
Abstract
Cluster of differentiation (CD)8+ T cells are like a double edged sword during chronic viral infections because they not only promote virus elimination but also induce virus-mediated immunopathology. Elevated levels of reactive oxygen species (ROS) have been reported during virus infections. However, the role of ROS in T-cell-mediated immunopathology remains unclear. Here we used the murine lymphocytic choriomeningitis virus to explore the role of ROS during the processes of virus elimination and induction of immunopathology. We found that virus infection led to elevated levels of ROS producing granulocytes and macrophages in virus-infected liver and spleen tissues that were triggered by the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. Lack of the regulatory subunit p47phox of the NADPH oxidase diminished ROS production in these cells. While CD8+ T cells exhibited ROS production that was independent of NADPH oxidase expression, survival and T-cell function was elevated in p47phox-deficient (Ncf1−/−) mice. In the absence of p47phox, enhanced T-cell immunity promoted virus elimination and blunted corresponding immunopathology. In conclusion, we find that NADPH-mediated production of ROS critically impairs the immune response, impacting elimination of virus and outcome of liver cell damage.
Similar content being viewed by others


Main
Chronic virus infections are a major health problem worldwide. About 500 million people currently suffer from chronic infections of hepatitis B or C (HCV) virus,1, 2 whereas 34 million people are infected with human immunodeficiency virus (HIV).3 Virus-specific T cells are usually able to kill virus-infected cells, but during chronic infections cluster of differentiation (CD)8+ T cells loose their function.4, 5, 6, 7, 8, 9 Moreover, during prolonged virus infection, consistent T-cell activation can cause destructive immunopathology.10, 11, 12
Mechanisms of virus elimination and virus-induced immunopathology have been well studied in mice using the non-cytopathic RNA lymphocytic choriomeningitis virus (LCMV). In this model, CD8+ T cells exert their cytotoxic effects mainly via the effector molecules perforin and interferon (IFN)-γ.13, 14, 15 Liver pathology in this model is primarily mediated by CD8+ T-cell cytotoxicity against virus-infected hepatocytes. Once hepatocytes are infected with LCMV, liver damage occurs following infiltration of virus-specific CD8+ T cells into the liver. Complete depletion of CD8+ T cells therefore reduces immunopathology in this context.16 However, partial reduction in CD8+ T-cell activity results in enhanced virus propagation, leading to elevated liver cell damage caused by infiltration of remaining CD8+ T cells.10, 11, 12, 17 T cells thus have dichotomous functions during viral infection, limiting viral replication on the one hand and promoting virus-induced pathology on the other.
Reactive oxygen species (ROS) are defined as oxidized molecules containing an oxygen atom, such as O2−, hydrogen peroxide (H2O2) and HO−.18 Although ROS is involved in cell signaling and homeostasis at physiological levels, excess ROS formation leads to cellular stress and death. ROS can be generated by endogenous processes, or via exogenous stimuli such as irradiation and chemical exposure.19 The major source of endogenous cellular ROS is the mitochondrial respiration chain.20 In addition, ROS can be generated by the NADPH (nicotinamide adenine dinucleotide phosphate) oxidase, which produces O2− via oxidation of NADPH to NADP+. ROS production, mediated by high expression of NADPH oxidase, is of critical importance to innate immune cells such as granulocytes, which migrate to inflamed areas and utilize ROS as a defense mechanism against pathogens.21 The NADPH oxidase consists of the membrane-bound components cytochrome b558, gp91phox and p22phox, as well as the cytosolic components p47phox, p67phox and the Rho guanosine triphosphatase Rac1.22 Following granulocyte stimulation, the cytosolic subunit p47phox is heavily phosphorylated,23 a prerequisite for NADPH oxidase activity.24
Oxidative stress plays a critical role in liver, cardiovascular, Alzheimer’s and autoimmune diseases.25, 26, 27, 28 Neutrophil infiltrates can be observed during autoimmunity such as rheumatoid arthritis,29 and a recent report shows that neutrophil p47phox phosphorylation is elevated in patients with rheumatoid arthritis.30 Surprisingly, p47phox-deficient mice have shown higher incidences of autoimmunity such as rheumatoid arthritis.31, 32 Production of ROS by macrophages could prevent incidence of arthritis in this context.31 Moreover, in an experimental model of liver fibrosis, p47phox-deficient animals (also known as Ncf1−/− mice) showed attenuated disease development.33 Interestingly, some studies also report elevated levels of ROS in the liver during chronic infection with HCV, a phenomenon that is triggered by viral proteins and is partially dependent on NADPH synthesis.34, 35 Consistently, ROS was able to promote virus binding and growth of LCMV in vitro.36 Although there is mounting evidence for the strong importance of ROS in different immune responses, the specific role of ROS during virus-mediated immunity remains to be clarified.
Here we report that ROS impairs immunity to LCMV. We show that oxidative stress was capable of inhibiting virus-specific CD8+ T-cell function. ROS levels were elevated during virus-induced inflammation, and elevated ROS was dependent on the regulatory subunit of the NADPH oxidase, p47phox. P47phox-deficient mice showed reduced ROS production during virus infection and consequently generated stronger virus-specific T-cell responses. These Ncf1−/− mice were able to eliminate the virus earlier than controls, and therefore had limited immunopathology.
