechnology), caspase-8 (sc-6136; Santa Cruz Biotechnology); caspase-3 (no. 9662; Cell Signaling Technology, Danvers, MA, USA) and FLIP Alx-804-428 (Enzo Life Sciences, Farmingdale, NY, USA).
Primary antibody probed blots were visualised with appropriate horseradish peroxidase-coupled secondary antibodies (Santa Cruz Biotechnology) using enhanced chemiluminescence (34078; Thermo Scientific, Waltham, MA, USA) for detection.
Cytochrome c release
Selective permeabilisation with digitonin was used to analyse the release of cytochrome c from mitochondria. Cytosolic and mitochondrial fractions were prepared according to Ndozangue-Touriguine et al.33 Briefly, after harvesting, 6 × 106 cells were washed twice with ice-cold PBS and resuspended for 30 min on ice in cytosolic lysis buffer. After centrifugation (1000 × g) the supernatant was collected as cytosolic fraction and the pellet was resuspended for 5 min on ice in mitochondrial lysis buffer. The supernatant after centrifugation at 10 000 × g was collected as mitochondrial fraction. Equal amounts of protein were analysed by western blot as described above. For densitometric analysis, the public domain (http://rsbweb.nih.gov/ij/) Java image processing program ImageJ was used. Each signal has been evaluated in comparison with the control and equalised for the loading control.
Annexin V staining and flow cytometric analysis
A total of 7.5 × 104 cells were cultured in six-well plates and allowed to grow overnight. The cells were then incubated as indicated and harvested by trypsinisation at the indicated time points. Expression of CD95 was analysed by staining of 1 × 105 cells with FITC-conjugated mouse monoclonal anti-human CD95 (FAB142F; R&D Systems, Minneapolis, MN, USA) and – as a control of analysis – with FITC-conjugated mouse IgG1 antibody (555748; BD Biosciences,) by a FACS Canto II flow cytometer (BD Biosciences) according to the manufacturer’s recommendations. Apoptosis was determined by staining with annexin V/ 7-AAD (550475/559925; BD Biosciences) according to the recommendations of the manufacturer. Briefly, 1 × 105 cells were washed with ice-cold annexin buffer (HEPES (10 mM), NaCl (140 mM) and CaCl2 (5 mM)) and stained with 5 μl of annexin V and 5 μl of 7-AAD for 15 min at room temperature in the dark. After adding annexin, buffer cells were analysed by a FACS Canto II flow cytometer (BD Biosciences). The percentage of apoptotic population (annexin V-positive cells) was calculated using BD FACS Diva software (BD Biosciences). Untreated cells were used to define basal levels of apoptotic and death cells.
Abbreviations
- AhR:
-
aryl hydrocarbon receptor
- BP:
-
benzo[a]pyrene
- BPDE:
-
(+)-anti-BP-7,8-diol-9,10-epoxide
- β-NF:
-
β-naphtoflavone
- CD95:
-
cluster of differentiation 95 (Fas)
- CD95L:
-
CD95 ligand
- CYP1A1:
-
cytochrome P450 1A1
- CYP1B1:
-
cytochrome P450 1B1
- e.v.:
-
empty vector
- k.d.:
-
knockdown
- NHEK:
-
normal human epithelial keratinocyte
- PAH:
-
polycyclic aromatic hydrocarbon
- shRNA:
-
small hairpin RNA
- TCDD:
-
2,3,7,8-tetrachlorodibenzo-p-dioxin
- TRAIL:
-
TNF-related apoptosis-inducing ligand
References
Hall M, Grover PL . Chemical carcinogenesis and mutagenesis In: Cooper CS, Grover PL, (eds). Handbook of Experimental Pharmacology, Vol I. Springer-Verlag: Berlin, 1990, pp 327–372.
Luch A, Baird WM . Carcinogenic polycyclic aromatic hydrocarbons In: McQueen CA, Roberts R, (eds). Comprehensive Toxicology, Vol 14. Elsevier: Oxford, 2010, pp 85–124.
Zhu H, Li Y, Trush MA . Characterization of benzo[a]pyrene quinone-induced toxicity to primary cultured bone marrow stromal cells from DBA/2 mice: potential role of mitochondrial dysfunction. Toxicol Appl Pharmacol 1995; 130: 108–120.
