
Abstract
Lead (Pb) is a critical industrial and environmental contaminant that can cause pathophysiological changes in several cellular and organ systems and their processes, including cell proliferation, differentiation, apoptosis, and survival. The skin is readily exposed to and damaged by Pb, but the mechanisms through which Pb damages cells are not fully understood. We examined the apoptotic properties of Pb in mouse skin fibroblast (MSF) in vitro. Treatment of fibroblasts with 40, 80, and 160 μM Pb for 24 h revealed morphological alterations, DNA damage, enhanced caspase-3, -8, and -9 activities, and apoptotic cell population. Furthermore, apoptosis was dosage (0–160 μM) and time (12–48 h) dependent. Concentrations of intracellular calcium (Ca2+) and reactive oxygen species were increased, and the mitochondrial membrane potential was decreased in exposed cells. Cell cycle arrest was evident at the G0/G1 phase. The Bax, Fas, caspase-3 and -8, and p53 transcript levels were increased, whereas Bcl-2 gene expression was decreased. Based on our analysis, Pb triggers MSF apoptosis bydisrupting intracellular homeostasis. Our findings enrich the knowledge about the mechanistic function of Pb-induced cytotoxicity on human skin fibroblasts and could potentially guide future Pb health risk assessments.
Similar content being viewed by others

Introduction
Heavy metal pollution threatens human survival and development. Lead (Pb), one of the most common toxic heavy metals, is a ubiquitous environmental and occupational pollutant that can accumulate in the environment over a prolonged period1. Pb is widely used in batteries, paints, varnishes, pigments, and other daily-use products. Occupational Pb exposure, associated with dry cell manufacturing, mining activities, metallurgy, automotive machinery, and welding, has resulted in concerning Pb concentrations in air, water, soil, and food chains globally2. Pb has a long biological half-life and is non-degradable; hence, its excretion rate from the body is extremely slow. In the human body, once Pb is absorbed via inhalation, ingestion, or dermal contact, it accumulates in the bone, blood, and soft tissue, with a half-life of 20–30 years in bone and 35 days in blood3. Pb is a toxin that can cause various biochemical, physiological, and behavioral dysfunctions4. Pb exposure can induce pathological alterations in multiple organ systems, including the digestive5, hemopoietic6, urinary7, nervous8, immune9, respiratory10, reproductive11, circulatory12, endocrine13, and kinetic14 systems. Furthermore, Pb can cause apoptosis or necrosis of various cells in vitro, such as rat pheochromocytoma cells (PC12), rat retinal rod cells15, chicken liver cells16, rat hepatic and renal cells, and germ cells17.
As a widespread environmental toxin, Pb can exert a broad range of toxic effects on cells and organs. Pb can induce tissue damage and oxidative stress in animal models. Moreover, Pb can induce deleterious effects, such as reactive oxygen species (ROS) production, direct loss of antioxidant stores, cell membrane breakdown, enzymatic degradation, and DNA damage18. At the cellular level, Pb can reduce the mitochondrial membrane potential (MMP), consume the adenosine triphosphate (ATP) pool, and increase ROS production, resulting in ultrastructural and functional disturbances in mitochondrial metabolism19. In the early stage, Pb directly affects the expression of Fos and Jun and inhibits DNA repair, and indirectly plays a genotoxic role as a co-mutagen, causing chromatin compaction and aggregation, as well as impairing DNA and RNA synthesis20. Growing evidence has revealed that Pb can activate/inhibit several intracellular signaling pathways associated with cell survival and death21.
Typical apoptosis is mainly activated by two pathways, namely the extrinsic and intrinsic pathways. The extrinsic pathway is activated via death receptors on the cell surface and causes the activation of caspase-8 or -10, which initiates the caspase-3 cascade, leading to apoptosis. The endogenous pathway is divided into the mitochondrial and ER pathways; the mitochondrial pathway originates from the release of mitochondrial cytochrome C and is associated with the activation of caspase-9, which activates the downstream caspase-3 enzyme series and initiates the caspase cascade leading to apoptosis. The endoplasmic reticulum pathway results from the activation of apoptotic enzymes in the endoplasmic reticulum. Disruption of endoplasmic reticulum calcium homeostasis can activate caspase-12, and activated caspase-12 can further cleave caspase-3 and participate in cell apoptosis caused by the endoplasmic reticulum pathway.
Environmental Pb pollution seriously harms human health, and Pb can induce tissue and cell apoptosis through various pathways22. Apoptosis maintains homeostasis by removing excess and abnormal cells. Although the highest and most direct Pb exposure occurs through the skin, Pb-mediated induction of apoptosis in skin cells and the underlying mechanism have rarely been reported23. Pb-induced apoptosis is considered a possible mechanism of Pb toxicity24. Pb exposure-induced excessive or disordered apoptosis is a major factor in the development and spread of autoimmune and neurodegenerative neurological diseases8. Although the apoptotic potential of Pb has been well documented, it remains unclear whether Pb can induce apoptosis in skin fibroblasts. In the present study, mouse skin fibroblasts (MSFs) were incubated with various Pb concentrations to determine Pb-mediated toxic effects by analyzing the impact on cell survival, morphology, proliferation, and apoptosis. The cell cycle, MMP, intracellular Ca2+ homeostasis, ROS, caspase-3, -8, and -9 activities, and gene expression levels of Bax, Bcl-2, Fas, caspase-3 and -8, and p53 were analyzed to comprehensively clarify the toxicity mechanism of Pb on human skin fibroblasts and enrich the general knowledge of Pb toxicity mechanisms.
Methods
Materials
Herein, mice were not involved in the isolation of MSFs. We employed first to third passage MSFs from the animal genetic resources laboratory cell bank, Institute of Animal Science, Chinese Academy of Agricultural Sciences. The supplier authenticated the fibroblasts, which tested negative for contamination. Lead acetate (Pb(Ac)2; purity, 99.999%), 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT), propidium iodide (PI), acridine orange (AO), and ethidium bromide (EB) were acquired from Sigma Chemical Co. (St. Louis, MO). Annexin V-FITC Apoptosis Detection Kit I was obtained from Becton Dickinson Company (Becton Drive, Franklin Lakes, NJ). Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco (Gibco BRL Life Technologies, Grand Island, NY).