Results
ROS is produced during virus infection
LCMV leads to a strong activation of the innate immune system.37 To determine whether ROS-producing cells are activated during LCMV infection, we infected C57BL/6 mice with 2 × 106 plaque-forming units (PFU) of LCMV WE. We found a strong accumulation of granulocytes in spleen and liver tissues 6 days after virus infection (Figure 1a and b). This was confirmed by immunohistochemistry of snap frozen sections of spleen and liver tissues of infected animals (Figure 1c). To explore whether granulocyte infiltration affects ROS production, C57Bl/6 mice were analyzed for ROS production after infection using dihydroethidium (DHE). The amount of ROS-producing granulocytes was significantly higher in spleen and liver tissues from infected animals (Figure 1d). As expected, granulocytes were absent in the liver tissue from naïve animals (Figure 1b). These data suggest that LCMV infection results in the recruitment of activated granulocytes into liver and spleen tissues. As p47phox is a regulatory subunit of the NADPH oxidase, which is largely responsible for ROS production in granulocytes,38, 39 we tested whether ROS production in granulocytes was dependent on Ncf1. Therefore, Ncf1−/− mice were infected with LCMV and ROS production in infiltrating granulocytes was analyzed. Infiltration of granulocytes into spleen and liver tissues did not differ between wild-type (WT) and p47phox-deficient animals at days 2, 4 and 6 after infection with LCMV (Supplementary Figure S1a). However, production of ROS in Ncf1−/− mice was significantly reduced at days 4 and 6 after infection (Figure 1d and Supplementary Figure S1b). Additionally, macrophages in spleen and liver (Kupffer cells) tissues showed positive staining for DHE. While Kupffer cells exhibited limited ROS production in the liver, macrophages in the spleen also trigger p47phox-dependent ROS production after infection (Figure 1e). Although the macrophage population did not increase (Supplementary Figure S1c), ROS-producing macrophages were elevated in WT animals when compared with p47phox-deficient mice (Supplementary Figure S1d). To further analyze the role of p47phox during innate immunity, bone marrow-derived macrophages were stimulated with LPS and nuclear factor-κB activation was assessed. We did not observe any differences in inhibitor of κBα (IκBα) phosphorylation and degradation or in p65 phosphorylation between WT and p47phox-deficient macrophages (Supplementary Figure 2a). When tumor necrosis factor (TNF)-α production was measured in the supernatant of cultured macrophages, we observed no significant difference at different doses of LPS (Supplementary Figure S2b). Next, the innate immune response to virus infection was assessed by measurement of IFN-α concentration in the serum. When WT and Ncf1−/− mice were infected with 2 × 106 PFU of LCMV WE, similar levels of IFN-α were induced in the serum after 24 and 48 h (Supplementary Figure S2c). In conclusion, ROS production in granulocytes and macrophages was increased after virus infection and dependent on the regulatory subunit of the NADPH oxidase, p47phox.

ROS is produced during virus infection. (a–c) C57BL/6 mice were infected with 2 × 106 PFU LCMV WE. On day 6, single-cell suspensions from spleen and liver tissues were stained with an anti-CD8 and anti-Gr-1 antibodies and then analyzed by FCM and compared with samples from naïve mice. (a) One set of representative contour plots of n=6 is shown (spleen: upper panels; liver: lower panels; naïve: left panels; day 6 post infection (p.i.): right panels). (b) Absolute numbers are shown from two pooled experiments (n=6). (c) Sections from snap frozen spleen (upper panels) and liver (lower panels) tissues from naïve (left panels) and infected (right panels) mice were stained with an anti-Gr-1 antibody (red staining). One of three representative sections is shown. (d and e) WT and Ncf1−/− mice were infected with 2 × 106 PFU LCMV WE. Single-cell suspensions from spleen (left panels) and liver (right panels) tissues of naïve and infected animals were stained with DHE, an anti-F4/80 antibody and an anti-Gr-1 antibody. (d) MFI of DHE on Gr-1+ cells was analyzed (upper panels, n=4). Absolute numbers of Gr-1+DHE+ cells are shown (lower panels, n=4). (e) MFI of DHE was analyzed on F4/80+ cells in the spleen (left panel) and liver (right panel) tissues. ***P<0.001, **P<0.01, *P<0.05 and NS, not significant using the one-way ANOVA
ROS inhibited CD8+ T-cell viability in vitro
To test the influence of oxidative stress on T-cell survival and function, splenocytes from LCMV-infected mice were re-stimulated in vitro with the LCMV-specific epitope gp33. As expected, epitope-specific CD8+ T cells responded with rapid production of IFN-γ and exhibited baseline viability of approximately 50%. Exposure to H2O2 reduced both the viability of virus-specific T cells and IFN-γ production in a concentration-dependent manner (Figure 2a and Supplementary Figure S3a). Coincubation of T cells with another oxidative agent, diamide, similarly induced cell death in LCMV-specific CD8+ T cells and resulted in the loss of gp33-mediated cytokine production, even at doses which were not lethal to cells (Figure 2b and Supplementary Figure S3a). Endogenous glutathione protects cells from the toxic effects of ROS. To test whether the CD8+ T-cell function and viability are dependent on glutathione, the glutathione inhibitor 2-phenylethyl isothiocyanate (PIETC)40 was added to splenocytes during restimulation with LCMV-specific epitopes. Similar to H2O2 and diamide, PIETC was also able to inhibit CD8+ T-cell viability and IFN-γ production, even at sub-lethal concentrations (Figure 2c and Supplementary Figure S3a). Next, to test the consequences of exposure to steady-state oxidative stress, we coincubated T cells with galactose oxidase in the presence or absence of galactose during restimulation with gp33. Similar to the above used agents, cell viability and IFN-γ production of CD8+ T cells was limited after exposure to a combination of galactose and galactose oxidase (Figure 2d and Supplementary Figure S3b). These data indicate that increasing ROS in vitro has a major effect not only in limiting T-cell survival but also in reducing T-cell function.

