
Abstract
Background:
Vorinostat, a histone deacetylase (HDAC) inhibitor, was investigated in combination with capecitabine plus cisplatin (XP) as a first-line chemotherapy for patients with unresectable or metastatic gastric cancer (GC).
Methods:
Eligible patients received 400 mg vorinostat once daily on days 1–14, 1000 mg m−2 capecitabine twice daily on days 1–14, and 60 mg m−2 cisplatin on day 1 every 3 weeks. Plasma levels of acetyl-H3, HDAC2, and p21 were measured for correlative analysis. The primary end point was the 6-month progression-free survival (PFS) rate. Secondary end points included the response rate, PFS, overall survival (OS), and safety profile.
Results:
A total of 45 patients with HER2-negative GC were included in this study. The objective response rate was 42%. The median PFS was 5.9 months, and the 6-month PFS rate was 44.4%. The median OS was 12.7 months. Most common grade 3–4 toxicities were neutropenia (41%), fatigue (34%), anorexia (32%), thromboembolism (27%), stomatitis (14%), and thrombocytopenia (11%). High plasma acetyl-H3 and p21 levels were significantly associated with a poor OS (P=0.02 and P=0.03, respectively).
Conclusions:
Vorinostat-XP is a feasible first-line chemotherapy for patients with advanced GC. However, this trial did not meet its primary end point, and more adverse events were observed in comparison with the historical data of flouropyrimidine–platinium doublet regimens.
Similar content being viewed by others
Main
Gastric cancer (GC) is one of the major leading causes of cancer-related deaths worldwide (Jung et al, 2014; Siegel et al, 2014). For patients with unresectable or metastatic disease, palliative chemotherapy improves quality of life and survival. Fluoropyrimidine–platinum combination chemotherapy has been established as a standard first-line chemotherapy for these patients (Koizumi et al, 2008; Kang et al, 2009; Ajani et al, 2010). With a better understanding of GC biology and the development of novel agents, the era of targeted therapies for GC has begun. For HER2-positive GC, trastuzumab has demonstrated improved survival outcomes when combined with chemotherapy (Bang et al, 2010). Anti-angiogenic agents, such as ramucirumab and apatinib, have proven survival benefits in unselected GC patients when used as second- or third-line therapies (Li et al, 2013; Fuchs et al, 2014; Qin et al, 2014; Wilke et al, 2014). However, the overall prognosis of metastatic GC patients remains dismal, and chemotherapies with greater efficacy are needed.
Vorinostat (Zolinza; Merck & Co., Inc., Kenilworth, NJ, USA) is an orally bioavailable histone deacetylase (HDAC) inhibitor that alters the level of histone and nonhistone protein acetylation, and thereby regulates gene expression, angiogenesis, cell proliferation, and cell survival (Bolden et al, 2006). Vorinostat monotherapy is approved for the treatment of refractory cutaneous T-cell lymphoma (Duvic et al, 2007). For GC, previous studies have suggested that HDAC expression and global histone modification may be associated with the prognosis of GC patients (Park et al, 2008; Weichert et al, 2008). Multiple preclinical and clinical studies reported that this HDAC inhibitor may enhance the anticancer activities of cytotoxic chemotherapy (Dokmanovic et al, 2007). This synergism was also observed in our previous in-house in vivo analysis of a murine GC xenograft model; when capecitabine or cisplatin was combined with vorinostat, their activities were enhanced (Supplementary Figures 1 and 2). Based on these results, we performed a phase I/II study of vorinostat in combination with capecitabine plus cisplatin (XP) to explore the role of HDAC inhibition in treating GC. The results of our phase I dose-finding study have been previously published elsewhere (Yoo et al, 2014). In our current report, we present the results of our phase II study and biomarker analysis.