Cell culture
MSFs were cultured in DMEM with 10% FBS under 5% CO2 and 95% air at 37 °C in a humid chamber. The medium was refreshed every 2 days. The cells were digested, dissociated, and divided into two sterilized flasks upon reaching 80–90% confluence.
Growth dynamics
In total, 2 × 104 cells/mL were seeded in a 24-well microporous plate. The cell growth rate was measured by counting three wells daily for 8 days using a hemocytometer. The growth curve was plotted, and the population doubling time (PDT) was assessed depending on the growth curve.
Drug preparation and cell exposure
Pb2+ was provided to cells as Pb(Ac)2, dissolved and diluted to the desired concentration with DMEM, and filtered for sterilization. Upon entering the logarithmic growth phase, cells were exposed to multiple Pb2+ concentrations (0, 40, 80, and 160 μM) for prespecified durations (12, 24, 36, and 48 h).
Cell survival assay
Cell survival was evaluated via MTT assay. In total, 1 × 105 cells/well cell suspensions (200 μL) were added to 96-well microplates. The old medium was removed, and 180 μL of fresh complete medium with varying Pb2+ concentrations was added when cells entered the logarithmic phase. Simultaneously, corresponding controls and replicates were established. Cells were cultured for 12, 24, 36, and 48 h. MTT solution (20 μL, 5 mg/mL) was introduced, and cells were incubated for 4 h. The supernatant was removed, and the precipitate was resuspended in dimethyl sulfoxide (DMSO; 200 μL). Absorbance was detected at 490 nm using a microplate reader (Elx 800; BioTek Instruments, Winooski, VT, USA).
AO/EB double staining
Cells were exposed to Pb2+ for 24 h, and cell suspensions were collected prior to staining with AO and EB solutions (2 mg/mL in ethanol). The cells were incubated at 25 °C in the absence of light for 6 min, followed by imaging with a confocal microscope (Nikon TE-2000-E, Tokyo, Japan).
Hoechst 33258 staining
Briefly, coverslips were sterilized and added to culture plates. Cells were allowed to grow on coverslips, treated with Pb2+ for 24 h, and subsequently entered the logarithmic growth phase. The control group was established simultaneously. After discarding the culture medium, a fixative was introduced prior to rinsing and staining with Hoechst 33258. Anti-fluorescence quenching sealing solution was dripped onto the glass slide, and coverslips with cells were covered on the glass slide, ensuring that cells were fully in contact with the sealing solution while avoiding bubble formation. Cells were observed and photographed under a confocal microscope.
Comet assay
Upon cell harvest, cells were rinsed in cold phosphate-buffered saline (PBS), and 1 × 105 cells/mL was suspended in PBS prior to the comet assay as described in the Alkaline Comet Assay kit protocol (Trevigen Inc., Gaithersburg, MD, USA). The cell suspension was combined with low-melting-point agarose, and the samples were transferred with a pipette onto comet slides prior to storage in ice-cold lysis buffer (Trevigen Inc.) at 4 °C for 35 min. The comet slides were electrophoresed with alkaline electrophoresis buffer and treated with 70% ethanol for 6 min before drying. DNA was stained with SYBR green I (Sigma) and photographed under a confocal microscope.
Transmission electron microscopy (TEM)
The cells were collected, rinsed twice with PBS, fixed in 2.5% glutaraldehyde and then in 1% osmium tetroxide, and rinsed using 0.1 mol/L of PBS. An ethanol series (30%, 50%, 70%, 80%, 90%, and 100%) was used for cell dehydration, followed by cell embedding in an epoxy resin using acetone, which was left to solidify. Ultrathin sections (50 nm) of the resin were cut using an ultrathin microtome (LEICAUC6i) and stained using uranyl acetate and lead citrate prior to imaging using a transmission electron microscope (JEM-2000Ex; JEOL Ltd., Tokyo, Japan).
Annexin V‑fluorescein isothiocyanate (FITC)/PI double‑labeling
Apoptosis was assessed based on the Annexin V‑FITC Apoptosis Detection kit directions. Upon harvest, cells were diluted to 5 × 105 cells/mL with binding buffer. Next, 5 μL each of PI and FITC were introduced to each 100 μL cell suspension sample, followed by incubation in 400 μL binding buffer for 10 min. Cells were analyzed via flow cytometry (FC500, Beckman Coulter, Brea, CA). A minimum of 10,000 cells were assessed per sample.
Cell cycle analysis
Upon harvest, cells were resuspended in cold 70% ethanol, incubated at 4 °C for 12 h, rinsed in PBS, incubated in RNase A (0.02 mg/mL), and stained with PI solution (sodium citrate: 1 mg/mL, PI: 0.05 mg/mL, NaCl: 0.585 g/mL, pH: 7.2–7.6) for 30 min at 4 °C. Cells were analyzed via flow cytometry. A minimum of 10,000 cells were assessed per sample.
MMP
Following harvest, cells were diluted to 1 × 106 cells/mL; 0.5 mL 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1) working solution (5 mg/mL) was introduced to each sample and then maintained in a CO2 incubator for 10 min, followed by twice rinsing with prewarmed PBS. Cells were resuspended in 0.5 mL PBS before analysis via flow cytometry. Overall, 10,000 cells were assessed per sample.
Intracellular Ca2+ homeostasis
DMSO was used to dissolve Fluo3-AM (Invitrogen, Carlsbad, CA, USA) to form a 2 mM solution before cryogenization at − 70 °C. Following harvest, cells were diluted to 1 × 106–2 × 106 cells/mL using serum-free culture media and loaded with Fluo3-AM (final concentration of 5 μM). Negative controls (without adding Fluo3-AM) were prepared prior to incubation under 5% CO2 at 37 °C without light for 35 min. Images were captured under a confocal microscope.
ROS analysis
Cells were harvested, concentration-adjusted to 1 × 106–2 × 106 cells/mL, attached to 2′,7′-dichlorodihydrofluorescein-diacetate (DCFH‑DA) (Molecular Probes, Eugene, OR, USA) to a final concentration of 8 μM, prior to incubation at 37 °C under 5% CO2 for 15 min. Images were captured under a confocal microscope.