ROS inhibited CD8+ T-cell viability in vitro. (a–d) C57BL/6 mice were infected with 200 PFU of LCMV WE. After 8 days, splenocytes were restimulated in vitro with gp33 in the absence or presence of H2O2, diamide, PIETC or with galactose oxidase (with or without 20 mg/ml galactose) at the indicated concentrations. (a) Titration of H2O2 is shown (n=3). (b) Titration of diamide is shown (n=3). (c) Titration of PIETC is shown (n=3). (d) Titration of galactose oxidase is shown (n=3; ***P<0.001 using the one-way ANOVA)
ROS inhibits CD8+ T-cell function in vivo
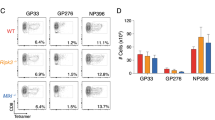
To investigate whether oxidative stress affected the virus-specific CD8+ T-cell response in vivo, WT and Ncf1−/− mice were infected with 2 × 106 PFU of LCMV WE, and infiltration of virus-specific CD8+ T cells in spleen and liver tissues was analyzed. The number of virus-specific CD8+ T cells was enhanced in spleen and liver tissues of Ncf1−/− mice (Figure 3a). This suggests that ROS may trigger death of virus-specific CD8+ T cells, a phenomenon typically observed during T-cell exhaustion.41, 42 Next, IFN-γ production by LCMV-specific T cells was analyzed. We found that the number of IFN-γ-producing CD8+ T cells was increased in spleen and liver tissues of Ncf1−/− mice (Figure 3b). We were further interested to know whether the function, and also the number of activated CD8+ T cells, was affected by ROS. As gauged from ratios of gp33-reactive IFN-γ-positive cells to gp33-tetramer+ cells, T-cell survival was reduced by ROS (Figure 3c). Consistently, 51Cr release assays also showed that cytotoxic T-cell function in single-cell suspended splenocytes was enhanced during infection with LCMV in Ncf1−/− animals when compared with WT mice (Supplementary Figure S4a). The production of other cytokines such as TNF-α by virus-specific CD8+ T cells was enhanced in the absence of Ncf1 (Figure 3d). However, the ratio of TNF-α+ to gp33-tetramer+ cells did not significantly differ between WT and Ncf1−/− mice (Figure 3e). Furthermore, the ratio of TNF-α+ to IFN-γ+ cells was similar in WT and p47phox-deficient animals (Figure 3e). IL-2 expression was also found to be comparable between WT and p47phox-deficient animals (Supplementary Figure S4b and c). Recently, it was reported that CD8+ T cells can produce ROS, which contributes to their proper function.43 However, DHE staining in virus-specific CD8+ T cells showed that this ROS production of LCMV-specific CD8+ T cells was not Ncf1 dependent (Supplementary Figures S5a and b). In summary, LCMV-specific CD8+ T-cell survival was enhanced in the absence of the NADPH oxidase regulating subunit p47phox.

NADPH oxidase-mediated ROS inhibits CD8+ T-cell immunity in vivo. Ncf1−/− and corresponding WT control mice were infected with 2 × 106 PFU LCMV WE. (a) On day 8, virus-specific tet-gp33+ CD8+ T cells were analyzed in the spleen and liver tissues. Data are presented as absolute numbers (n=6). (b) On day 8, single-cell suspensions from the spleen (left panel) and liver (right panel) tissues were restimulated with the LCMV-specific epitopes gp33 or np396. Cells were stained with an anti-CD8 and anti-IFN-γ antibodies. Absolute numbers of IFN-γ+CD8+ cells are presented (n=3, one out of two independent experiment is shown). (c) Ratios were calculated from absolute numbers of IFN-γ+ cells/Tet-gp33+ cells (n=3–6). (d) Single-cell suspensions from the spleen (left panel) and liver (right panel) tissues were stained for anti-CD8 and intracellular TNF-α. Data are presented as absolute numbers of TNF-α+ CD8+ cells (n=6). (e) Ratios of TNF-α+/Tet-gp33+ (left panels) and ratios of TNF-α+/IFN-γ+ (right panels) are shown after restimulation with gp33 or np396 (n=5 for spleen tissue and n=6 for liver tissue)
Accelerated LCMV elimination in Ncf1–/– mice
To explore whether the more robust CD8+ T-cell response measured in Ncf1−/− mice also enhanced virus elimination, virus titers in spleen, liver, kidney and lung tissues were measured after infection with 2 × 106 PFU LCMV WE. At early time points (days 2 and 4), WT and Ncf1−/− mice showed similar LCMV titers (Supplementary Figure S6a and b). Accordingly, immunohistology of spleen and liver tissues revealed comparable distribution of LCMV nucleoprotein (NP) 4 days after infection (Supplementary Figure S6c). We therefore concluded that LCMV was able to replicate in Ncf1−/− cells and that early (CD8+ T-cell independent) virus replication was not affected by p47phox. These data emphasize that anti-viral innate immunity is not abnormal in p47phox-deficient animals. Similarly, there was no significant difference detectable between Ncf1−/− and WT mice in viral titer 6 days after infection (Figure 4a). However, 8 days after LCMV infection, Ncf1−/− animals displayed significantly lower virus titers (Figure 4a). While LCMV persisted particularly in kidney and lung tissues of WT mice, most Ncf1−/− mice had reduced the LCMV titer below the limit of detection in these organs (Figure 4a). Accordingly, liver sections obtained 8 days after LCMV infection showed less LCMV NP in Ncf1−/− mice (Figure 4b). At 12 days after infection, the virus NP was virtually eliminated in p47phox-deficient animals, whereas WT mice continued to show virus-infected cells in the liver (Figure 4b). Additionally, we could observe less virus-infected hepatocytes in liver sections from Ncf1−/− animals when compared with control sections from WT mice. After depletion of CD8+ T cells, both WT and Ncf1−/− animals exhibited a large number of infected hepatocytes, suggesting virus control is dependent on CD8+ T cells (Figure 4c). At 12 days after infection, LCMV NP was readily detected in the splenic marginal zone of WT mice. In contrast, Ncf1−/− mice had almost completely eliminated virus antigen from the spleen (Figure 4d). In conclusion, Ncf1−/− mice showed lower virus titers and eliminated the virus faster than corresponding control mice, demonstrating a critical negative role for p47phox in anti-viral immunity.

