
Abstract
Background:
Enzastaurin (Enz) is a serine/threonine kinase inhibitor blocking protein kinase C (PKC)β/AKT pathway. However, an ability of this compound to inhibit cancer invasion and metastasis is not yet clearly elucidated.
Methods:
The ability of Enz to inhibit invasion and metastasis, and to target molecules was investigated in non-small cell lung cancer (NSCLC) by RT–PCR validated microarray, Matrigel, and in vivo chorionallantoic membrane (CAM) assays.
Results:
Enzastaurin significantly reduced migration, invasion, and in vivo metastasis to lungs and liver (CAM assay) of diverse NSCLC cell lines. Genes promoting cancer progression (u-PAR, VEGFC, and HIF1α) and tumour suppression (VHL, RASSF1, and FHIT) of NSCLC were significantly (P<0.05) down- or upregulated after Enz treatment in H460, A549, and H1299 cells, respectively. Luciferase/chromatin immunoprecipitation analysis showed that Enz transcriptionally controls urokinase-type plasminogen activator receptor (u-PAR) expression by promoter inhibition through Sp1, Sp3, and c-Jun(AP-1). Moreover, siRNA knockdown of u-PAR re-sensitised Enz-resistant cells and induced apoptosis, suggesting u-PAR as a marker of Enz resistance.
Conclusion:
This study shows that Enz inhibits migration, invasion, and in vivo metastasis by targeting u-PAR, besides further targeting progression-related and tumour-suppressor genes in NSCLC. Together with u-PAR being a novel putative marker of Enz response, these data encourage molecularly tailored clinical studies on Enz in NSCLC therapy.
Similar content being viewed by others


Main
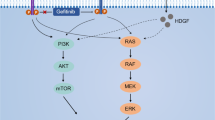
Non-small cell lung cancer (NSCLC) accounts for ∼85% of lung cancer cases and is the leading cause of tumour-related death (Jemal et al, 2005; Gridelli et al, 2007), progressive stages and metastasis being the most frequent cause for the high NSCLC lethality. Protein kinase C (PKC) isoforms have been shown to be highly expressed in NSCLC as compared to lung epithelial cells (Clark et al, 2003). Activation of these isoforms contributes to patient survival, proliferation, and the malignant progression of human cancers including breast carcinoma (Ali et al, 2009), B-cell lymphoma (Shipp et al, 2002), glioblastoma (da Rocha et al, 2002), and colorectal carcinoma (Gokmen-Polar et al, 2001). Protein kinase C signalling, which functions through serine/threonine kinase activity, is involved in tumour-induced angiogenesis, tumour growth, differentiation, cytokine secretion, migration, and apoptosis, and is a prominent target for anticancer therapy (Carducci et al, 2006). Protein kinase C activation can trigger ERK and PI3K/AKT pathways, promoting cell proliferation and apoptosis (Balendran et al, 2000; Goekjian and Jirousek, 2001). Moreover, both PKC and AKT phosphorylate glycogen synthase kinaseβ (GSK3β) at Ser9, supporting the notion that these signalling pathways overlap (Fang et al, 2002).
Enzastaurin (Enz), an acyclic bisindolylmaleimide, was initially developed as a specific inhibitor of PKCβ (Faul et al, 2003). In addition to this major target, Enz also inhibits other PKC isoforms (δ, α, ɛ, and γ) (Graff et al, 2005). Enzastaurin was initially developed for anti-angiogenic cancer therapy (Keyes et al, 2002), and recent preclinical studies have shown that Enz exerts proapoptotic, growth inhibitory, and anti-angiogenic properties in a variety of human cancers (Keyes et al, 2002; Graff et al, 2005; Querfeld et al, 2006; Willey et al, 2009). Moreover, in clinical studies, Enz was well tolerated and has shown encouraging activity in a variety of tumours (Carducci et al, 2006; Watkins et al, 2006; Hanauske et al, 2009). Enzastaurin was shown to target the PI3K/AKT pathway, to inhibit GSK3β and ribosomal protein S6 phosphorylation, to decrease the expression of VEGF (Keyes et al, 2002; Graff et al, 2005) and also to show a synergism with SUI174 and pemetrexed (Tekle et al, 2008; Faoro et al, 2009; Giovannetti et al, 2010).