Caspase-3, -8, and -9 activity assay
Caspase‑3, ‑8, and ‑9 activities were analyzed using a caspase activity kit (Beyotime Institute of Biotechnology, Haimen, China) based on kit directions and as reported before25.
Real-time quantitative polymerase chain reaction (qPCR)
Total RNA was isolated, and complementary DNA (cDNA) was synthesized with a Primer Script TM RT kit (Takara, Dalian, China). Targeted GAPDH (housekeeping gene), Bcl-2, Bax, Fas, caspase-3, caspase-8, and p53 primers were prepared using the Primer Premier 5.0 software (PREMIER Biosoft, Palo Alto, CA, USA) and generated by Shanghai Biotechnology Co. Ltd (Shanghai, PR China). qPCR was performed using an ABI 7500 qPCR system (Applied Biosystems, Foster City, CA, USA) and qPCR Detection Kit (Takara) as per kit instructions under the following conditions: denaturation (94 °C, 3 min), denaturation (40 cycles, 94 °C, 30 s), annealing (52–55 °C, 30 s), and elongation (72 °C, 30 s). Each experiment was repeated three times and performed in duplicate in 96-well plates. Relative gene levels were computed using the 2−△△Ct formula, and alterations in gene levels were evaluated via the Student’s t-test (P < 0.05 was set as the significance threshold).
Statistical analysis
All experiments were performed in triplicate to verify reproducibility. Data are presented as mean ± standard deviation (SD), and analysis was carried out using the Statistical Analysis System (SAS) software (SAS Institute Inc., Cary, NC, USA) and comparison via a multiple comparison test (DUNCAN). P < 0.05 was set as the significance threshold.
Results
Growth dynamics
The MSF growth curve presented a strong “S” outline, and the PDT was approximately 32 h. The cells entered a latent phase from days 1–2, entering the logarithmic phase from days 2–5. The density peaked on day 5, reached a plateau on day 6, and subsequently declined (Fig. 1a).

Growth dynamics, cytotoxicity analysis, and AO/EB double staining. (a) Mouse skin fibroblast (MSF) growth curve. (b) Cytotoxicity assessment. c Morphological evaluation of nuclear damage in MSFs after Pb2+ exposure for 24 h. (a) Control, (b) 40 μM, (c) 80 μM, and (d) 160 μM; scale bar = 50 μm. Statistically different values relative to matched controls are labeled with * (P < 0.05) and ** (P < 0.01; n = 3).
Cell survival assay
The MTT assay was utilized to determine the mitochondrial succinate dehydrogenase activity and detect mitochondrial function, which is widely used to evaluate cell viability. Pb2+ reduces the viability of MSFs. Cell survival decreased considerably with increased Pb2+ concentration and duration of exposure; in other words, cell survival reduced in a dose‑ and time‑dependent manner (Fig. 1b).
AO/EB double staining
In normal cells, the nuclear chromatin is pyknotic or beaded. The 40 µM treatment group showed numerous early apoptotic cells with pale yellow nuclei. In the 80 µM treatment group, some cells exhibited uneven morphology and late apoptosis. The 160 µM treatment group exhibited a considerable number of late apoptotic cells, with orange-red nucleosomes, concentrated chromatin, variable sizes, and relatively thick staining. Some late apoptotic cells were close to death (Fig. 1c).
Hoechst 33258 staining
The nuclear chromatin of control cells was uniformly stained, displaying round or ovoid nuclei, and emitting strong fluorescence. The treatment group showed cell chromatin concentration, uneven coloring, nuclear pyknosis, and bright spots, with nuclear pyknosis worsening with increasing Pb2+ concentration (Fig. 2a).

Hoechst 33258 staining, comet assays, and transmission electron microscopy (TEM). (a) Morphological evaluation of mouse skin fibroblasts (MSFs) 24 h after Pb2+ exposure using Hoechst 33258 staining by confocal microscopy. (a) Control, (b) 40 μM, (c) 80 μM, and (d) 160 μM; scale bar = 10 μm. (b) and (d) Comet assay showed DNA damage in MSF exposed to Pb2+ for 24 h. Significantly different values relative to matched controls are labeled with ** (P < 0.01; n = 3). (c) Subcellular assessment via TEM. (a) Control and (b) 160 μM; scale bar = 2 μm.
Comet assay
To analyze DNA damage, 100 fibroblasts were randomly selected from each group. The DNA of control cells was confined to the nucleus, while DNA strands of Pb2+-treated cells were broken, with DNA fragments traveling along the electric field, forming a bright fluorescent trail (Fig. 2b). The trail size indicated injury severity, which increased with increasing Pb2+ doses (Fig. 2d), thereby indicating that Pb2+ could disrupt the DNA integrity of MSFs.
TEM
The control cells exhibited abundant cell surface microvilli, uniform cytoplasm, intact nuclear membrane structure, clear and complete nucleoli, and uniform staining. Considering Pb2+-treated cells, cell surface microvilli disappeared, vacuoles appeared in the cytoplasm, and the nuclear membrane structure was damaged. The chromatin of apoptotic cells exhibited chromatin was condensed, marginalized, and segmented into clumps (Fig. 2c).
Annexin V-fluorescein isothiocyanate (FITC)/PI double labeling
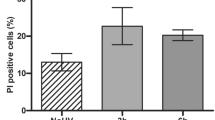
Following exposure of MSFs to 0, 40, 80, and 160 μM of Pb2+ for 12 to 48 h, the apoptotic rate (P < 0.01) significantly increased from 0.4 to 9.7% when compared with that of the control group (12, 36, and 48 h; histogram plots not shown; Fig. 3a). Furthermore, apoptosis was dose- and time-dependent (Fig. 3b).

Annexin V-FITC/PI double labeling. (a) Scatter plot of the apoptotic rates of mouse skin fibroblasts (MSFs) treated with 0, 40, 80, or 160 μM Pb2+ for 24 h (12, 36, and 48 h plots not shown). (b) Apoptotic rates of MSFs exposed to 0, 40, 80, or 160 μM Pb2+ for 12, 24, 36, and 48 h. Significantly different values relative to matched controls are labeled with ** (P < 0.01; n = 3).