Accelerated LCMV elimination in p47phox-deficient mice. (a and b) Ncf1−/− and corresponding WT control mice were infected with 2 × 106 PFU LCMV WE. (a) Virus titers were determined in the spleen, liver, kidney and lung tissues on days 6, 8, 12 and 16 (n=3−6). (b) Liver immunohistochemistry of LCMV-infected C57BL/6 and Ncf1−/− mice were examined for LCMV NP expression. One representative of n=3 is shown. (c) CD8+ T cells were depleted in WT and Ncf1−/− mice using an anti-CD8-depleting antibody (clone: YST-169). On day 6, livers were stained for LCMV-NP (clone: VL-4). One representative out of n=3 of is shown. Infected hepatocytes were highlighted with arrows. (d) Ncf1−/− and corresponding WT control mice were infected with 2 × 106 PFU LCMV WE. Liver immunohistochemistry of LCMV-infected C57BL/6 and Ncf1−/− mice was examined for LCMV NP expression. One of three representative slides is shown
NADPH oxidase enhances LCMV-induced immunopathology
The more robust T-cell response observed following LCMV infection in p47phox-deficient animals caused rapid virus elimination; we next tested whether this response also caused increased immunopathology. After infection of WT mice with 2 × 106 PFU LCMV-WE, mice develop virus-induced hepatitis, which is dependent on CD8+ T cells.16, 17 Liver cell damage can be detected by the enzyme alanine aminotransferase (ALT), which is present in hepatocytes. Strikingly, opposed to WT animals p47phox-deficient animals did not show signs of liver cell damage after infection with LCMV (Figure 5a). Similarly, bilirubin concentrations were elevated during virus-induced hepatitis in WT but not in Ncf1−/− mice (Figure 5b). To examine whether immunopathology was associated with the induction of liver fibrosis, we tested collagen RNA formation by real-time polymerase chain reaction (RT-PCR). After 8 days of infection, WT mice showed significantly increased collagen mRNA levels when compared with Ncf1−/− mice (Figure 5c). In conclusion, we found that ROS production delayed CD8+ T-cell-dependent virus control and enhanced immunopathology in the liver.

NADPH oxidase triggers LCMV-induced immunopathology. Ncf1−/− and corresponding WT control mice were infected with 2 × 106 PFU LCMV WE. (a) ALT activity was determined in the serum of C57BL/6 (closed circles) and Ncf1−/− mice (open squares) at the indicated time points (n=7). (b) Bilirubin concentrations were determined in the serum of C57BL/6 (closed circles) and Ncf1−/− mice (open squares) at the indicated time points (n=7). (c) Relative collagen mRNA copies (relative to naïve collagen mRNA levels) were analyzed in liver samples 8 days after infection (n=3)
Glutathione depletion by buthionine sulfoximine influences CD8+ T-cell-dependent virus control
The antioxidant glutathione is part of one of the most important redox system in mammalian cells.44 Buthionine sulfoximine (BSO) is known to deplete glutathione,45, 46 which in turn increases ROS levels. Consistently, when mice were treated with BSO, elevated ROS production could be observed in granulocytes (Figures 6a). BSO treatment did not affect CD8+ T-cell numbers in the spleen before infection (Supplementary Figure S7). However, treatment with BSO reduced virus-specific T-cell frequencies after infection with LCMV (Figure 6b). Additionally, the total number of functional IFN-γ-producing CD8+ T cells was decreased in BSO-treated mice (Figure 6c). Furthermore, the ratio of IFN-γ+/LCMV-specific T cells was decreased in animals that received BSO treatment, indicating that not only T-cell numbers but also T-cell function is reduced after BSO treatment (Figure 6d). Our in vitro data indicated that excessive ROS not only limits survival of virus-specific T cells but may also affect T-cell function. BSO treatment provides excess ROS and therefore may cause loss of T-cell function in addition to cell death of virus-specific T cells. Conclusively, the reduced T-cell response led to delayed control of LCMV (Figure 6e). These data support our main conclusions that ROS accumulation decreases virus-specific CD8+ T-cell function, resulting in delayed elimination of virus.

Glutathione depletion by BSO influences CD8+ T-cell-dependent virus control. C57BL/6 mice were treated with 200 μmol BSO daily starting a week before infection to 12 days after infection. Control mice were treated with saline. Mice were infected with 2 × 106 PFU LCMV WE. (a) On day 6 after infection, Gr-1+ cells were analyzed for DHE fluorescence. MFI of DHE was analyzed (n=4). (b) Tet-gp33+CD8+ and tet-np396+ CD8+ virus-specific T cells were analyzed on day 12 in the spleen (left panel) and liver (right panel) tissues (n=6–7). (c) Single-cell suspensions generated from spleen (left panel) and liver (right panel) tissues were restimulated with gp33 and np396. Intracellular IFN-γ was analyzed (n=6 and 7). (d) Ratio of the number of IFN-γ+ stimulated by gp33 divided by the number of gp33-tetramer+ cells is presented for spleen (left panel) and liver (right panel) tissues (n=6 and 7, paired t-test) (e) Virus titers were analyzed in spleen, liver, lung and kidney tissues 12 days after infection (n=5 and 6)
Discussion
In this study, we investigated the role of ROS in virus-induced immunity in the murine LCMV model. ROS production was elevated during virus infection in vivo, caused by NADPH oxidase activation of granulocytes and macrophages. Oxidative stress resulted in direct reduction in the survival of virus-specific CD8+ T cells. Consistently, Ncf1−/− mice, in which ROS production is partially blocked, had a more robust virus-induced CD8+ T-cell response, which resulted in rapid virus elimination and decreased immunopathology relative to WT animals.
