
Abstract
MicroRNAs (miRNAs) are small non-protein-coding RNAs that function as endogenous negative gene regulators. Dysfunctions of miRNAs are frequently found in malignancies, including lung cancer. In this review, we summarise the current understanding of miRNAs in lung cancer tumourigenesis, and highlight their potential in overcoming drug resistance, abetting histological sub-classification techniques, and serving as biomarkers for lung cancer risk stratification and outcome prediction.
Similar content being viewed by others


Main
Lung cancer is the leading cause of cancer mortality worldwide, and 80% of lung cancers are non-small cell lung cancers (NSCLCs) (Jemal et al, 2009). Despite improvements in early diagnosis made possible by emerging technologies and newly developed chemo/targeted therapies that improve treatment responses, the overall 5-year survival for NSCLC patients remains low (15%) and the recurrence rate is high, even in early-stage groups (Miller, 2005). The poor prognosis is due to late disease presentation, tumour heterogeneities within histological subtypes, and our relatively limited understanding of tumour biology. Emerging targeted therapies directed against specific cellular alterations require precise sub-classification of NSCLCs that is beyond the capabilities of standard histopathological diagnostic techniques. However, knowledge accumulated through genomic medicine creates the possibility of unravelling the remaining mysteries of lung cancer oncogenesis, and opens the door to molecular classification and risk stratification based on gene expression profiles and microRNA (miRNA) signatures.
MicroRNAs are small non-coding, endogenous, single-stranded RNAs that regulate gene expression (Bartel, 2004). Mature miRNAs and Argonaute (Ago) proteins form the RNA-induced silencing complex (RISC), which mediates post-transcriptional gene silencing through induction of messenger RNA (mRNA) degradation or translational inhibition (Liu et al, 2004; Pillai et al, 2004). Target mRNA specification is determined by sequence complementarity between the seed sequence of an individual miRNA and the target mRNAs (Baek et al, 2008; Eulalio et al, 2008; Selbach et al, 2008; Bartel, 2009). Recent proteomic studies have revealed a broad spectrum of targets for each individual miRNA (Baek et al, 2008; Selbach et al, 2008). By regulating gene expression at the post-transcriptional level, miRNAs profoundly influence a wide variety of pathways, and their greatest impact is on developmental and oncogenic pathways (Lee et al, 1993; Wightman et al, 1993; He et al, 2005, 2007; Johnson et al, 2005; Lu et al, 2005; Calin and Croce, 2006).
Half of all miRNA genes are found within or near chromosomal fragile sites, common breakpoints, or minimal regions of loss-of-heterozygosity or amplification (Calin et al, 2004). Accumulating evidence shows that miRNAs are grossly dysregulated in human cancers, including NSCLC, and may serve as oncogenes or tumour suppressors (Croce, 2009). Recent studies have shown that not only can miRNAs be used to sub-classify NSCLCs (Bishop et al, 2010) but specific miRNA profiles may also predict prognosis and disease recurrence in early-stage NSCLCs (Yanaihara et al, 2006; Yu et al, 2008; Raponi et al, 2009; Seike et al, 2009; Patnaik et al, 2010).
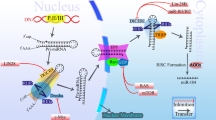
In this review, we briefly describe the biogenesis of miRNAs, their roles in lung cancer pathogenesis, and their potential use in NSCLC subgroup classification, risk stratification, and therapy. The stratification of lung cancer based on miRNA information is illustrated in Figure 1.

Stratification of lung cancer based on microRNA information. NSCLC, non-small-cell lung cancer.