Materials and methods
Patients
Patients with histologically documented unresectable or metastatic gastric adenocarcinoma were eligible for inclusion in this study if they met the following criteria: age between 18 and 70 years; Eastern Cooperative Oncology Group (ECOG) performance status of 0–2; completion of adjuvant chemotherapy 6 months before entry into the study, or no history of chemotherapy; adequate bone marrow, renal, and liver function; no prior radiotherapy; and estimated life expectancy of >3 months. For the phase II part of the study, a measurable lesion according to RECIST (version 1.1) was required for inclusion. Patients who had previously received capecitabine or platinum as an adjuvant treatment were excluded. Prior exposure to any HDAC inhibitor was not allowed, except valproic acid with a 30-day washout period. The study protocol was approved by the institutional review board of Asan Medical Center, Seoul, Korea, and all patients provided written informed consent before enrolment. The study was conducted in accordance with the Declaration of Helsinki and the Guidelines for Good Clinical Practice (ClinicalTrial.gov Identifier: NCT01045538).
Study design and treatment
This study was originally designed as a single-centre, phase I/II study. The maximum tolerated dose and recommended phase II dose (RP2D) of vorinostat-XP administered to patients with advanced GC were defined in the phase I part of the study that has been previously published (Yoo et al, 2014). In brief, the dose-limiting toxicities of vorinostat-XP included thrombocytopenia, fatigue, stomatitis, and anorexia. The RP2D for the phase II part of the study was 400 mg vorinostat once daily on days 1–14, 1000 mg m−2 capecitabine twice daily on days 1–14, and 60 mg m−2 cisplatin on day 1 every 3 weeks. This RP2D was used in the current study. The dose modification scheme for treatment-related advent adverse events was as follows. For grade 4 neutropenia >5 days, grade 4 thrombocytopenia, or grade ⩾3 febrile neutropenia, the doses of capecitabine and cisplatin were reduced by 25% in the subsequent cycle. Capecitabine was interrupted for grade ⩾2 nonhaematological toxicities with appropriate therapy. If nonhaematological toxicities were resolved or decreased to grade 1, capecitabine was rechallenged with a dose reduced up to 50%. The dose of cisplatin was modified if the creatinine clearance decreased or grade ⩾3 nausea/vomiting and ⩾grade 2 neurotoxicity occurred. For haematological toxicities, the dose of vorinostat was reduced (100 mg reduction) if grade 3 toxicity occurred; for grade 4 toxicity, vorinostat was interrupted until recovery to grade ⩽2 and restarted at a reduced dose. For nonhaematological toxicities, vorinostat was delayed and restarted at a reduced dose when the toxicity improved to grade ⩽1 for grade 3 toxicity or discontinued for grade 4 toxicity. If more than two dose reductions were required, vorinostat was discontinued.
Assessment
At baseline, patients underwent a review of their medical history, physical examination, complete blood count (CBC) with differential counts, chemistry, electrolytes, coagulation battery, urinalysis, electrocardiography, chest X-ray, and computed tomography (CT) scanning of the abdomen and pelvis. Physical examinations, chest X-rays, CBC, chemistry, and electrolytes were repeated before each chemotherapy cycle. Tumour response was graded every two cycles according to the RECIST criteria (version 1.1) by the investigators. Toxicities were evaluated according to the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03).
Circulating biomarker analysis
A peripheral blood sample (10 ml) was obtained from patients before study treatment and 2 h after ingesting vorinostat capsules at post-treatment day 8 of cycle 1. Peripheral blood mononuclear cells (PBMCs) were isolated by centrifugation using Lymphoprep (AXIS-SHIELD PoC, Oslo, Norway). Isolated PBMCs were lysed in M-PER buffer (Pierce, Rockford, IL, USA). The protein content was quantified using the Bradford assay (Bio-Rad, Hercules, CA, USA). Cell extracts were separated by SDS–PAGE and transferred to nitrocellulose membranes (Millipore, Billerica, MA, USA) for western blot analysis. The blots were incubated overnight at 4 °C with primary antibodies against histone acetyl-H3 (Cell Signaling, Danvers, MA, USA), HDAC2 (Cell Signaling), p21 (Cell Signaling), and β-actin (Sigma, St Louis, MO, USA). The blots were stripped and reprobed for histone H3 (Cell Signaling), and images were quantified using Multi-Gauge v2.3 software (Fujifilm, Tokyo, Japan). Histone H3 acetylation was measured by quantifying acetyl-H3 band pixel intensity and normalising it to the respective H3 band in a representative immunoblot. The changes between the pre- and post-vorinostat acetyl-H3, HDAC2, and p21 levels were calculated for each patient.