Infections with HCV and HIV can become chronic. These viruses result in a virus-specific CD8+ T-cell response, which limits virus production at early stages after infection. However, at later stages virus-specific CD8+ T cells exhaust and/or trigger immunopathology.9, 17 Elevated levels of ROS have been reported for infections with HCV.34, 35 However, the role of virus-induced ROS, and especially the influence of ROS on the adaptive T-cell response is unclear. Our model suggests that virus-induced ROS inhibits virus-specific T-cell survival. Inhibition of the NADPH oxidase, which contributed in our model to virus-induced ROS production, could therefore provide a new therapeutic approach to investigate in order to enhance T-cell-mediated immunity.
ROS is produced by multiple mechanisms in mammalian cells. For instance, H2O2 production occurs as a product of normal mitochondrial energy metabolism in all cells, whereas specific cells such as granulocytes employ ROS in a directed manner for anti-bacterial defense. Cellular responses to ROS are also varied, and likely depend on cell type and redox status. Decreasing cellular ROS with antioxidant treatment has been previously shown to block virus binding and growth at early time points after infection.36 However, this mechanism alone is unlikely to explain the enhanced control of LCMV in Ncf1−/− mice as early virus titers were comparable between WT and Ncf1−/−.
When we infected Ncf1−/− mice with LCMV, we observed a more robust virus induced CD8+ T-cell response compared with WT animals. However, NADPH oxidase-deficient T cells showed similar ROS production to WT cells, suggesting that the main source of T-cell intrinsic ROS is not from NADPH oxidase activity but from metabolism.20 Clonal expansion requires high metabolic activity providing an explanation for why CD8+ T cells appear ROS positive after infection.47 Interestingly, recent discussions focus on metabolic pathways as major mechanism for limiting T-cell function.48 Our data support the concept that metabolic ROS affects T-cell immunity. In the presence of NADPH oxidase, which is highly expressed in granulocytes,38 T-cell intrinsic metabolic ROS likely adds to an existing oxidative environment fueled by neutrophil-generated ROS, resulting in a toxic combination. This effect apparently limited T-cell survival in the presence of WT Ncf1. On the other hand, the more robust T-cell response in Ncf1−/− mice, which exhibit lower total ROS production, was associated with more rapid and more complete virus elimination, along with little T-cell-mediated liver cell damage or signs of disease.
Interestingly, a recent study has reported a critical influence of ROS in autoimmunity, in that T-cell-mediated arthritis was inhibited by elevated ROS levels.31 Our data suggest that ROS may trigger death of T cells offering an explanation for protection from autoimmunity. However, enhanced ROS production during autoimmune disease could also lead to direct tissue damage; therefore, the potential utility and toxicity of inducing oxidative stress to prevent autoimmunity should be investigated in future studies.
Elevated ROS levels are observed in many tumor types. Cancers with high ROS production display aggressive growth characteristics and are correlated with poor prognosis.49 ROS is elevated in tumors not only as a consequence of the high metabolic activity of cancer cells20 but also due to NADPH oxidase activity.50 In view of our findings, the elevated ROS levels in tumors may contribute to rapid tumor growth and disease progression in part by limiting immune response against cancer cells.
In summary, our data have revealed an influence of ROS on virus-induced immunity and immunopathology. Elevated ROS levels in granulocytes decreased survival of CD8+ T cells. Lack of NADPH oxidase resulted in more robust virus-induced immunity and faster elimination of the virus. As a result, virus-induced immunopathological disease was dramatically reduced in the absence of NADPH oxidase activity.
Materials and Methods
Mice, viruses and virus titration
LCMV strain WE was originally obtained from F Lehmann-Grube (Heinrich Pette Institute, Hamburg, Germany) and was propagated in L929 cells as described.51 Virus titers were measured using a plaque-forming assay as described previously.52 Briefly, organs were harvested into Hank’s balanced salt solution (HBSS) and homogenized using a Tissue Lyser (Qiagen, Hilden, Germany). A total of 0.8 × 106 MC57 cells were added to titrated virus samples on 24-well plates. After 3 h, 1% methylcellulose-containing medium was added. After 48 h, plates were fixed (4% formalin), permeabilized (1% Triton X-100 in HBSS), and stained with anti-LCMV NP (clone VL4) antibody, peroxidase anti-rat antibody, and o-phenlyenediamine dihydrochloride (Sigma, Heidenheim, Germany) dissolved in 50 mM Na2HPO4 and 25 mM citric acid. Mice were infected intravenously with 2 × 106 PFU of LCMV WE. All mice used in this study were maintained on the C57BL/6 genetic background. All experiments were performed in single ventilated cages. Animal experiments were carried out with authorization of the Veterinäramt of the Kanton Zurich and in accordance with the Swiss law for animal protection, the guidelines of the Ontario Cancer Institute and the German law for animal protection.
Treatment with BSO
BSO was purchased from Sigma. BSO was prepared in 0.5 M solution according to the manufacturer’s instructions. Mice were treated daily by intraperitoneal injection of 400 μl.