Bock KW, Köhle C . The mammalian aryl hydrocarbon (Ah) receptor: from mediator of dioxin toxicity toward physiological functions in skin and liver. Biol Chem 2009; 390: 1225–1235.
Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008; 453: 106–109.
Esser C, Rannug A, Stockinger G . The aryl hydrocarbon receptor in immunity. Trends Immunol 2009; 30: 449–455.
Esser C . The immune phenotype of AhR null mouse mutants: not a simple mirror of xenobiotic receptor over-activation. Biochem Pharmacol 2009; 77: 597–607.
Puga A, Ma C, Marlowe JL . The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem Pharmacol 2009; 77: 713–722.
Ohtake F, Baba A, Takada I, Okada M, Iwasaki K, Miki H et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 2007; 446: 562–666.
Ko CB, Kim SJ, Park C, Kim BR, Shin CH, Choi S et al. Benzo[a]pyrene-induced apoptotic death of mouse hepatoma Hepa1c1c7 cells via activation of intrinsic caspase cascade and mitochondrial dysfunction. Toxicology 2004; 199: 35–46.
Kim SJ, Ko CB, Park C, Kim BR, Sung TH, Koh DH et al. p38 MAP kinase regulates benzo[a]pyrene-induced apoptosis through the regulation of p53 activation. Arch Biochem Biophys 2005; 444: 121–129.
Tampio M, Loikkanen J, Myllynen P, Mertanen A, Vähäkangas KH . Benzo[a]pyrene increases phosphorylation of p53 at serine 392 in relation to p53 induction and cell death in MCF-7 cells. Toxicol Lett 2008; 178: 152–159.
Xiao H, Rawal M, Hahm ER, Singh SV . Benzo[a]pyrene-7,8-diol-9,10-epoxide causes caspase-mediated apoptosis in H460 human lung cancer cell line. Cell Cycle 2007; 6: 2826–2834.
Brinkmann J, Hutzler C, Trappe S, Stolpmann K, Bock U, Liebsch M et al. Comparative studies on the metabolism of benzo[a]pyrene in keratinocytes in vitro skin models and human skin ex vivo — metabolite patterns as indicator for the metabolic capacity of skin equivalents? Skin-In-Vitro Conference Frankfurt/Main 2010.
Leverkus M, Trautmann A . CD95-mediated signals in the skin: going out with an (inflammatory) bang? J Invest Dermatol 2006; 126: 2364–2366.
Wehrli P, Viard I, Bullani R, Tschopp J, French LE . Death receptors in cutaneous biology and disease. J Invest Dermatol 2000; 115: 141–148.
Kalmes M, Neumeyer A, Rio P, Hanenberg H, Fritsche E, Blömeke B . Impact of the arylhydrocarbon receptor on eugenol- and isoeugenol-induced cell cycle arrest in human immortalized keratinocytes (HaCaT). Biol Chem 2006; 387: 1201–1207.
Sheard MA, Vojtesek B, Janakova L, Kovarik J, Zaloudik J . Up-regulation of Fas (CD95) in human p53 wild-type cancer cells treated with ionizing radiation. Int J Cancer 1997; 73: 757–762.
Leverkus M, Sprick MR, Wachter T, Denk A, Bröcker EB, Walczak H et al. induced apoptosis and gene induction in HaCaT keratinocytes: differential contribution of TRAIL receptors 1 and 2. J Invest Dermatol 2003; 121: 149–155.
Jansen BJ, van Ruissen F, Cerneus S, Cloin W, Bergers M, van Erp PE et al. Tumor necrosis factor related apoptosis inducing ligand triggers apoptosis in dividing but not in differentiating human epidermal keratinocytes. J Invest Dermatol 2003; 121: 1433–1439.
Davarinos NA, Pollenz RS . Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. J Biol Chem 1999; 274: 28708–28715.
Scaffidi C, Schmitz I, Krammer PH, Peter ME . The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem 1999; 274: 1541–1548.