Cell cycle analysis
With increasing Pb2+ concentration, the proportion of G0/G1 cells increased, and the proportion of S and G2 phase cells decreased accordingly. Based on these results, there was a dose-dependent association between Pb2+ levels and cell cycle dysregulation, indicating that Pb2+ inhibited DNA synthesis in the G0/G1 phase (Fig. 4a). In the diploid DNA peak (sub-G1 population), the proportion of apoptotic cells increased in a dose-dependent fashion (Fig. 4b).

Cell cycle progression. (a) Cell cycle progression and sub-G1 levels in mouse skin fibroblasts (MSFs) following Pb2+ exposure for 24 h. (b) Cell distribution in the sub-G1, G0/G1, S, and G2/M cell cycle phases. Significantly different values relative to matched controls are labeled with * (P < 0.05) and ** (P < 0.01; n = 3).
MMP
JC-1 is a lipophilic cationic stain that exclusively enters mitochondria and reversibly changes it from a green to a red color with increasing MMP. In normal cells, JC-1 accumulates in the mitochondria and shows red fluorescence. In apoptotic cells, JC-1 exists as a monomer with green cytoplasm, indicating a low MMP. The proportion of C-gated cells indicates the alterations in the MMP; a larger quantity represents a decrease in the MMP (Fig. 5a). Accordingly, the MMP of treated cells was significantly reduced when compared with that of control cells (P < 0.01) (Fig. 5b).

Mitochondrial membrane potential (MMP). (a) Scatter plot of mouse skin fibroblast (MSF) MMP following treatment with 0, 40, 80, or 160 μM Pb2+ for 24 h. (b) The ratio of fluorescence intensity. Significantly different values relative to matched controls are labeled with ** (P < 0.01; n = 3).
Intracellular Ca2+ homeostasis
Upon treatment with higher Pb2+ concentrations, apoptotic cells showed a higher proportion and intensity of green fluorescence, indicating an increase in free Ca2+ levels. The green fluorescence observed in 160 μM Pb2+-treated cells was evenly distributed, suggesting a severe disruption in intracellular Ca2+ homeostasis (Fig. 6a).

Intracellular Ca2+ homeostasis and ROS analysis. (a) Effect of Pb2+ exposure on intracellular Ca2+ homeostasis in mouse skin fibroblasts (MSFs) after treatment for 24 h. (b) Effect of Pb2+ exposure on intracellular ROS levels in MSFs after treatment for 24 h. Both (a) and (b) show (a) Control, (b) 40 μM, (c) 80 μM, and (d) 160 μM; scale bar = 50 μm. ROS, reactive oxygen species.
ROS analysis
The higher the Pb2+ concentration, the greater the number of apoptotic cells and the stronger the green fluorescence (Fig. 6b). The green fluorescence intensity increased, suggesting that ROS concentration also increased. The green fluorescence was distributed throughout the cells in the 80 and 160 μM Pb2 + treatment groups.
Caspase-3, -8, and -9 activity analyses
Caspase-3, -8, and -9 are critical for cell apoptosis, serving as active proteases. Based on our analysis, the caspase-3 activity differed significantly in the 40 and 80 μM Pb2+ treated cells when compared with that in control cells (P < 0.05). The cell number was significantly increased in the 160 μM Pb2+-treated group when compared with that in the control group (P < 0.01). Caspase-8 and -9 activities notably differed in cells of all three treatment groups when compared with those in control cells (P < 0.01, Fig. 7a). The caspase-3, -8, and -9 activities increased with increasing Pb2+ concentrations in a dose-dependent fashion.

Caspase-3, -8, and -9 activity assays and gene expression levels. (a) Caspase-3, -8, and -9 activities were assessed in cytosolic extracts of mouse skin fibroblasts (MSFs) after Pb2+ treatment for 24 h. (b) Evaluation of Bax, Bcl-2, Fas, caspase-3, caspase-8, and p53 transcript expression at 24 h (adjusted to GAPDH). Significantly different values relative to matched controls are labeled with * (P < 0.05) and ** (P < 0.01; n = 3), which applies to (a) and (b).
qPCR
Considering glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the endogenous control, Bax and Bcl-2 are expressed in the outer mitochondrial membrane, and the Bax/Bcl-2 ratio strongly regulates apoptosis. Based on the qPCR analysis, treatment with 160 µM Pb2+ significantly increased Bax, Fas, caspase-3, caspase-8, and p53 levels in mouse fibroblasts (P < 0.01) but significantly decreased Bcl-2 expression (P < 0.01, Fig. 7b). these results indicate that Pb2+ treatment increased the Bax/Bcl-2 ratio in mouse fibroblasts.
Discussion
Environmental pollution attributed to heavy metals caused by human activities is a global concern. Pb, a heavy metal responsible for environmental pollution, is a non-biodegradable inorganic pollutant widely distributed in soil–water–air ecosystems owing to its easy migration and transformation26. Pb can cause a series of behavioral, biochemical, and physiological dysfunctions in organisms27. Toxicological and epidemiological studies have documented Pb-induced toxicity in several cells, organs, and organ systems28. Pb accumulation in living organisms has deleterious long-term effects, given that Pb induces cytotoxicity in various cell types through distinct cytotoxic pathways29. For instance, Pb suppresses the proliferation of human leukemia cells by inducing cell cycle arrest, DNA damage, and accelerating apoptosis via caspase-3 activation30. Pb mediates mitochondrial permeability transition pore (MPTP) opening, releases mitochondrial cytochrome c, and induces apoptosis of proximal tubule cells. Furthermore, Pb can induce apoptosis of rat proximal tubular cells through endoplasmic reticulum stress31. The skin is one of the major target organs for Pb poisoning. However, the Pb toxicity pathway in the skin remains elusive. Herein, we present evidence to suggest that Pb exposure suppresses MSF cell survival and promotes cell apoptosis.