Statistical analysis
The 6-month progression-free survival (PFS) rate with XP was ∼40% (P0) in a previous phase III study (Kang et al, 2009). With the addition of vorinostat to XP, we hypothesised that the 6-month PFS rate would improve to 60% (P1) with a hazard ratio of 0.56. Using the log-rank method with a type I error of 0.1 and a power of 0.85, a total of 45 patients was necessary. According to the prespecified protocol, the outcomes of 7 patients who had measurable disease and were treated with RP2D in the phase I study were included in the current analysis. Therefore, 38 patients were additionally enrolled in this phase II study.
The primary end point of our current phase II study was the 6-month PFS rate. Secondary end points included the response rate, PFS, overall survival (OS), and safety profile. The Kaplan–Meier method was used to estimate the PFS and OS. The PFS was measured from the start of treatment to documented tumour progression or death from any cause, whichever occurred first. Overall survival was calculated from the start of treatment to the date of death from any cause. Efficacy parameters were analysed with inclusion of all patients who received at least one dose of study medication. A two-sided P-value of <0.05 was considered statistically significant, and SPSS 20.0 (IBM SPSS Inc., Chicago, IL, USA) was used for all statistical analyses.
Results
Patients
Between 9 March 2012 and 4 July 2014, 38 patients were enrolled in the phase II part of this study. With the inclusion of 7 patients treated with RP2D in the phase I part, a total of 45 patients were included in this analysis. The baseline characteristics of the patients are summarised in Table 1. The median patient age was 56 years (range=36–77 years), and 34 (76%) patients were male. There was a protocol violation that included one patient older than 70 years. However, all efficacy and safety analyses were done with inclusion of this patient according to the protocol. More than two-thirds of the study patients (n=35; 78%) initially presented with metastatic disease. The most frequent metastatic sites were the lymph nodes (87%), liver (56%), and peritoneum (40%). The HER2 positivity was evaluated in all patients using immunohistochemistry and fluorescence in situ hybridisation (FISH), and there were no HER2-positive GC patients. The median follow-up duration was 8.6 months (range=5.4–32.9 months) in the surviving patients at the time of the analysis.
Efficacy
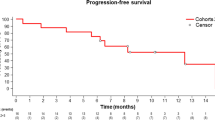
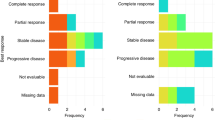
The response evaluation was available in all but two patients who were lost to follow-up before the first response assessment. There were no patients who achieved a complete response. The confirmed partial response was achieved in 19 (42%) patients, and stable disease was shown in 23 (51%) patients (Table 2). Figure 1 shows the waterfall plot of the changes in the sizes of target lesions. The 6-month PFS rate – the primary end point of this study – was 44.4% (95% confidence interval (CI)=28.7–60.1%), and the median PFS was 5.9 months (95% CI=3.9–7.9 months; Figure 2). The median OS was 12.7 months (95% CI=8.8–16.6 months).

Waterfall plot of the changes in the target lesion.PD=progressive disease; PR=partial response; SD=stable disease.

Progression-free survival (A) and overall survival (B) outcomes. The median progression-free survival was 5.9 months (95% CI=3.9–7.9) and the median overall survival was 12.7 months (95% CI=8.8–16.6). The 6-month progression-free survival rate was 44.4%.
After progression on vorinostat-XP, second-line chemotherapy was administered to 20 patients (44%). Taxanes (paclitaxel or docetaxel) and irinotecan-based chemotherapy were the most commonly administered second-line regimens (n=12 (60%) and n=5 (25%), respectively). The investigational targeted agents, including everolimus, were used in 3 (15%) patients. Five patients (11%) received subsequent third or later lines of chemotherapy.
Safety
A median of 6 cycles (range=1–24 cycles) of vorinostat-XP were administered, and 4 patients were still receiving treatment with the study chemotherapy at the time of analysis. Study treatments were discontinued because of adverse events in 6 patients (13%) that included cerebral infarction (n=3), intracerebral haemorrhage (n=1), prolonged grade 3 thrombocytopenia (n=1), and prolonged grade 3 fatigue and anorexia (n=1). Doses of vorinostat, capecitabine, and cisplatin were reduced in 21 (47%), 33 (74%), and 26 (58%) patients, respectively. The study chemotherapy regimen was delayed in 30 patients (67%) because of toxicity. Adverse events developed in ⩾10% of patients are summarised in Table 3. There were no treatment-related deaths. The most common grade 3–4 adverse event was neutropenia (41%), followed by fatigue (34%), anorexia (32%), thromboembolism (27%), stomatitis (14%), and thrombocytopenia (11%). Febrile neutropenia occurred in 1 patient (2%).