MicroRNA biogenesis
MicroRNA genes are evolutionarily conserved and are located within the introns or exons of protein-coding genes, as well as in intergenic areas (Rodriguez et al, 2004). Canonically, miRNA genes are transcribed by RNA polymerase II or III into kilobase-long primary miRNA transcripts (pri-miRNAs). Pri-miRNAs are next cleaved into ∼70 nucleotide-long precursor miRNAs (pre-miRNAs) by the nuclear microprocessor complex formed by the RNase III Drosha and DiGeorge syndrome critical region gene 8 (DGCR8). The average human pre-miRNA contains a 33-base-pair hairpin stem, a terminal loop, and two single-stranded flanking regions upstream and downstream of the hairpin. Pre-miRNAs are next transported by the exportin-5/Ran GTPase complex into the cytoplasm, where miRNAs undergo maturation (Lee et al, 2003, 2004; Yi et al, 2003; Denli et al, 2004). In the cytoplasm, pre-miRNAs are cleaved by RNase III Dicer into an ∼22 nucleotide-long miRNA duplex and are unwound by helicase. The passenger strand is degraded, and the selected guide strand together with Ago protein activates RISC, resulting in mRNA degradation or translational inhibition, depending on the percentage of sequence complementarity between the miRNA 5′-seed and mRNA 3′-UTR element (Hammond et al, 2000; Diederichs and Haber, 2007). Alternatively, pre-miRNAs are derived directly from size-matched introns. These so-called ‘mitrons’ skip the Drosha–DGCR8 processing step and are spliced out of their host genes. These lariats are debranched, refolded into the stem-loop structure of typical pre-miRNAs, and then enter the canonical pathway (Okamura et al, 2007; Ruby et al, 2007).
A recent study by Suzuki et al demonstrated that p53 interacts with the Drosha microprocessor complex through DEAD-box RNA helicase p68 (DDX5) and facilitates the processing of pri-miRNAs into pre-miRNAs. This study describes the p53-mediated post-transcriptional maturation of miRNAs, linking the core tumour suppressor p53 to the miRNA biogenesis pathway (Suzuki et al, 2009). These findings may provide an explanation for the widespread miRNA downregulation observed in human cancers, in which p53 is often dysfunctional (Lu et al, 2005).
Defects in the miRNA biogenesis pathway and lung cancer
Drosha, DGCR8, and Dicer are the three best-established regulators of miRNA processing. Defects in the miRNA biogenesis machinery may be closely related to oncogenesis. Deletion of Dicer abrogates the production of mature miRNAs (Bernstein et al, 2003), and conditional deletion of Dicer1 enhances lung tumour development in a K-Ras-induced lung cancer mouse model (Kumar et al, 2007). In 2005, Karube et al (2005) reported that reduced Dicer expression levels were correlated with poor survival in a cohort of 67 surgically resected NSCLC patients. This correlation has been confirmed in an ovarian cancer, three breast cancers, and a lung cancer cohort in which high Dicer and Drosha mRNA expression is associated with better overall and disease-free survival (Merritt et al, 2008).
MicroRNAs function as tumour suppressors or oncogenes in lung cancer
Let-7 miRNAs as tumour suppressors
Let-7 was first identified in Caenorhabditis elegans as a regulator of the timing of cell fate determination (Reinhart et al, 2000). In C. elegans with mutant let-7, stem cells fail to exit the cell cycle and differentiate, but continue to divide, a hallmark of cancer cells (Reinhart et al, 2000). In humans, the let-7 family is a cluster of miRNAs whose genes map to different chromosomal regions that are frequently deleted in lung cancer (Calin et al, 2004). Cell studies have shown that let-7 miRNA overexpression in the A549 cell line inhibits cell growth and reduces cell-cycle progression (Johnson et al, 2007). In mouse NSCLC xenograft and orthotopic models, ectopic let-7g expression reduces tumour burden (Kumar et al, 2008), and intranasal let-7 administration represses lung adenocarcinoma (AD) formation (Esquela-Kerscher et al, 2008). Furthermore, reduced let-7 gene expression in NSCLC patients is correlated with poor prognosis (Takamizawa et al, 2004; Yanaihara et al, 2006), and a single nucleotide polymorphism in let-7 complementary site 6 of the K-RAS mRNA 3′-UTR is significantly associated with increased risk for NSCLC among moderate smokers (Chin et al, 2008). Collectively, these observations suggest a role for let-7 family miRNAs as tumour suppressors. In addition, let-7 miRNAs negatively regulate multiple oncogenes, including the RAS (Johnson et al, 2005), MYC (Kumar et al, 2007), and HMGA2 (Lee and Dutta, 2007), and cell-cycle progression regulators, such as CDC25A, CDK6, and cyclin D2 (Johnson et al, 2007). Although a corresponding knockout mouse would be invaluable for in-depth studies of let-7 tumour suppressor functions, multiple copies of let-7 in the genome make generating such a model somewhat problematic (Calin et al, 2004).