Pb has been shown to significantly reduce TM3 cell proliferation in a concentration- and time/dependent manner29. Following treatment of HK-2 cells with increasing concentrations of lead nitrate (0–50 µM) for 24 h, intracellular ROS levels increased, whereas glutathione (GSH) levels decreased substantially in a dose-dependent manner32. Our findings are consistent with prior toxicological investigations involving other cell lines, showing a dose-dependent decrease in MSF survival, as evidenced by the MTT assay and Annexin V-FITC/PI double labeling. Annexin V- FITC/PI double labeling showed that Pb strongly promoted apoptosis, and the apoptotic-promoting effect was more notable with increasing Pb concentrations. AO/EB staining, Hoechst 33258 staining, and comet assay were used to demonstrate that Pb could induce MSF apoptosis. The membrane permeability of early apoptotic cells was relatively low, and ethidium bromide did not easily enter the cell interior; the cells were light yellow, with pyknotic or bead-like nuclear chromatin. The membrane permeability of late apoptotic cells was higher than that of early-stage apoptotic cells, but differed in terms of membrane rupture, with cells appearing orange-red and relatively more stained; some late apoptotic cells were close to death to a certain extent (Fig. 1c). Our assay results indicated that Pb could induce DNA damage, which is a unique change in apoptotic cells. The comet assay is a sensitive method for assessing individual cell nucleosomal DNA fragmentation. Ultrastructure analysis by electron microscopy is considered the optimal approach for identifying cell apoptosis. Based on our TEM analysis, Pb could induce typical apoptotic morphological changes, including cell shrinking, mitochondrial depolarization, subcellular structure destruction, and chromatin condensation in MSFs. In addition, flow cytometric analysis revealed that Pb treatment considerably increased the proportion of apoptotic cells in a concentration-dependent manner. Our results are consistent with those of previous studies, in which Pb accelerated apoptosis in TM3 cells33, cardiomyocytes34, and human monocyte-macrophage (THP-1) cells35.
Apoptosis, gene-controlled programmed cell death, is crucial for maintaining the internal environmental balance and normal body development. The cell cycle is a highly regulated process that modulates cell development and differentiation36. Given that apoptosis stimulation affects the cell cycle, alterations in cell cycle distribution may be related to apoptosis and differentiation. Hence, cell cycle analysis is among the most informative assessments for determining apoptosis. Pb was found to strongly trigger cell cycle arrest in the G0/G1 phase in the HL-60 cell lines30. To verify whether the Pb-mediated MSF suppression is related to cell cycle arrest, we examined cell cycle progression using flow cytometry. Based on our analysis, Pb caused G0/G1 phase cycle arrest in MSFs; the proportion of G1 cells and the level of hypodiploid DNA (sub-G1 population) were increased, the amount of S-phase cells was decreased, and cell proliferation was inhibited. Therefore, we hypothesized that Pb interferes with DNA replication and cell cycle distribution, resulting in MSF apoptosis. Mitochondria are necessary for apoptosis and are considered central regulators of apoptosis in vertebrates. One early event of apoptosis involves a reduction in MMP past a particular point. Pb reportedly binds to mitochondria at ion sites, resulting in a decrease in the membrane potential, shifting mitochondrial channels to an active open state37. Therefore, cell apoptosis is initiated when mitochondrial polarization, swelling, and membrane rupture occur along with the release of cytochrome C and other cytokines into the cytoplasm38. Pb treatment can stimulate the mitochondria-based apoptotic network39. Pb-driven mitochondria-based primary cultured rat proximal tubular cell apoptosis was shown to depend on the opening of the MPTP, and the three regulatory components of MPTP (CYPD, ANT, and VDAC) play roles in Pb-linked MPTP opening34. We demonstrated that the MMP of MSFs decreased dose-dependently with increased Pb exposure, indicating that Pb-driven MSF apoptosis is associated with mitochondria-based apoptosis. The Pb-based toxic sites in the mitochondria and the relative sensitivity of multiple mitochondria-mediated pathways remain poorly elucidated.
Under normal conditions, the body has a certain amount of ROS. As the messenger of various metabolic and signaling pathways within the body, ROS participates in the transcription of various genes and the expression of related functional proteins by activating and regulating various transcription factors, participating in cell proliferation, differentiation and maintenance of cells and tissues, and body metabolism40. ROS has strong reactive activity and oxidation. High ROS concentrations can directly or indirectly cause oxidative damage to lipid substances such as DNA, protein and cell membranes and induce cell apoptosis, senescence, and death41. In mouse renal proximal tubular cells as target cells, Pb exposure caused cell apoptosis, necrosis, mitochondrial damage and other cell damage, accompanied by increased intracellular ROS content42. Long-term low-dose Pb exposure can impact the structure and function of mouse renal proximal tubule cells. In addition, Pb exposure was found to alter cell morphology and hinder the ability of cells to take up calcium ions. These physiological changes are related to the content of intracellular ROS and occur in a dose-dependent manner43. Low concentration, acute Pb exposure could activate the activity of protein kinase C (PKC) in cells, which, in turn, regulated the calcium transport related N-methyl-d-aspartic acid receptor (NMDA) receptor channel, thereby increasing intracellular Ca2+ content. However, NMDA receptor activation will promote the release of excitatory amino acid glutamate, subsequently triggering an increase in intracellular ROS content, resulting in oxidative stress44. Oxidative stress and cell apoptosis are the two main pathways through which Pb exerts its toxic effects. Pb exposure-induced oxidative stress can accelerate the process of cell apoptosis, as well as impact ROS production, consequently affecting the process of oxidative stress45.
Ca2+, a secondary messenger, is necessary for cell metabolism; an imbalance in Ca2+ homeostasis can induce apoptosis through various pathways. The initial increase in Ca2+ may be attributed to the influx of Ca2+ in the extracellular space or redistribution of intracellular storage, resulting in irreversible or reversible changes in several major pathways46. Increased intracellular free Ca2+ activates multiple signals and events involved in almost all aspects of cellular activity, ultimately inducing apoptosis47. Based on our analysis, Pb treatment increased cytoplasmic free Ca2+ concentration, disrupting intracellular Ca2+ homeostasis. In workers exposed to Pb, eryptosis associated with oxidative damage seems to be characterized by a rise in free intracellular Ca2+, owing to enhanced Ca2+ channel permeability and diminished Ca2+ Mg2+ (ATPase) activity48.