Regarding thromboembolic events, embolic cerebral infarction occurred in four patients within the second cycles of study treatment, and all patients recovered without significant neurological deficit. There was a single case of popliteal artery thrombosis that occurred during the first cycle, and 4 patients with pulmonary embolism (1 patient in the first cycle, 2 patients in the fourth cycle, and 1 patient in the seventh cycle). Arterial thromboembolism was well managed with anticoagulation therapy, and there were no life-threatening cases that required management in the intensive care unit. Deep vein thrombosis developed in 6 patients after a median 4 cycles of chemotherapy (range=1–7 cycles), and 3 of these patients simultaneously developed arterial thromboembolism.
Circulating biomarkers
For the biomarker analysis, blood samples were collected from all patients; however, analyses of acetyl-H3, HDAC2, and p21 levels were available for 40 patients (89%). In cycle 1 day 8 (C1D8), the levels of acetyl-H3, HDAC2, and p21 were significantly increased in comparison with those at cycle 1 day 1 (C1D1). The ratios of the biomarker levels (C1D8/C1D1) were a mean value of 4.31 (s.d.=4.96; P<0.001) for acetyl-H3, 5.08 (s.d.=6.80; P<0.001) for HDAC2, and 5.32 (s.d.=12.28; P=0.03) for p21. For the correlative analysis with survival outcomes, patients were grouped according to the median level of each biomarker. There were no significant associations between PFS and baseline acetyl-H3 (P=0.13), HDAC2 (P=0.16), and p21 (P=0.22). Baseline acetyl-H3 and p21 were significantly associated with OS (P=0.02 and P=0.03, respectively; Figure 3), but there was no relationship between baseline HDAC2 and OS (P=0.72). Patients with high baseline acetyl-H3 and p21 (>median) showed poorer OS in comparison with patients with low values (⩽median); for acetyl-H3, the median 8.8 months (95% CI=5.5–12.1 months) vs 15.9 months (95% CI=9.0–22.7 months; P=0.02); and for p21, the median 9.4 months (95% CI=5.9–12.9 months) vs 15.9 months (95% CI=8.4–23.4 months; P=0.03). The changes in acetyl-H3, HDAC2, and p21 after study treatment were not significantly correlated with PFS or OS.

Association between overall survival and baseline serum acetyl-H3 (A) and p21 (B).
Discussion
In our current study, vorinostat-XP was feasible as a first-line chemotherapy for patients with advanced GC. However, this study did not meet its primary end point (6-month PFS rate of 60%), and is likely to be associated with increased adverse events, particularly haematological toxicities and thromboembolism.
With vorinostat-XP, the objective response rate was 42%, and the median PFS and OS rates were 5.9 and 12.7 months, respectively. The addition of vorinostat did not therefore appear to improve the clinical outcomes of GC patients, considering that the standard 3-weekly XP, which consisted of 1000 mg m−2 capecitabine on days 1–14 and 80 mg m−2 cisplatin on day 1, demonstrated an objective response rate of 46%, median PFS of 5.6 months, and median OS of 10.5 months in a previous phase III trial (Kang et al, 2009). Compared with the standard XP regimen, there was only a mild dose reduction in cisplatin (60 mg m−2) when administering XP as part of our current experimental regimen. In a recent phase III trial that compared different dosing schedules of cisplatin plus S-1 (another fluoropyrimidine derivative), a higher dose of cisplatin was associated with significantly increased PFS; however, the magnitude of PFS improvement was slight and there were no differences in OS (Ryu et al, 2015). This suggests that the comparable efficacy of vorinostat-XP to standard XP may be related to the inactivity of vorinostat towards GC or insufficient synergism between vorinostat and cytotoxic chemotherapeutic agents in the clinical setting rather than the reduced dose of cisplatin.