All members of the miR-17-92 cluster and miR-31 are oncogenes
All members of the miR-17-29 cluster (miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, miR-92-1) are oncogenes that reside in 13q31.3 (He et al, 2005). These miRNAs cooperate with c-Myc to accelerate tumour development and help promote tumour neovascularisation (O’Donnell et al, 2005; Dews et al, 2006). The miR-17-92 cluster is overexpressed in small-cell lung cancer (Hayashita et al, 2005). Ebi et al (2009) have confirmed this relationship and reported the association of miR-17-92 overexpression with RB inactivation. Their results suggest that this miRNA cluster may be a potential therapeutic target in lung cancer.
miR-31 is another example of an miRNA with oncogenic properties (termed an oncomir) in lung cancer. As reported by Liu et al, knockdown of miR-31 represses lung cancer cell clonal growth and in vivo tumourigenicity. Their data show that miR-31 functions as an oncomir by directly repressing the tumour suppressors LATS2 and PPP2R2A (Liu et al, 2010). This miR-31/LATS2/PPP2R2A pathway constitutes a new growth regulator in lung cancer.
MicroRNAs and conventional chemotherapy for NSCLCs
The selection for chemotherapy-resistant cells is often observed in platinum-based chemotherapy, currently the main regimen in NSCLC treatment (Seve and Dumontet, 2005), and is a key cause of chemotherapeutic failure. A recent in vitro study by Galluzzi et al (2010) showed that miR-630 inhibits p53-regulated pro-apoptotic signalling pathways that are specifically induced by cisplatin and carboplatin. This is one example demonstrating that the role of miRNAs may involve chemo-sensitivity/-resistance determination and suggesting the possibility that manipulating miRNAs may be potentially useful to modulate the cancer chemoresistance.
miRNAs and targeted therapies
Epidermal growth factor receptor (EGFR) signalling and EGFR mutations have been a major focus of lung cancer studies conducted during the past 5 years. Epidermal growth factor receptor is one of the most common proto-oncogenes in lung cancer. Leucine-to-arginine substitution at position 858 (L858R) and deletion mutants in exon 19 constitute 90% of lung cancer-specific EGFR-activating mutations. These mutations induce lung AD in a mouse model and confer hypersensitivity to EGFR–tyrosine kinase inhibitors (EGFR–TKIs) in humans (Rosell et al, 2009).
Several recent studies have uncovered a relationship between the EGFR signalling pathway and miRNAs. Weiss et al (2008) showed that miR-128b is a direct regulator of EGFR. miR-128b loss-of-heterozygosity is frequently found in NSCLC patients and is positively correlated with clinical response and survival after gefitinib treatment. In addition, Cho et al (2009) showed that restoration of the tumour suppressor miR-145 inhibits cancer cell growth in lung AD patients with EGFR-activating mutations. Furthermore, miR-7, an miRNA frequently downregulated in lung cancer, has been shown to suppress EGFR and Raf1 mRNA expression. It also attenuates the activation of Akt and ERK, two key players in the EGFR signalling pathway, suggesting that miR-7 negatively regulates the EGFR pathway (Webster et al, 2009). Finally, miR-21 is upregulated under conditions in which EGFR signalling is activated, especially in the context of EGFR-activating mutations, and is suggested to be related to lung carcinogenesis in never smokers (Seike et al, 2009). Growing evidence from miRNA studies may help clarify the role of the EGFR network in lung cancer oncogenesis and provide a clue to solve EGFR—TKI resistance problems.
The Apo2L/tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) is a member of the TNF family known to induce apoptosis in a variety of cancers (Schaefer et al, 2007). Treatment with TRAIL induces programmed cell death in cancer cells. However, a significant proportion of cancers are resistant to TRAIL-induced apoptosis through different mechanisms. Garofalo et al (2009) showed that miR-221 and miR-222 contribute to lung cancer resistance to TRAIL therapy by downregulating PTEN and TIMP3 tumour suppressors. These observations hint at the extent to which a greater knowledge of miRNAs might bring a deeper understanding of drug-resistance mechanisms, and suggest a future role for miRNAs as a solution to the drug resistance problem.