Apoptosis may be related to an imbalance in calcium homeostasis, oxidative damage, and mitochondrial damage49. Under physiological conditions, the production and clearance of oxygen free radicals are in a dynamic equilibrium. Several factors may reduce the ability of the antioxidant system to scavenge oxygen free radicals, thereby upregulating oxygen free radicals. Pb exposure can induce the generation of oxygen free radicals, and oxidative stress directly damages cell membranes, leading to increased cell membrane permeability50. Herein, Pb treatment resulted in increased ROS levels in MSFs. Our results corroborate the findings of a previous study50 and suggest that Pb can induce oxidative stress in MSFs in vitro. Excessive ROS production has consistently been shown to induce apoptosis.
Caspases are a family of proteases, and the caspase-induced cascade is critical for the stimuli-induced apoptotic process. Caspase-3 is primarily activated by cytotoxic drugs51, and it strongly regulates apoptotic DNA damage. In human leukemia cells, Pb was shown to modulate apoptosis via caspase-3 activation30. Herein, Pb-exposed MSFs showed increased caspase-3, -8, and -9 activities. Our results corroborate those of a previous study, which demonstrated that Pb exposure could promote renal cell apoptosis and impair cell function by activating caspase-352.
Bcl-2 is a major contributor to cell death signaling, and the protein family has both anti- and pro-apoptotic members. Bcl-2 regulates the balance between pro-apoptotic members like Bax and pro-survival Bcl-2-like proteins and functions as an anti-apoptotic protein that modulates multiple apoptotic networks. The Bax protein is considered to be oligomeric and can enhance mitochondrial outer membrane permeability53. A decrease in the Bax/Bcl-2 ratio suppresses apoptosis, whereas an increase in the Bcl-2 ratio enhances apoptosis54. Therefore, the Bax to Bcl-2 ratio strongly modulates cell survival and apoptosis. In addition, p53 inhibits the anti-apoptotic protein Bcl-2 and promotes the pro-apoptotic protein Bax55. Bax and Bcl-2 regulate the mitochondrial outer membrane permeability, stimulate cytochrome C release, activate the caspase cascade, and induce apoptosis56. The Fas gene is also important in mediating apoptosis. Based on our findings, increasing Pb concentrations enhanced expression levels of Fas, Bax, p53, caspase-3, and caspase-8 and reduced those of Bcl-2. In addition, the Bax to Bcl-2 ratio was increased. Therefore, Pb can interfere with the levels of multiple apoptotic genes in MSFs. In PC12 cells, Pb triggers apoptosis, accompanied by increased caspase-3 and Bax levels and an associated increase in p53 expression, elevated Bax expression, and reduced Bcl-2 levels57. Furthermore, Pb can stimulate Fas gene expression and promote testicular germ cell apoptosis in a dose-dependent manner58.
Pb affects the levels of oxidants in MSFs and enhances ROS production; in turn, increased ROS concentration leads to the dysregulation of the cellular lipid bilayer and disrupts Ca2+ transport. Pb leads to instability in calcium homeostasis and destroys membrane ion channels by disrupting electron transport and reducing ATP concentration, ultimately leading to cell apoptosis59. Pb-induced apoptosis of MSFs also appears to depend on the mitochondria, given that Pb can affect the MMP. Damaged cell membranes are characterized by a decrease in MMP and interference in the balance of molecules such as Bax and Bcl-260. Alterations in the Bax/Bcl-2 ratio can trigger cytochrome C release and caspase activation and, ultimately, cell apoptosis61.
However, given that the present study was an in vitro assessment, limitations are inevitable. Considering one such limitation, it should be noted that the results do not necessarily represent real-world effects of Pb on MSFs in vivo, as various other factors affect cell proliferation and apoptosis in vivo. Another limitation of the study is that protein levels of apoptotic markers were not quantified, which can be considered a future research direction.
Conclusions
In conclusion, Pb induces considerable toxic effects on MSFs, such as decreasing cell survival, accelerating cell morphological alterations, and apoptosis. Treatment with Pb treatment arrested the cell cycle, decreased the MMP, increased the Bax/Bcl-2 ratio, and enhanced caspase-3, -8, and -9 activation, suggesting that Pb can induce MSF apoptosis. These intracellular processes were related to Ca2+ overload, suppression of DNA formation, oxidative stress, mitochondrial dysfunction, and altered gene expression. Our findings provide useful regarding the etiology of Pb toxicity in the skin and indicate the undesirable outcomes of Pb exposure in humans. This evidence offers a basis for further studies on the Pb pollutant-based toxicity mechanisms to improve health risk assessments.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Miao, Z. et al. The antagonistic effect of selenium on lead-induced apoptosis and necroptosis via P38/JNK/ERK pathway in chicken kidney. Ecotoxicol. Environ. Saf. 231, 113176. https://doi.org/10.1016/j.ecoenv.2022.113176 (2022).
Wu, H. Y. et al. Heavy metal pollution and health risk assessment of agricultural soil near a smelter in an industrial city in China. Int. J. Environ. Health Res. 30, 174–186 (2019).
Wirth, J. J. & Mijal, R. S. Adverse effects of low level heavy metal exposure on male reproductive function. Syst. Biol. Reprod. Med. 56, 147–167 (2010).
Singh, N., Kumar, A., Gupta, V. K. & Sharma, B. Biochemical and molecular bases of lead-induced toxicity in mammalian systems and possible mitigations. Chem. Res. Toxicol. 31, 1009–1021 (2018).
Jalali, S. M., Najafzadeh, H. & Bahmei, S. Protective role of silymarin and d-penicillamine against lead-induced liver toxicity and oxidative stress. Toxicol. Ind. Health. 33, 512–518 (2017).
Liu, J. et al. Mitochondria defects are involved in lead-acetate-induced adult hematopoietic stem cell decline. Toxicol. Lett. 235, 37–44 (2015).
Song, X., Li, Z., Liu, F., Wang, Z. & Wang, L. Restoration of autophagy by puerarin in lead-exposed primary rat proximal tubular cells via regulating AMPK-mTOR signaling. J. Biochem. Mol. Toxicol. 31, e21869. https://doi.org/10.1002/jbt.21869 (2017).
Chibowska, K. et al. Effect of lead (Pb) on inflammatory processes in the brain. Int. J. Mol. Sci. 17, 2140 (2016).
Fenga, C., Gangemi, S., Di Salvatore, V., Falzone, L. & Libra, M. Immunological effects of occupational exposure to lead (review). Mol. Med. Rep. 15, 3355–3360 (2017).