Although there were no treatment-related deaths in our current cohort, vorinostat-XP seems to be associated with greater toxicities in comparison with the standard fluoropyrimidine–platinum doublet regimens (Koizumi et al, 2008; Kang et al, 2009; Ajani et al, 2010). Grade 3–4 neutropenia, fatigue, anorexia, stomatitis, and thrombocytopenia occurred in 41%, 34%, 32%, 14% and 11% of our patients, respectively, and these rates are notably higher in comparison with the results of standard XP in previous phase III trials that reported rates of 16–30%, <1–2%, 2–6%, 2%, and 3%, respectively (Kang et al, 2009; Bang et al, 2010). The increase in toxicities with the addition of vorinostat was inevitable in some aspects because the backbone XP in the current study regimen has only a mild dose modification from the standard XP. This is also consistent with the previous phase I trial on colorectal cancer patients who received vorinostat in combination with 5-FU plus oxaliplatin, in which haematologic toxicities and fatigue were increased in frequency and severity with the addition of vorinostat (Fakih et al, 2009). Consequently, the increase in toxicities led to frequent dose modification and, in particular, study treatment was discontinued because of the treatment-related advent adverse events in 13% of the study population. This limited administration of study treatments might have had a negative impact on the efficacy outcomes with vorinostat-XP in our present study.
In our current patient series, thromboembolism developed in 27% (n=12) during study treatment. Of note, embolic cerebral infarction and pulmonary embolism – which are potentially life-threatening events – occurred in 4 patients (9%) each, although all of these patients recovered without clinically significant sequelae. In previous Korean studies on unresectable or metastatic GC patients, the 6-month and 1-year incidences of thromboembolism were reportedly 2.5–9.4% and 3.5%, respectively (Lee et al, 2009; Kang et al, 2012). Considering that all patients included in our present study were Korean and the timing of development of thromboembolism was mostly within 4 months from the start of treatment, the high incidence of thromboembolism in our study cohort is likely vorinostat related, although a clear attribution to either vorinostat itself or its combination with capecitabine or cisplatin cannot be made because of the lack of a control arm in our trial. However, because not all previous studies on vorinostat reported an increased incidence of thromboembolism (Fakih et al, 2009; Munster et al, 2009;, 2011), our results might be due to the synergism between vorinostat and capecitabine or cisplatin on hypercoagulability. Further investigations are needed to evaluate the impact of the HDAC inhibitor on thromboembolism in cancer patients, and meticulous work-up for thromboembolism may be needed in clinical trials on HDAC inhibitors, at least when administered to GC patients or combined with capecitabine or cisplatin.
There is no established biomarker for predicting the efficacy of HDAC inhibitors (Stimson and La Thangue, 2009). Our present biomarker analysis showed that acetyl-H3, HDAC2, and p21 are useful pharmacodynamic markers for vorinostat. Although these results did not correlate with the PFS outcomes, the baseline plasma levels of acetyl-H3 and p21 were significantly associated with OS, suggesting their relevance as prognostic factors in patients with GC. A recent report suggested that HR23B might be the potential predictive factor for HDAC inhibitors (Khan et al, 2010; Ihle et al, 2015); however, this was not evaluated in our present study.
In conclusion, the combination of vorinostat with XP is not likely to enhance efficacy in comparison with standard fluoropyrimidine–platinum doublet regimens in patients with GC. Considering the accompanying higher toxicities, further clinical trials on the current study regimen do not seem to be warranted, at least for GC patients. Because our results may be due to the inactivity of vorinostat on GC or the irrelevant effects of its combination with XP, further investigations using different HDAC inhibitors in different settings (i.e., second- or third-line therapies) or with different backbone cytotoxic chemotherapeutics may be acceptable.
Change history
24 May 2016
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ajani JA, Rodriguez W, Bodoky G, Moiseyenko V, Lichinitser M, Gorbunova V, Vynnychenko I, Garin A, Lang I, Falcon S (2010) Multicenter phase III comparison of cisplatin/S-1 with cisplatin/infusional fluorouracil in advanced gastric or gastroesophageal adenocarcinoma study: the FLAGS trial. J Clin Oncol 28: 1547–1553.
Bang Y-J, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, Aprile G, Kulikov E, Hill J, Lehle M, Rüschoff J, Kang Y-K ToGA Trial Investigators (2010) Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376: 687–697.