Bilirubin, alkaline phosphatase and ALT
Serum levels were measured using a serum multiple biochemical analyzer (Ektachem DTSCII; Johnson & Johnson Inc., Rochester, MN, USA).
Quantitative RT-PCR measurement
RT-PCR analysis was performed as described previously.53 Briefly, mRNA was extracted from livers stored in 1 ml Trizol (Invitrogen, Grand Island, NY, USA) according to the manufacturer’s instructions. After homogenization using a Tissue lyser (Qiagen), 200 μl chloroform was added and samples were vortexed for at least 15 s. Then, samples were centrifuged at 12 000 r.p.m. at 4 °C for 10 min, supernatant transferred into a new tube and mRNA precipitated with 2-isopropanol for 20 min at room temperature. After one wash with 75% ethanol, pellets were air dried and resuspended in DEPC-treated water (Invitrogen). Gene expression of collagen and GAPDH was performed using kits from Applied Biosystems, Darmstadt, Germany. For analysis, the expression levels of all target genes were normalized to GAPDH expression (ΔCt). Gene expression values were then calculated based on the ΔΔCt method, using the mean of three naive mice as a standard to which all other samples were compared. Relative quantities (RQ) were determined using the equation: RQ=2−ΔΔCt.
Histology
Histological analysis was performed on snap frozen tissue as described.54 Antibodies against mouse CD8 (clone YTS169.4), LCMV-NP (clone VL4) and anti-Gr-1 were used. Histology was developed using an alkaline phosphatase system.
Flow cytometry analysis
Tetramer production, surface and intracellular flow cytometry measurement (FCM) staining was performed as described previously.55 Briefly, single-cell suspensions from spleen and liver tissues, and peripheral blood lymphocytes were stained using PE-labeled gp33 major histocompability complex (MHC) class I tetramers (gp33/H-2Db) for 15 min at 37 °C, followed by staining with anti-CD8-PerCP (BD Biosciences, San Jose, CA, USA) for 30 min at 4 °C. For determination of their activation status, lymphocytes were stained with anti-PD-1 and anti-IL-7Rα (BD Biosciences) for 30 min at 4 °C. The T-cell function was tested by performing an intracellular cytokine staining using splenocytes from WT mice infected with 102 PFU of LCMV WE 8 days post infection in the presence of H2O2, diamide, PIETC, galatose oxidase and galatose (Sigma). For intracellular cytokine stain, single-cell suspended splenocytes or liver tissue were incubated with the LCMV-specific peptides gp33 and np396. After 1 h, brefeldin A (eBiosciences, San Diego, CA, USA) was added, followed by additional 2-h incubation (IL-2 restimulation); 4-h incubation (TNF-α restimulation); and 5-h incubation (IFN-γ restimulation) at 37 °C. After surface stain with anti-CD8 (eBiosciences), cells were fixed with 2% formalin and permeabilized with PBS containing 1% FCS and 0.1% saponin, and stained with anti-IL-2, anti-TNF-α or anti-IFN-γ (eBiosciences) antibodies for 30 min at 4 °C. Oxidative stress was measured following 20 min staining with 10 μM DHE (Invitrogen) and was determined by quantifying the mean fluorescence intensity (MFI) shown in arbitrary units. Anti-Gr-1, anti-CD4, anti-B220, anti-MHCII, anti-CD11c and anti-F4/80 antibodies were from eBiosciences.
Western blotting
Bone marrow-derived macrophages, generated as described previously,56 were stimulated with 200 ng/ml LPS (026:B6; Sigma) harvested at the indicated time points and blots were probed anti-p-p65 (Ser536), p-IκBα (Ser32), IκBα, p-ERK, ERK, actin (Cell Signaling Technology, Frankfurt, Germany) and p65 (Santa Cruz Biotechnology, Heidelberg, Germany).
51Cr release assay
51Cr (chromium) release assay was performed as described previously.17
Statistical analysis
Data are expressed as mean±S.E.M. Statistically significant differences between two different groups were analyzed using Student’s t-test. Statistical differences between several groups were tested using one-way ANOVA with additional Bonferroni or Dunnett’s post-tests. Statistically significant differences between groups in experiments involving more than one analysis time point were calculated using two-way ANOVA (repeated measurements). P–values <0.05 were considered as statistically significant.
Abbreviations
- ALT:
-
alanine aminotransferase
- BSO:
-
buthionine sulfoximine
- CD:
-
cluster of differentiation
- Cr:
-
chromium
- DHE:
-
dihydroethidium
- FCM:
-
flow cytometry measurement
- HBSS:
-
Hank’s balanced salt solution
- HCV:
-
hepatitis C virus
- HIV:
-
human immunodeficiency virus
- IFN:
-
interferon
- IκBα:
-
inhibitor of κBα
- LCMV:
-
lymphocytic choriomeningitis virus
- MFI:
-
mean fluorescence intensity
- NADPH:
-
nicotinamide adenine dinucleotide phosphate
- NP:
-
nucleoprotein
- MHC:
-
major histocompability complex
- p.i.:
-
post infection
- PIETC:
-
2-phenylethyl isothiocyanate
- RNA:
-
ribonucleic acid
- ROS:
-
reactive oxygen species
- RT-PCR:
-
real-time polymerase chain reaction
- S.E.M.:
-
standard error of the mean
- TNF:
-
tumor necrosis factor
- WT:
-
wild type
References
Rehermann B, Nascimbeni M . Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol 2005; 5: 215–229.
Rehermann B . Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009; 119: 1745–1754.
From the Centers for Disease Control and Prevention The global HIV and AIDS epidemic, 2001. JAMA 2001; 285: 3081–3083.