Boskabady, M. H., Tabatabai, S. A. & Farkhondeh, T. Inhaled lead affects lung pathology and inflammation in sensitized and control guinea pigs. Environ. Toxicol. 31, 452–460 (2016).
Shen, W., Chen, J., Yin, J. & Wang, S. L. Selenium protects reproductive system and foetus development in a rat model of gestational lead exposure. Eur. Rev. Med. Pharmacol. Sci. 20, 773–780 (2016).
Wildemann, T. M., Siciliano, S. D. & Weber, L. P. The mechanisms associated with the development of hypertension after exposure to lead, mercury species or their mixtures differs with the metal and the mixture ratio. Toxicology 339, 1–8 (2016).
Doumouchtsis, K. K., Doumouchtsis, S. K., Doumouchtsis, E. K. & Perrea, D. N. The effect of lead intoxication on endocrine functions. J. Endocrinol. Investig. 32, 175–183 (2009).
Pounds, J. G., Long, G. J. & Rosen, J. F. Cellular and molecular toxicity of lead in bone. Environ. Health Perspect. 91, 17–32 (1991).
He, L. H. et al. Bcl-xL overexpression blocks bax-mediated mitochondrial contact site formation and apoptosis in rod photoreceptors of lead-exposed mice. Proc. Natl. Acad. Sci. USA 100, 1022–1027 (2003).
Chi, Q. et al. Involvement of mitochondrial pathway in environmental metal pollutant lead-induced apoptosis of chicken liver: Perspectives from oxidative stress and energy metabolism. Environ. Sci. Pollut. Res. Int. 24, 28121–28131 (2017).
Yedjou, C. G., Tchounwou, C. K., Haile, S., Edwards, F. & Tchounwou, P. B. N-acetyl-cysteine protects against DNA damage associated with lead toxicity in HepG2 cells. Ethnicity Dis. 20, 101 (2010).
Matovic, V., Buha, A., Ethukic-Cosic, D. & Bulat, Z. Insight into the oxidative stress induced by lead and/or cadmium in blood, liver and kidneys. Food Chem. Toxicol. 78, 130–140 (2015).
Baranowska-Bosiacka, I. et al. Altered energy status of primary cerebellar granule neuronal cultures from rats exposed to lead in the pre- and neonatal period. Toxicology 280, 24–32 (2011).
Narayana, K. & Raghupathy, R. DNA damage in lead-exposed hepatocytes: Coexistence of apoptosis and necrosis?. Drug Chem. Toxicol. 35, 208–217 (2012).
Mujaibel, L. M. & Kilarkaje, N. Mitogen-activated protein kinase signaling and its association with oxidative stress and apoptosis in lead-exposed hepatocytes. Environ. Toxicol. 30, 513–529 (2015).
Yin, K. et al. Antagonistic effect of selenium on lead-induced neutrophil apoptosis in chickens via miR-16-5p targeting of PiK3R1 and IGF1R. Chemosphere 246, 125794. https://doi.org/10.1016/j.chemosphere.2019.125794 (2020).
He, X. Y. et al. Cytotoxic responses and apoptosis in rat kidney epithelial cells exposed to lead. Biomed. Environ. Sci. 29, 529–533 (2016).
Wang, X. et al. Selenium protects against lead-induced apoptosis via endoplasmic reticulum stress in chicken kidneys. Biol. Trace Elem. Res. 182, 354–363 (2018).
Young, M. M. et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J. Biol. Chem. 287, 12455–12468 (2017).
Chu, B. X. et al. Interplay between autophagy and apoptosis in lead(II)-induced cytotoxicity of primary rat proximal tubular cells. J. Inorg. Biochem. 182, 184–193 (2018).
Tchounwou, P. B., Yedjou, C. G., Patlolla, A. K. & Sutton, D. J. Heavy metal toxicity and the environment. Exp. Suppl. 101, 133–164 (2012).
Kim, H. C. et al. Evaluation and management of lead exposure. Ann. Occup. Environ. Med. 27, 1–9 (2015).
Zhou, L. et al. Lead acetate induces apoptosis in Leydig cells by activating PPARγ/caspase-3/PARP pathway. Int. J. Environ. Health Res. 31, 34–44 (2021).
Yedjou, C. G., Tchounwou, H. M. & Tchounwou, P. B. DNA damage, cell cycle arrest, and apoptosis induction caused by lead in human leukemia cells. Int. J. Environ. Res. Public Health. 13, 56 (2015).
Mao, W. et al. The calmodulin kinase II (CaMKII)-p38 MAPK-ATF6 pathway is involved in the endoplasmic reticulum (ER) stress and myocyte apoptosis in ß1-adrenergic receptor (AR) peptide induced cardiomyopathy. Circulation 114, II_242 (2006).
Siddarth, M. et al. Lead-induced DNA damage and cell apoptosis in human renal proximal tubular epithelial cell: Attenuation via N-acetyl cysteine and tannic acid. J. Biochem. Mol. Toxicol. 32, e22038. https://doi.org/10.1002/jbt.22038 (2018).
He, X. et al. Lead induces apoptosis in mouse TM3 Leydig cells through the Fas/FasL death receptor pathway. Environ. Toxicol. Pharmacol. 56, 99–105 (2017).
Li, C. et al. Lead-induced cardiomyocytes apoptosis by inhibiting gap junction intercellular communication via autophagy activation. Chem. Biol. Interact. 337, 109331. https://doi.org/10.1016/j.cbi.2020.109331 (2021).
Metryka, E. et al. Lead (Pb) accumulation in human THP-1 monocytes/macrophages in vitro and the influence on cell apoptosis. Biol. Trace Elem. Res. 199, 955–967 (2021).
Darzynkiewicz, Z. et al. DNA damage signaling assessed in individual cells in relation to the cell cycle phase and induction of apoptosis. Crit. Rev. Clin. Lab. Sci. 49, 199–217 (2011).
Kowluru, R. A., Kowluru, V., Xiong, Y. & Ho, Y. S. Overexpression of mitochondrial superoxide dismutase in mice protects the retina from diabetes induced oxidative stress. Free Radic. Biol. Med. 41, 1191–1196 (2006).