Siegmund D, Wicovsky A, Schmitz I, Schulze-Osthoff K, Kreuz S, Leverkus M et al. Death receptor-induced signalling pathways are differentially regulated by γ-interferon upstream of caspase 8 processing. Mol Cell Biol 2005; 25: 6363–5379.
Kreuz S, Siegmund D, Scheurich P, Wajant H . NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signalling. Mol Cell Biol 2001; 21: 3964–3973.
Levine-Fridman A, Chen L, Elferink CJ . Cytochrome P4501A1 promotes G1 phase cell cycle progression by controlling aryl hydrocarbon receptor activity. Mol Pharmacol 2004; 65: 461–469.
Tauchi M, Hida A, Negishi T, Katsuoka F, Noda S, Mimura J et al. Constitutive expression of aryl hydrocarbon receptor in keratinocytes causes inflammatory skin lesions. Mol Cell Biol 2005; 25: 9360–9368.
Ikuta T, Namiki T, Fujii-Kuriyama Y, Kawajiri K . AhR protein trafficking and function in the skin. Biochem Pharmacol 2009; 77: 588–596.
Ehrenschwender M, Wajant H . The role of FasL and Fas in health and disease. Adv Exp Med Biol 2009; 647: 64–93.
Kerstan A, Leverkus M, Trautmann A . Effector pathways during eczematous dermatitis: where inflammation meets cell death. Exp Dermatol 2009; 18: 893–899.
Harr T, French LE . Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis 2010; 5: 35.
Viard I, Wehrli P, Bullani R, Schneider P, Holler N, Salomon D et al. Inhibition of toxic epidermal necrolysis by blockade of CD95 with human intravenous immunoglobulin. Science 1998; 282: 490–493.
Park KT, Mitchell KA, Huang G, Elferink CJ . The aryl hydrocarbon receptor predisposes hepatocytes to Fas-mediated apoptosis. Mol Pharmacol 2005; 67: 612–622.
Ndozangue-Touriguine O, Sebbagh M, Mérino D, Micheau O, Bertoglio J, Bréard J . A mitochondrial block and expression of XIAP lead to resistance to TRAIL-induced apoptosis during progression to metastasis of a colon carcinoma. Oncogene 2008; 27: 6012–6022.
Iordanov MS, Sundholm AJ, Simpson EL, Hanifin JM, Ryabinina OP, Choi RJ et al. Cell death-induced activation of epidermal growth factor receptor in keratinocytes: implications for restricting epidermal damage in dermatitis. J Invest Dermatol 2005; 125: 134–412.
Farley SM, Dotson AD, Purdy DE, Sundholm AJ, Schneider P, Magun BE et al. Fas ligand elicits a caspase-independent proinflammatory response in human keratinocytes: implications for dermatitis. J Invest Dermatol 2006; 126: 2438–151.
Farley SM, Purdy DE, Ryabinina OP, Schneider P, Magun BE, Iordanov MS . Fas ligand-induced proinflammatory transcriptional responses in reconstructed human epidermis. Recruitment of the epidermal growth factor receptor and activation of MAP kinases. J Biol Chem 2008; 283: 919–928.
Peter ME, Budd RC, Desbarats J, Hedrick SM, Hueber AO, Newell MK et al. The CD95 receptor: apoptosis revisited. Cell 2007; 129: 447–450.
Acknowledgements
This work was partly supported by the Federal Ministry of Education and Research (0315226A) and the DFG (He 5275/2-1). We are grateful to the German Federal Institute for Risk Assessment for funding of this project.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Stephanou
Supplementary Information accompanies the paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Stolpmann, K., Brinkmann, J., Salzmann, S. et al. Activation of the aryl hydrocarbon receptor sensitises human keratinocytes for CD95L- and TRAIL-induced apoptosis. Cell Death Dis 3, e388 (2012). https://doi.org/10.1038/cddis.2012.127
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2012.127
Keywords
This article is cited by
-
Immunoproteomic identification and characterization of Ni2+-regulated proteins implicates Ni2+ in the induction of monocyte cell death
Cell Death & Disease (2017)
-
Cigarette smoke-induced cell death of a spermatocyte cell line can be prevented by inactivating the Aryl hydrocarbon receptor
Cell Death Discovery (2015)