Bolden JE, Peart MJ, Johnstone RW (2006) Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5: 769–784.
Dokmanovic M, Clarke C, Marks PA (2007) Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res 5: 981–989.
Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM, Frankel SR (2007) Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 109: 31–39.
Fakih MG, Pendyala L, Fetterly G, Toth K, Zwiebel JA, Espinoza-Delgado I, Litwin A, Rustum YM, Ross ME, Holleran JL, Egorin MJ (2009) A phase I, pharmacokinetic and pharmacodynamic study on vorinostat in combination with 5-fluorouracil, leucovorin, and oxaliplatin in patients with refractory colorectal cancer. Clin Cancer Res 15: 3189–3195.
Fuchs CS, Tomasek J, Yong CJ, Dumitru F, Passalacqua R, Goswami C, Safran H, Santos dos LV, Aprile G, Ferry DR, Melichar B, Tehfe M, Topuzov E, Zalcberg JR, Chau I, Campbell W, Sivanandan C, Pikiel J, Koshiji M, Hsu Y, Liepa AM, Gao L, Schwartz JD, Tabernero J REGARD Trial Investigators (2014) Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 383: 31–39.
Ihle MA, Merkelbach-Bruse S, Hartmann W, Bauer S, Ratner N, Sonobe H, Nishio J, Larsson O, Åman P, Pedeutour F, Taguchi T, Wardelmann E, Buettner R, Schildhaus H-U (2016) HR23b expression is a potential predictive biomarker for HDAC inhibitor treatment in mesenchymal tumours and is associated with response to vorinostat. J Pathol Clin Res 2: 59–71.
Jung K-W, Won Y-J, Kong H-J, Oh C-M, Lee DH, Lee JS (2014) Cancer statistics in Korea: incidence, mortality, survival, and prevalence in 2011. Cancer Res Treat 46: 109–123.
Kang MJ, Ryoo B-Y, Ryu M-H, Koo DH, Chang H-M, Lee J-L, Kim TW, Kang Y-K (2012) Venous thromboembolism (VTE) in patients with advanced gastric cancer: an Asian experience. Eur J Cancer 48: 492–500.
Kang YK, Kang WK, Shin DB, Chen J, Xiong J, Wang J, Lichinitser M, Guan Z, Khasanov R, Zheng L, Philco-Salas M, Suarez T, Santamaria J, Forster G, McCloud PI (2009) Capecitabine/cisplatin versus 5-fluorouracil/cisplatin as first-line therapy in patients with advanced gastric cancer: a randomised phase III noninferiority trial. Ann Oncol 20: 666–673.
Khan O, Fotheringham S, Wood V, Stimson L, Zhang C, Pezzella F, Duvic M, Kerr DJ, La Thangue NB (2010) HR23B is a biomarker for tumor sensitivity to HDAC inhibitor-based therapy. Proc Natl Acad Sci USA 107: 6532–6537.
Koizumi W, Narahara H, Hara T, Takagane A, Akiya T, Takagi M, Miyashita K, Nishizaki T, Kobayashi O, Takiyama W, Toh Y, Nagaie T, Takagi S, Yamamura Y, Yanaoka K, Orita H, Takeuchi M (2008) S-1 plus cisplatin versus S-1 alone for first-line treatment of advanced gastric cancer (SPIRITS trial): a phase III trial. Lancet Oncol 9: 215–221.
Lee KW, Bang SM, Kim S, Lee HJ, Shin DY, Koh Y, Lee YG, Cha Y, Kim YJ, Kim JH, Park DJ, Kim HH, Oh D, Lee JS (2009) The incidence, risk factors and prognostic implications of venous thromboembolism in patients with gastric cancer. J Thromb Haemost 8: 540–547.
Li J, Qin S, Xu J, Guo W, Xiong J, Bai Y, Sun G, Yang Y, Wang L, Xu N, Cheng Y, Wang Z, Zheng L, Tao M, Zhu X, Ji D, Liu X, Yu H (2013) Apatinib for chemotherapy-refractory advanced metastatic gastric cancer: results from a randomized, placebo-controlled, parallel-arm, phase II trial. J Clin Oncol 31: 3219–3225.