MicroRNAs in NSCLC histological differentiation, risk stratification, and outcome prediction
Histological differentiation
With the emergence of targeted therapies directed against specific molecular events or entities, accurate classification of tumours into AD and squamous cell carcinoma (SCC) becomes a necessity. However, this can be challenging, especially in cases in which biopsy/aspirate specimens are small or tumours are poorly differentiated. miR-205 is reported to be a useful marker for differentiating SCC from non-SCC NSCLCs, with a sensitivity of 96% and specificity of 90%, even in small biopsies from poorly differentiated tumours (Lebanony et al, 2009; Bishop et al, 2010). Landi et al (2010) also reported a five-miRNA signature (miR-25, miR-34c-5p, miR-191, let-7e, and miR-34a) that accurately differentiated SCC from AD, and the lower expression level of this signature correlated with poor overall survival among SCC patients. This is a further step beyond the traditional protein markers used in immunohistochemical diagnosis of equivocal cases. It is anticipated that miRNA markers will ultimately also be found for AD.
Risk stratification and outcome prediction
Risk stratification and prognosis assessment have become a major concern in the era of personalised medicine. Gene expression profiling has reached a plateau in this regard (Garber et al, 2001; Shedden et al, 2008), although recent miRNA studies show great promise (Yanaihara et al, 2006; Yu et al, 2008; Raponi et al, 2009).
Yanaihara et al (2006) reported that high miR-155 and low miR-let7a-2 expression correlated with poor overall survival in lung AD patients. Our group also identified a five-miRNA signature (miR-137, miR-372, miR-182*, miR-221, and let-7a) that correlated with disease-free survival in a cohort of 122 NSCLC patients (Yu et al, 2008). In addition, Raponi et al. reported that miR-146b is a robust predictor of overall survival in SCC. High miR-146 expression correlated with a poor overall survival in SCC patients and the same trend was also observed in the expression level of miR-155 among the same group of patients (Raponi et al, 2009). The pathway prediction of miR-146b-targeted genes revealed a significant overlap of biological pathways in their previously reported 50-gene expression signature (Beer et al, 2002; Raponi et al, 2009). Patnaik et al (2010) also defined an miRNA signature that predicted post-operative recurrence of stage I NSCLCs. It is clearly evident from these studies that, as is the case with gene expression profiling, miRNA signatures suggested by different groups are almost non-overlapping. This could be because of the different experimental platforms used (qRT–PCR versus different miRNA array systems), batch effects inherent in the microarray experiment itself, and potential ethnic differences in the study populations, as observed in the EGFR mutation story. More detailed studies are needed to clarify these issues. In addition, new modalities, such as next-generation sequencing, may provide tools to enhance the prospects of miRNA research.
A recent study by Hu et al (2010) reported a four-miRNA signature (miR-486, miR-30d, miR-1, and miR-499) that predicted survival of stage I to IIIa NSCLCs. Unlike all previous studies, their miRNAs were identified in serum using a Solexa platform (Nanjing Medical University, Nanjing University and Jiangsu Cancer Hospital, Nanjing, China). Although this represents a relative non-invasive approach, questions remain as to the representativeness of serum miRNA profiles in solid cancers, given that this approach fails to identify miRNAs commonly found in lung cancer tissues.
Perspectives
Numerous miRNAs are dysregulated in cancers, and a single miRNA can have multiple targets involved in different oncogenic pathways. Accumulating evidence also suggests a role for miRNAs in fighting drug resistance. These properties make miRNAs attractive targets in cancer therapy. However, the fact that one miRNA may target hundreds of mRNAs deserves special consideration, insofar as it implies the possibility of unpredictable side effects, even if a specific miRNA is effectively targeted. A greater understanding of miRNA biology and the development of suitable delivery systems are required to translate these basic research results into clinical practice. In addition, miRNAs may abet the histological characterisation of NSCLC differentiation, especially in cases in which biopsy/aspiration specimens are inadequate or tumours are poorly differentiated. This is especially important when targeted therapy is