Sobao Y, Tomiyama H, Sugi K, Tokunaga M, Ueno T, Saito S et al. The role of hepatitis B virus-specific memory CD8 T cells in the control of viral replication. J Hepatol 2002; 36: 105–115.
Thimme R, Oldach D, Chang KM, Steiger C, Ray SC, Chisari FV . Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med 2001; 194: 1395–1406.
Rehermann B, Fowler P, Sidney J, Person J, Redeker A, Brown M et al. The cytotoxic T lymphocyte response to multiple hepatitis B virus polymerase epitopes during and after acute viral hepatitis. J Exp Med 1995; 181: 1047–1058.
Ferrari C, Penna A, Bertoletti A, Valli A, Antoni AD, Giuberti T et al. Cellular immune response to hepatitis B virus-encoded antigens in acute and chronic hepatitis B virus infection. J Immunol 1990; 145: 3442–3449.
Porichis F, Kaufmann DE . Role of PD-1 in HIV pathogenesis and as target for therapy. Curr HIV/AIDS Rep 2012; 9: 81–90.
Wherry EJ . T cell exhaustion. Nat Immunol 2011; 12: 492–499.
Kurokohchi K, Masaki T, Arima K, Miyauchi Y, Funaki T, Yoneyama H et al. CD28-negative CD8-positive cytotoxic T lymphocytes mediate hepatocellular damage in hepatitis C virus infection. J Clin Immunol 2003; 23: 518–527.
Leroy V, Vigan I, Mosnier JF, Dufeu-Duchesne T, Pernollet M, Zarski JP et al. Phenotypic and functional characterization of intrahepatic T lymphocytes during chronic hepatitis C. Hepatology 2003; 38: 829–841.
Napoli J, Bishop GA, McGuinness PH, Painter DM, McCaughan GW . Progressive liver injury in chronic hepatitis C infection correlates with increased intrahepatic expression of Th1-associated cytokines. Hepatology 1996; 24: 759–765.
Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen KJ et al. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 1994; 369: 31–37.
Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV . Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity 1996; 4: 25–36.
Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV . Viral clearance without destruction of infected cells during acute HBV infection. Science 1999; 284: 825–829.
Zinkernagel RM, Haenseler E, Leist T, Cerny A, Hengartner H, Althage A . T cell-mediated hepatitis in mice infected with lymphocytic choriomeningitis virus. Liver cell destruction by H-2 class I-restricted virus-specific cytotoxic T cells as a physiological correlate of the 51Cr-release assay? J Exp Med 1986; 164: 1075–1092.
Lang PA, Contaldo C, Georgiev P, El-Badry AM, Recher M, Kurrer M et al. Aggravation of viral hepatitis by platelet-derived serotonin. Nat Med 2008; 14: 756–761.
El-Benna J, Dang PM, Gougerot-Pocidalo MA, Marie JC, Braut-Boucher F . p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp Mol Med 2009; 41: 217–225.
Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P . Redox regulation of cell survival. Antioxid Redox Signal 2008; 10: 1343–1374.
Le Bras M, Clement MV, Pervaiz S, Brenner C . Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol Histopathol 2005; 20: 205–219.
Babior BM . NADPH oxidase: an update. Blood 1999; 93: 1464–1476.
Ushio-Fukai M . Localizing NADPH oxidase-derived ROS. Sci STKE 2006; 2006 re8.
Segal AW, Heyworth PG, Cockcroft S, Barrowman MM . Stimulated neutrophils from patients with autosomal recessive chronic granulomatous disease fail to phosphorylate a Mr-44,000 protein. Nature 1985; 316: 547–549.
Faust LR, el Benna J, Babior BM, Chanock SJ . The phosphorylation targets of p47phox, a subunit of the respiratory burst oxidase. Functions of the individual target serines as evaluated by site-directed mutagenesis. J Clin Invest 1995; 96: 1499–1505.
Parola M, Robino G . Oxidative stress-related molecules and liver fibrosis. J Hepatol 2001; 35: 297–306.
Wingler K, Hermans JJ, Schiffers P, Moens A, Paul M, Schmidt HH . NOX1, 2, 4, 5: counting out oxidative stress. Br J Pharmacol 2011; 164: 866–883.
Axelsen PH, Komatsu H, Murray IV . Oxidative stress and cell membranes in the pathogenesis of Alzheimer's disease. Physiology 2011; 26: 54–69.
Victor VM, Rocha M, Herance R, Hernandez-Mijares A . Oxidative stress and mitochondrial dysfunction in type 2 diabetes. Curr Pharm Des 2011; 17: 3947–3958.
Firestein GS . The T cell cometh: interplay between adaptive immunity and cytokine networks in rheumatoid arthritis. J Clin Invest 2004; 114: 471–474.
Dang PM, Stensballe A, Boussetta T, Raad H, Dewas C, Kroviarski Y et al. A specific p47phox -serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. J Clin Invest 2006; 116: 2033–2043.
Gelderman KA, Hultqvist M, Pizzolla A, Zhao M, Nandakumar KS, Mattsson R et al. Macrophages suppress T cell responses and arthritis development in mice by producing reactive oxygen species. J Clin Invest 2007; 117: 3020–3028.
Hultqvist M, Backlund J, Bauer K, Gelderman KA, Holmdahl R . Lack of reactive oxygen species breaks T cell tolerance to collagen type II and allows development of arthritis in mice. J Immunol 2007; 179: 1431–1437.
Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest 2003; 112: 1383–1394.
Okuda M, Li K, Beard MR, Showalter LA, Scholle F, Lemon SM et al. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology 2002; 122: 366–375.