Kulms, D. & Schwarz, T. Independent contribution of three different pathways to ultraviolet-B-induced apoptosis. Biochem. Pharmacol. 64, 837–841 (2002).
Liu, G. et al. Mitochondrial permeability transition and its regulatory components are implicated in apoptosis of primary cultures of rat proximal tubular cells exposed to lead. Arch. Toxicol. 90, 1193–1209 (2016).
Januszyk, S. et al. Expression profile of mRNAs and miRNAs related to the oxidative-stress phenomenon in the Ishikawa cell line Treated either cisplatin or salinomycin. Biomedicines. 10, 1190. https://doi.org/10.3390/biomedicines10051190 (2022).
Folkes, L. K., Christlieb, M., Madej, E., Stratford, M. R. & Wardman, P. Oxidative metabolism of combretastatin A-1 produces quinone intermediates with the potential to bind to nucleophiles and to enhance oxidative stress via free radicals. Chem. Res. Toxicol. 20, 1885–1894 (2007).
Stacchiotti, A. et al. Stress proteins and oxidative damage in renal derived cell line exposed to inorganic mercury and lead. Toxicology 264, 215–224 (2009).
Navarro-Moreno, L. G. et al. Effects of lead intoxication on intercellular junctions and biochemical alteration of the renal proximal tubule cells. Toxicol. In Vitro. 23, 1298–1304 (2009).
Jadhav, A. L., Ramesh, G. T. & Gunasekar, P. G. Contribution of protein kinase C and glutamate in Pb2+-induced cytotoxicity. Toxicol. Lett. 115, 89–98 (2000).
Ziech, D. et al. The role of reactive oxygen species and oxidative stress in environmental carcinogenesis and biomarker development. Chem. Biol. Interact. 188, 334–339 (2010).
Ma, Y., Fu, D. & Liu, Z. Effect of lead on apoptosis in cultured rat primary osteoblasts. Toxicol. Ind. Health. 28, 136–146 (2012).
Lemasters, J. J., Theruvath, T. P., Zhong, Z. & Nieminen, A. L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta. 1787, 1395–1401 (2019).
López-Vanegas, N. C., Hernández, G., Maldonado-Vega, M. & Calderón-Salinas, J. V. Leukocyte apoptosis, TNF-α concentration and oxidative damage in lead-exposed workers. Toxicol. Appl. Pharmacol. 391, 114901. https://doi.org/10.1016/j.taap.2020.114901 (2020).
Suriyo, T., Watcharasit, P., Thiantanawat, A. & Satayavivad, J. Arsenite promotes apoptosis and dysfunction in microvascular endothelial cells via an alteration of intracellular calcium homeostasis. Toxicol. In Vitro. 26, 386–395 (2012).
Hernández-Franco, P., Silva, M., Valverde, M. & Rojas, E. Induction of oxidative stress by low doses of lead in human hepatic cell line WRL-68. Biometals 24, 951–958 (2011).
Xu, L. H. et al. Lead induces apoptosis and histone hyperacetylation in rat cardiovascular tissues. PLoS ONE 10, e0129091. https://doi.org/10.1371/journal.pone.0129091 (2015).
Yedjou, C. G., Tchounwou, H. M. & Tchounwou, P. B. DNA damage, cell cycle arrest, and apoptosis induction caused by lead in human leukemia cells. Int. J. Environ. Res. Public Health. 13(1), 56 (2016).
Yuan, G. et al. Sub-chronic lead and cadmium co-induce apoptosis protein expression in liver and kidney of rats. Int. J. Clin. Exp. Pathol. 7(6), 2905–2914 (2014).
Liu, Z. et al. Lead induces Siberian tiger fibroblast apoptosis by interfering with intracellular homeostasis. Drug Chem. Toxicol. 41, 188–198 (2018).
Luna, C. et al. c-Jun N-terminal kinase and p38 mitogen-activated protein kinase pathways link capacitation with apoptosis and seminal plasma proteins protect sperm by interfering with both routes. Biol. Reprod. 96, 800–815 (2017).
Kim, H. et al. Id-1 regulates Bcl-2 and Bax expression through p53 and NF-kB in MCF-7 breast cancer cells. Breast Cancer Res. Treat. 112, 287–296 (2008).
Deng, S. et al. Furazolidone induces apoptosis through activating reactive oxygen species-dependent mitochondrial signaling pathway and suppressing PI3K/Akt signaling pathway in HepG2 cells. Food Chem. Toxicol. 75, 173–186 (2015).
Xu, J., Ji, L. D. & Xu, L. H. Lead-induced apoptosis in PC12 cells: Involvement of p53, Bcl-2 family and caspase-3. Toxicol. Lett. 166, 160–167 (2006).
Dong, S. et al. The role of MAPK and FAS death receptor pathways in testicular germ cell apoptosis induced by lead. Acta Bioch. Biophys. Sin. 41, 800–807 (2009).
Antonsson, B., Montessuit, S., Lauper, S., Eskes, R. & Martinou, J. C. Bax oligomerization is required for channel-forming activity in liposomes and trigger cytochrome c release from mitochondria. Biochem. J. 345, 271–278 (2000).
He, L. et al. Bcl-XL overexpression blocks bax-mediated mitochondrial contact sites formation and apoptosis in rod photoreceptors of lead exposed mice. Proc. Natl. Acad. Sci. USA 100, 1022–1027 (2003).
Acknowledgements
We would like to thank Editage (http://www.editage.com) for editing and reviewing this manuscript for the English language.
Funding
This work was supported by the Natural Science Foundation of Liaoning Province (20180550694, 2023-MS-309), the Ministry of Science and Technology, National Science and Technology Basic Conditions Platform project (TZDWZYK2019-11).
Author information
Authors and Affiliations
Contributions
H.W. and Z.L. performed most of the experiments and drafted the manuscript. J.G. performed the remaining experiments with assistance from H.W. and Z.L. H.W. analyzed the data. Z.L. and W.G. conceived and designed the studies, oversaw the experiments, and were responsible for the decision to submit the article for publication. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, H., Wang, H., Guan, J. et al. Lead induces mouse skin fibroblast apoptosis by disrupting intracellular homeostasis. Sci Rep 13, 9670 (2023). https://doi.org/10.1038/s41598-023-36835-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-36835-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