Tumour progression is a multistep process including uncontrolled growth, invasion, and metastasis (Douglas and Robert, 2000), which, to a considerable part, is mediated through an abnormal regulation and function of genes. Several studies have analysed the involvement of individual genes (Müller-Tidow et al, 2001; Marchetti et al, 2002; Werle et al, 2004), gene expression differences between cell lines (Chen et al, 2001; Gemma et al, 2001), and gene expression differences between primary tumours and normal lung tissues in this context (Bhattacharjee et al, 2001; Hellmann et al, 2001; Beer et al, 2002; Heighway et al, 2002; Wikman et al, 2002; Zochbauer-Muller et al, 2002; Borczuk et al, 2003; Diederichs et al, 2004; Hayes et al, 2006). The urokinase-type plasminogen activator (u-PA) and its receptor (u-PAR) are essential for proteolysis in many solid tumours, the main mechanism being the activation of plasminogen-dependent proteolysis and a cascade of protolytic activities, leading to extracellular matrix degradation, invasion, intravasation, and metastasis establishment (Jo et al, 2003; Dass et al, 2008). Extensive clinical studies have shown that the expression of u-PAR, u-PA, and its specific inhibitor PAI-1 is associated with tumour recurrence and poor survival of diverse cancer types (Werle et al, 2004; Beyer et al, 2006). In addition to the u-PAR, an overexpression of further molecules such as hypoxia-inducible factor 1α (HIF1α), a transcription factor, and vascular endothelial growth factor C (VEGFC), a major inducer of angiogenesis, has been specifically shown to correlate with poor prognosis, tumour progression, and metastasis of NSCLC (Zhang et al, 2007; Wang et al, 2009). Genes u-PAR, HIF1α, and VEGFC are transcriptionally activated by, for example, MAPK- and/or PI3K/AKT-related pathways (Jo et al, 2003; Tsai et al, 2003; Phillips et al, 2005; Skurk et al, 2005). Tumour-suppressor genes VHL, RASSF1, and FHIT are downregulated in NSCLC (Zochbauer-Muller et al, 2002; Hayes et al, 2006), which are implicated in the ubiquitin ligase system (Karhausen et al, 2005), cell-cycle regulation (Dammann et al, 2000), and apoptosis (Ji et al, 1999), respectively.
Although Enz has been reported to reduce tumour growth and to enhance apoptosis, a potential ability of this compound to inhibit invasion and metastasis as the most important characteristics of malignant tumours has never been studied. Therefore, this study was conducted to (1) determine the effects of Enz on migration, invasion, and in vivo metastasis; and (2) determine and implicate first molecular targets mediating this ability of Enz in NSCLC. Furthermore, in a systematic profiling approach we sought to give general insights into important target molecules of Enz in NSCLC.
Our study reveals that PKC inhibition by Enz reduces NSCLC proliferation, migration, invasion, and in vivo metastasis. Moreover, we show that u-PAR, VEGFC, and HIF1α, among other targets, are being downregulated, and the tumour-suppressor genes FHIT, RASSF1, and VHL are upregulated upon Enz treatment. We further describe u-PAR as an essential target being transcriptionally regulated by Enz through Sp1, Sp3, and c-Jun(AP-1) binding to the promoter, and as a putative novel marker of Enz sensitivity.
Materials and methods
Materials, cell lines, and reagents
Enzastaurin (in DMSO) was a generous gift from Eli Lilly (Indianapolis, IN, USA). All cell lines (H460, LXF289, A427, H1395, A549, and H1299) were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA) and cultured in ATCC-recommended medium (10% FBS, 37°C, 5% CO2). Fertilised special pathogen-free eggs were from Charles River (Hilden, Germany). Antibodies used for supershift/chromatin immunoprecipitation analysis (ChIP) experiments were from Santa Cruz Biotechnology (Santa Cruz, CA, USA): p-c-Jun (sc-822x), JunD (sc-74x), JunB (sc-46x), c-Fos (sc-52x), FosB (sc-7203x), Fra-1 (sc-22794x), Fra-2 (sc-604x), Sp1 (sc-59x), Sp3 (sc-644x), and unspecific IgG (sc-2338). Western blot antibodies against phospho-PKC(PAN) 9371, phospho-p44/42(Thr202/Tyr204) 4370, p44/42 9102, phospho-GSK3β(Ser9) 9336, and phospho-Akt(Ser473) 4058 were from Cell Signaling, Danvers, MA, USA, and Akt1 (sc-1618) and β-Actin (sc-1616R) were from Santa Cruz Biotechnology.
Proliferation assay
Cells (3 × 103) per well were seeded in a 96-well plate in a total volume of 100 μl medium with 10% FBS. After 24 h, cells were treated either with DMSO or Enz at concentrations between 0 and 15 μ M. After 72 h, the growth inhibitory effect was evaluated using 20 μl per well of CellTiter96 AqueousOneSolution (Promega, Madison, WI, USA) according to the manufacturer's instructions.