the treatment of choice. Risk stratification and drug-response prediction are the central elements of personalised medicine. During recent years, research has focused mainly on gene expression profiling and a number of important studies have been published. As gene expression profiling reaches a plateau and begins to face limitations, miRNA signature is a rising star that may provide new resolutions to old problems. It is difficult to say whether miRNA signature is superior or inferior to gene expression profiling, with respect to risk stratification and outcome prediction. What is clear, however, is that the more we understand cancer biology, the more likely we are to translate these laboratory-oriented studies into clinical practice. Finally, miRNAs are much more stable in serum and plasma than in mRNAs, raising the exciting prospect that miRNAs might be used as non-invasive biomarkers for disease monitoring and histological classification under specific circumstances. Going forward, insights gained from miRNA studies may open a new era in lung cancer treatments, providing improved patient selection for targeted agents, and forming the basis for the development of novel therapeutics and/or early disease biomarkers.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP (2008) The impact of microRNAs on protein output. Nature 455: 64–71
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233
Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, Lizyness ML, Kuick R, Hayasaka S, Taylor JM, Iannettoni MD, Orringer MB, Hanash S (2002) Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med 8: 816–824
Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, Elledge SJ, Anderson KV, Hannon GJ (2003) Dicer is essential for mouse development. Nature Genet 35: 215–217
Bishop JA, Benjamin H, Cholakh H, Chajut A, Clark DP, Westra WH (2010) Accurate classification of non-small cell lung carcinoma using a novel microRNA-based approach. Clin Cancer Res 16: 610–619
Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, Croce CM (2004) Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA 101: 2999–3004
Calin GA, Croce CM (2006) MicroRNA signatures in human cancers. Nature Rev Cancer 6: 857–866
Chin LJ, Ratner E, Leng S, Zhai R, Nallur S, Babar I, Muller RU, Straka E, Su L, Burki EA, Crowell RE, Patel R, Kulkarni T, Homer R, Zelterman D, Kidd KK, Zhu Y, Christiani DC, Belinsky SA, Slack FJ, Weidhaas JB (2008) A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res 68: 8535–8540
Cho WC, Chow AS, Au JS (2009) Restoration of tumour suppressor hsa-miR-145 inhibits cancer cell growth in lung adenocarcinoma patients with epidermal growth factor receptor mutation. Eur J Cancer 45: 2197–2206
Croce CM (2009) Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet 10: 704–714
Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ (2004) Processing of primary microRNAs by the Microprocessor complex. Nature 432: 231–235
Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A (2006) Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet 38: 1060–1065
Diederichs S, Haber DA (2007) Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell 131: 1097–1108
Ebi H, Sato T, Sugito N, Hosono Y, Yatabe Y, Matsuyama Y, Yamaguchi T, Osada H, Suzuki M, Takahashi T (2009) Counterbalance between RB inactivation and miR-17-92 overexpression in reactive oxygen species and DNA damage induction in lung cancers. Oncogene 28: 3371–3379
Esquela-Kerscher A, Trang P, Wiggins JF, Patrawala L, Cheng A, Ford L, Weidhaas JB, Brown D, Bader AG, Slack FJ (2008) The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle 7: 759–764
Eulalio A, Huntzinger E, Izaurralde E (2008) Getting to the root of miRNA-mediated gene silencing. Cell 132: 9–14
Galluzzi L, Morselli E, Vitale I, Kepp O, Senovilla L, Criollo A, Servant N, Paccard C, Hupé P, Robert T, Ripoche H, Lazar V, Harel-Bellan A, Dessen P, Barillot E, Kroemer G (2010) miR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res 70: 1793–1803
Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen GD, Perou CM, Whyte RI, Altman RB, Brown PO, Botstein D, Petersen I (2001) Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci USA 98: 13784–13789
Garofalo M, Di Leva G, Romano G, Nuovo G, Suh SS, Ngankeu A, Taccioli C, Pichiorri F, Alder H, Secchiero P, Gasparini P, Gonelli A, Costinean S, Acunzo M, Condorelli G, Croce CM (2009) miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell 16: 498–509
Hammond S, Bernstein E, Beach D, Hannon GJ (2000) An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 404: 293–329
Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K, Tomida S, Yatabe Y, Kawahara K, Sekido Y, Takahashi T (2005) A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res 65: 9628–9632
He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM (2005) A microRNA polycistron as a potential human oncogene. Nature 435: 828–833
He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, Jackson AL, Linsley PS, Chen C, Lowe SW, Cleary MA, Hannon GJ (2007) A microRNA component of the p53 tumour suppressor network. Nature 447: 1130–1134
Hu Z, Chen X, Zhao Y, Tian T, Jin G, Shu Y, Chen Y, Xu L, Zen K, Zhang C, Shen H (2010) Serum microRNA signatures identified in a genome-wide serum microRNA expression profiling predict survival of non-small-cell lung cancer. J Clin Oncol 28: 1721–1726
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ (2009) Cancer statistics, 2009. CA Cancer J Clin 59: 225–249
Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ (2005) RAS is regulated by the let-7 microRNA family. Cell 120: 635–647
Johnson CD, Esquela-Kerscher A, Stefani G, Byrom M, Kelnar K, Ovcharenko D, Wilson M, Wang X, Shelton J, Shingara J, Chin L, Brown D, Slack FJ (2007) The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res 67: 7713–7722
Karube Y, Tanaka H, Osada H, Tomida S, Tatematsu Y, Yanagisawa K, Yatabe Y, Takamizawa J, Miyoshi S, Mitsudomi T, Takahashi T (2005) Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci 96: 111–115
Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T (2007) Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet 39: 673–677
Kumar MS, Erkeland SJ, Pester RE, Chen CY, Ebert MS, Sharp PA, Jacks T (2008) Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci USA 105: 3903–3908
Landi MT, Zhao Y, Rotunno M, Koshiol J, Liu H, Bergen AW, Rubagotti M, Goldstein AM, Linnoila I, Marincola FM, Tucker MA, Bertazzi PA, Pesatori AC, Caporaso NE, McShane LM, Wang E (2010) MicroRNA expression differentiates histology and predicts survival of lung cancer. Clin Cancer Res 16: 430–441
Lebanony D, Benjamin H, Gilad S, Ezagouri M, Dov A, Ashkenazi K, Gefen N, Izraeli S, Rechavi G, Pass H, Nonaka D, Li J, Spector Y, Rosenfeld N, Chajut A, Cohen D, Aharonov R, Mansukhani M (2009) Diagnostic assay based on hsa-miR-205 expression distinguishes squamous from nonsquamous non-small-cell lung carcinoma. J Clin Oncol 27: 2030–2037
Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843–854
Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Rådmark O, Kim S, Kim VN (2003) The nuclear RNase III Drosha initiates microRNA processing. Nature 425: 415–419
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN (2004) MicroRNA genes are transcribed by RNA polymerase II. EMBO J 23: 4051–4060
Lee YS, Dutta A (2007) The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev 21: 1025–1030
Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, Hammond SM, Joshua-Tor L, Hannon GJ (2004) Argonaute2 is the catalytic engine of mammalian RNAi. Science 305: 1437–1441
Liu X, Sempere LF, Ouyang H, Memoli VA, Andrew AS, Luo Y, Demidenko E, Korc M, Shi W, Preis M, Dragnev KH, Li H, Direnzo J, Bak M, Freemantle SJ, Kauppinen S, Dmitrovsky E (2010) MicroRNA-31 functions as an oncogenic microRNA in mouse and human lung cancer cells by repressing specific tumor suppressors. J Clin Invest 120: 1298–1309
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR (2005) MicroRNA expression profiles classify human cancers. Nature 435: 834–838
Merritt WM, Lin YG, Han LY, Kamat AA, Spannuth WA, Schmandt R, Urbauer D, Pennacchio LA, Cheng JF, Nick AM, Deavers MT, Mourad-Zeidan A, Wang H, Mueller P, Lenburg ME, Gray JW, Mok S, Birrer MJ, Lopez-Berestein G, Coleman RL, Bar-Eli M, Sood AK (2008) Dicer, Drosha, and outcomes in patients with ovarian cancer. N Engl J Med 359: 2641–2650
Miller YE (2005) Pathogenesis of lung cancer: 100 year report. Am J Respir Cell Mol Biol 33: 216–223
O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT (2005) c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435: 839–843
Okamura K, Hagen JW, Duan H, Tyler DM, Lai EC (2007) The mirtron pathway generates microrNA-class regulatory RNAs in Drosophila. Cell 130: 89–100
Patnaik SK, Kannisto E, Knudsen S, Yendamuri S (2010) Evaluation of microRNA expression profiles that may predict recurrence of localized stage I non-small cell lung cancer after surgical resection. Cancer Res 70: 36–45
Pillai RS, Artus CG, Filipowicz W (2004) Tethering of human Ago proteins to mRNA mimics the miRNA-mediated repression of protein synthesis. RNA 10: 1518–1525