Boudreau HE, Emerson SU, Korzeniowska A, Jendrysik MA, Leto TL . Hepatitis C virus (HCV) proteins induce NADPH oxidase 4 expression in a transforming growth factor beta-dependent manner: a new contributor to HCV-induced oxidative stress. J Virol 2009; 83: 12934–12946.
Michalek RD, Pellom ST, Holbrook BC, Grayson JM . The requirement of reactive oxygen intermediates for lymphocytic choriomeningitis virus binding and growth. Virology 2008; 379: 205–212.
Lang PA, Recher M, Haussinger D, Lang KS . Genes determining the course of virus persistence in the liver: lessons from murine infection with lymphocytic choriomeningitis virus. Cell Physiol Biochem 2010; 26: 263–272.
Volpp BD, Nauseef WM, Donelson JE, Moser DR, Clark RA . Cloning of the cDNA and functional expression of the 47-kilodalton cytosolic component of human neutrophil respiratory burst oxidase. Proc Natl Acad Sci USA 1989; 86: 7195–7199.
Jackson SH, Gallin JI, Holland SM . The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med 1995; 182: 751–758.
Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 2006; 10: 241–252.
Lang KS, Recher M, Navarini AA, Harris NL, Lohning M, Junt T et al. Inverse correlation between IL-7 receptor expression and CD8 T cell exhaustion during persistent antigen stimulation. Eur J Immunol 2005; 35: 738–745.
Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007; 27: 670–684.
Purushothaman D, Sarin A . Cytokine-dependent regulation of NADPH oxidase activity and the consequences for activated T cell homeostasis. J Exp Med 2009; 206: 1515–1523.
Miyamoto Y, Koh YH, Park YS, Fujiwara N, Sakiyama H, Misonou Y et al. Oxidative stress caused by inactivation of glutathione peroxidase and adaptive responses. Biol Chem 2003; 384: 567–574.
D‘Alessio M, Cerella C, Amici C, Pesce C, Coppola S, Fanelli C et al. Glutathione depletion up-regulates Bcl-2 in BSO-resistant cells. FASEB J 2004; 18: 1609–1611.
Drew R, Miners JO . The effects of buthionine sulphoximine (BSO) on glutathione depletion and xenobiotic biotransformation. Biochem Pharmacol 1984; 33: 2989–2994.
Pearce EL . Metabolism in T cell activation and differentiation. Curr Opin Immunol 2010; 3: 314–320.
Maciver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC . Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukocyte Biol 2008; 84: 949–957.
Trachootham D, Alexandre J, Huang P . Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 2009; 8: 579–591.
Ushio-Fukai M, Nakamura Y . Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett 2008; 266: 37–52.
Welsh RM, Seedhom MO . Lymphocytic choriomeningitis virus (LCMV): propagation, quantitation, and storage. Curr Protoc Microbiol 2008 Chapter 15: Unit 15A 1.
Battegay M, Cooper S, Althage A, Banziger J, Hengartner H, Zinkernagel RM . Quantification of lymphocytic choriomeningitis virus with an immunological focus assay in 24- or 96-well plates. J Virol Methods 1991; 33: 191–198.
Lang PA, Recher M, Honke N, Scheu S, Borkens S, Gailus N et al. Tissue macrophages suppress viral replication and prevent severe immunopathology in an interferon-I-dependent manner in mice. Hepatology 2010; 52: 25–32.
Recher M, Lang KS, Navarini A, Hunziker L, Lang PA, Fink K et al. Extralymphatic virus sanctuaries as a consequence of potent T-cell activation. Nat Med 2007; 13: 1316–1323.
Lang PA, Cervantes-Barragan L, Verschoor A, Navarini AA, Recher M, Pellegrini M et al. Hematopoietic cell-derived interferon controls viral replication and virus-induced disease. Blood 2009; 113: 1045–1052.
McIlwain DR, Lang PA, Maretzky T, Hamada K, Ohishi K, Maney SK et al. iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science 2012; 335: 229–232.
Acknowledgements
We thank Konstanze Schättel and Eugene Bäcker for technical support. This study was supported by the German Research Council through the Collaborative Research Center 974, TP-A10 (SFB974: Experimental Hepatology; Coordinator: DH), DFG LA1419/5-1 and DFG LA2558/3-1. KSL and PAL were supported by the Alexander von Humboldt Foundation (SKA2008 and 2010). DB was supported by a postdoctoral fellowship of the German Research Foundation (DFG). This work was also supported by Manchot Graduiertenschule (MOI) and the Strategic Research Fund of the Heinrich Heine University. Tetramers were kindly provided by the NIH. This work was also supported by a CIHR Grant to PSO, Swiss National Science Foundation Grants to HH (no. 3100A0-100779) and R.Z. (no. 3100A0-100068). MR was supported by Grant PASMP3-127678/1 from the Swiss National Science Foundation (SNF/SSMBS).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Lang, P., Xu, H., Grusdat, M. et al. Reactive oxygen species delay control of lymphocytic choriomeningitis virus. Cell Death Differ 20, 649–658 (2013). https://doi.org/10.1038/cdd.2012.167
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2012.167
Keywords
This article is cited by
-
Valosin-containing protein (VCP/p97) inhibition reduces viral clearance and induces toxicity associated with muscular damage
Cell Death & Disease (2022)
-
Slow viral propagation during initial phase of infection leads to viral persistence in mice
Communications Biology (2021)
-
The phosphatase PAC1 acts as a T cell suppressor and attenuates host antitumor immunity
Nature Immunology (2020)
-
Obesity and risk of respiratory tract infections: results of an infection-diary based cohort study
BMC Public Health (2018)
-
Cholestasis induced liver pathology results in dysfunctional immune responses after arenavirus infection
Scientific Reports (2018)