Munster PN, Marchion D, Thomas S, Egorin M, Minton S, Springett G, Lee J-H, Simon G, Chiappori A, Sullivan D, Daud A (2009) Phase I trial of vorinostat and doxorubicin in solid tumours: histone deacetylase 2 expression as a predictive marker. Br J Cancer 101: 1044–1050.
Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, Miller A, Melisko M, Ismail-Khan R, Rugo H, Moasser M, Minton SE (2011) A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer 104: 1828–1835.
Park YS, Jin MY, Kim YJ, Yook JH, Kim BS, Jang SJ (2008) The global histone modification pattern correlates with cancer recurrence and overall survival in gastric adenocarcinoma. Ann Surg Oncol 15: 1968–1976.
Qin S, Li J, Xu J, Xiong J, Wu C, Bai Y, Liu W, Tong J, Liu Y, Xu R, Wang Z, Wang Q, Ouyang X, Yang Y, Ba Y, Liang J, Lin X, Luo D, Zheng R, Wu K, Sun G, Wang L, Zheng L, Guo H, Wu J, Xu N, Yang J, Zhang H, Cheng Y, Wang N, Chen L, Fan Z, Yu H (2014) Phase III study of apatinib in advanced gastric cancer: A randomized, double-blind, placebo-controlled trial. J Clin Oncol 32 (Suppl): Abstract 4003.
Ryu MH, Baba E, Lee KH, Park YI, Boku N, Hyodo I, Nam BH, Esaki T, Yoo C, Ryoo BY, Song EK, Cho SH, Kang WK, Yang SH, Zang DY, Shin DB, Park SR, Shinozaki K, Takano T, Kang YK (2015) Comparison of two different S-1 plus cisplatin dosing schedules as first-line chemotherapy for metastatic and/or recurrent gastric cancer: a multicenter, randomized phase III trial (SOS). Ann Oncol 26: 2097–2101.
Siegel R, Ma J, Zou Z, Jemal A (2014) Cancer statistics, 2014. CA Cancer J Clin 64: 9–29.
Stimson L, La Thangue NB (2009) Biomarkers for predicting clinical responses to HDAC inhibitors. Cancer Lett 280: 177–183.
Weichert W, Röske A, Gekeler V, Beckers T, Ebert MP, Pross M, Dietel M, Denkert C, Röcken C (2008) Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: a retrospective analysis. Lancet Oncol 9: 139–148.
Wilke H, Muro K, Van Cutsem E, Oh S-C, Bodoky G, Shimada Y, Hironaka S, Sugimoto N, Lipatov O, Kim T-Y, Cunningham D, Rougier P, Komatsu Y, Ajani J, Emig M, Carlesi R, Ferry D, Chandrawansa K, Schwartz JD, Ohtsu A RAINBOW Study Group (2014) Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol 15: 1224–1235.
Yoo C, Ryu M-H, Na Y-S, Ryoo B-Y, Lee C-W, Maeng J, Kim S-Y, Koo DH, Park I, Kang Y-K (2014) Phase I and pharmacodynamic study of vorinostat combined with capecitabine and cisplatin as first-line chemotherapy in advanced gastric cancer. Invest New Drugs 32: 271–278.
Acknowledgements
This study was supported in part by Merck & Co., Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Yoo, C., Ryu, MH., Na, YS. et al. Vorinostat in combination with capecitabine plus cisplatin as a first-line chemotherapy for patients with metastatic or unresectable gastric cancer: phase II study and biomarker analysis. Br J Cancer 114, 1185–1190 (2016). https://doi.org/10.1038/bjc.2016.125
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.125
Keywords
This article is cited by
-
Current advances in understanding the molecular profile of hereditary diffuse gastric cancer and its clinical implications
Journal of Experimental & Clinical Cancer Research (2023)
-
Chromatin and noncoding RNA-mediated mechanisms of gastric tumorigenesis
Experimental & Molecular Medicine (2023)
-
Signaling pathways and therapeutic interventions in gastric cancer
Signal Transduction and Targeted Therapy (2022)
-
Advances in epigenetic therapeutics with focus on solid tumors
Clinical Epigenetics (2021)
-
Identification of HDAC9 as a viable therapeutic target for the treatment of gastric cancer
Experimental & Molecular Medicine (2019)