Raponi M, Dossey L, Jatkoe T, Wu X, Chen G, Fan H, Beer DG (2009) MicroRNA classifiers for predicting prognosis of squamous cell lung cancer. Cancer Res 69: 5776–6783
Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G (2000) The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403: 901–906
Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A (2004) Identification of mammalian microRNA host genes and transcription units. Genome Res 14: 1902–1910
Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M, Insa A, Massuti B, Gonzalez-Larriba JL, Paz-Ares L, Bover I, Garcia-Campelo R, Moreno MA, Catot S, Rolfo C, Reguart N, Palmero R, Sánchez JM, Bastus R, Mayo C, Bertran-Alamillo J, Molina MA, Sanchez JJ, Taron M, Spanish Lung Cancer Group (2009) Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 361: 958–967
Ruby JG, Jan CH, Bartel DP (2007) Intronic microRNA precursors that bypass Drosha processing. Nature 448: 83–86
Schaefer U, Voloshanenko O, Willen D, Walczak H (2007) TRAIL: a multifunctional cytokine. Front Biosci 12: 3813–3824
Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N (2008) Widespread changes in protein synthesis induced by microRNAs. Nature 455: 58–63
Seike M, Goto A, Okano T, Bowman ED, Schetter AJ, Horikawa I, Mathe EA, Jen J, Yang P, Sugimura H, Gemma A, Kudoh S, Croce CM, Harris CC (2009) MiR-21 is an EGFR-regulated anti-apoptotic factor in lung cancer in never-smokers. Proc Natl Acad Sci USA 106: 12085–12090
Seve P, Dumontet C (2005) Chemoresistance in non-small cell lung cancer. Curr Med Chem Anticancer Agents 5: 73–88
Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, Gerald WL, Eschrich S, Jurisica I, Giordano TJ, Misek DE, Chang AC, Zhu CQ, Strumpf D, Hanash S, Shepherd FA, Ding K, Seymour L, Naoki K, Pennell N, Weir B, Verhaak R, Ladd-Acosta C, Golub T, Gruidl M, Sharma A, Szoke J, Zakowski M, Rusch V, Kris M, Viale A, Motoi N, Travis W, Conley B, Seshan VE, Meyerson M, Kuick R, Dobbin KK, Lively T, Jacobson JW, Beer DG (2008) Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med 14: 822–827
Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K (2009) Modulation of microRNA processing by p53. Nature 460: 529–533
Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, Mitsudomi T, Takahashi T (2004) Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res 64: 3753–3756
Webster RJ, Giles KM, Price KJ, Zhang PM, Mattick JS, Leedman PJ (2009) Regulation of epidermal growth factor receptor signaling in human cancer cells by microRNA-7. J Biol Chem 284: 5731–5741
Weiss GJ, Bemis LT, Nakajima E, Sugita M, Birks DK, Robinson WA, Varella-Garcia M, Bunn Jr PA, Haney J, Helfrich BA, Kato H, Hirsch FR, Franklin WA (2008) EGFR regulation by microRNA in lung cancer: correlation with clinical response and survival to gefitinib and EGFR expression in cell lines. Ann Oncol 19: 1053–1059
Wightman B, Ha I, Ruvkun G (1993) Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75: 855–862
Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, Stephens RM, Okamoto A, Yokota J, Tanaka T, Calin GA, Liu CG, Croce CM, Harris CC (2006) Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 9: 189–198
Yi R, Qin Y, Macara IG, Cullen BR (2003) Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 17: 3011–3016
Yu SL, Chen HY, Chang GC, Chen CY, Chen HW, Singh S, Cheng CL, Yu CJ, Lee YC, Chen HS, Su TJ, Chiang CC, Li HN, Hong QS, Su HY, Chen CC, Chen WJ, Liu CC, Chan WK, Chen WJ, Li KC, Chen JJ, Yang PC (2008) MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell 13: 48–57
Acknowledgements
This work is supported by NRPGM (NSC98-3112-B-002-041) and NTU 97R0066-08.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Lin, PY., Yu, SL. & Yang, PC. MicroRNA in lung cancer. Br J Cancer 103, 1144–1148 (2010). https://doi.org/10.1038/sj.bjc.6605901
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6605901
Keywords
This article is cited by
-
miRNome profiling of lung cancer metastases revealed a key role for miRNA-PD-L1 axis in the modulation of chemotherapy response
Journal of Hematology & Oncology (2022)
-
Bioengineering of a single long noncoding RNA molecule that carries multiple small RNAs
Applied Microbiology and Biotechnology (2019)
-
miR-182 suppresses invadopodia formation and metastasis in non-small cell lung cancer by targeting cortactin gene
Journal of Experimental & Clinical Cancer Research (2018)
-
Multipronged activity of combinatorial miR-143 and miR-506 inhibits Lung Cancer cell cycle progression and angiogenesis in vitro
Scientific Reports (2018)
-
MicroRNA-29c functions as a tumor suppressor by targeting VEGFA in lung adenocarcinoma
Molecular Cancer (2017)