Wound-healing assay
Cells (3 × 105) per well were seeded in a 24-well plate in a total volume of 500 μl medium (10% FBS). After 24 h, the medium was changed to serum-free and a wound created in the monolayer by using a pipette tip. Cells were washed twice (PBS), and incubated with fresh medium supplemented with DMSO or Enz. In case of EGF stimulation, EGF (100 nM) was added 4 h after initial DMSO or Enz treatment. Digital pictures were taken at 0 and 48 h. To quantify migration, we calculated the diameter of the wound as the average of three different locations.
Matrigel invasion assay
Cells (1 × 105), starved in serum-free medium (24 h), were plated on transwell chambers precoated with 10 μg Matrigel. DMSO or Enz was applied to bottom and top chambers. In case of EGF, cells were pretreated (4 h) with DMSO or Enz, followed by EGF (100 nM). 10% FBS medium in the bottom chamber was used as a chemoattractant. After 24 h, noninvading cells were removed with cotton swabs. Invaded cells were trypsinised and counted using the ATP luminescence-based motility-invasion assay as described (Leupold et al, 2007).
cDNA labelling, array hybridisation, and data analysis
Total RNA was isolated as described (Mudduluru and Allgayer, 2008), and probe labelling and array hybridisation on Illumina Human Sentrix-8 BeadChip arrays (Illumina, San Diego, CA, USA) were conducted according to Illumina's recommended sample labelling procedure based on the modified Eberwine protocol (Eberwine et al, 1992). Scanning was carried out using a Beadstation array scanner, adjusted to a scaling factor of 1 and PMT settings at 430. Data extraction was carried out for all beads individually, and outliers were removed when median absolute deviation was >2.5. All remaining data points were used for the calculation of the mean average signal for a given probe, and standard deviation for each probe was calculated. Data analysis was carried out by normalisation of the signals using the quantile normalisation algorithm without background subtraction, and differentially regulated genes defined by calculating the standard deviation differences of a given probe in a one-by-one comparison of samples.
Luciferase reporter assay
Cells were transiently transfected with 500 ng of −398/+51 u-PAR WT/Sp1- or AP1-mutant plasmid, or with pGL3 basic (Allgayer et al, 1999), and 50 ng of Renilla-luciferase plasmid as a transfection control using Effectene (Qiagen, Hilden, Germany). After 24 h, cells were treated with DMSO or Enz for another 24 h, and reporter assays performed using the Dual-Luciferase reporter assay system (Promega).
Real-time PCR
Total RNA isolation and cDNA synthesis was performed as described (Mudduluru and Allgayer, 2008). The u-PAR (Hs00182181; Applied Biosystems, Carlsbad, CA, USA) mRNA was quantified using TaqMan Universal PCR Master Mix and β-Actin (Human ACTB 4333762F; Applied Biosystems) as housekeeping gene. Quantification of HIF-1α, VEGFC, VHL, RASSF1, FHIT, Sp1, Sp3, and c-Jun mRNA was performed using SYBR Green-PCR-Master-Mix (Applied Biosystems). Relative expression of these genes was calculated by the 2−ΔΔCT method. Primer sequences are provided in Supplementary Table 1.
ELISA and western blotting
ELISA and western blot analysis were performed as described (Leupold et al, 2007; Mudduluru and Allgayer, 2008). In brief, for ELISA, cells were either treated with DMSO or with Enz for 24 h. For western blot analysis, cells were serum starved for 24 h and treated with DMSO or Enz for another 24 h. Cells were stimulated with EGF (100 nM) for 15 min. Cells were washed twice with ice-cold PBS and lysed with lysis buffer (Biosource, Camarillo, CA, USA). Protein concentration was determined by BCA (Pierce, Rockford, IL, USA). The u-PAR protein was assayed using the Imubind-u-PAR-ELISA kit (American Diagnostica, Hauppauge, NY, USA).
Transfection of si-uPAR
si-u-PAR(ID289377; Ambion, Austin, TX, USA) was used to knockdown the expression of u-PAR. A nonspecific si-RNA (scrambled) served as a negative control. Sh-RNA vectors of sh-u-PAR (RHS4430-99290516), and non-silencing control (RHS4346) were purchased from Open Biosystems (Huntsville, AL, USA). A549 and H1299 cells were transiently transfected using RNAiFect or Effectene, respectively (Qiagen). At 24 h after transfection, cells were used for MTT and Matrigel assays as described.
Apoptosis assays
Cells (1 × 106 per ml) were treated with Enz (as the respective IC50 values) and harvested after 48 h either with or without transfections. Apoptosis induction by Enz was measured by the FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen, San Diego, CA, USA) as described (Querfeld et al, 2006). According to the manufacturer's instructions, cells were incubated with 5 μl per test FITC-conjugated annexin V in the presence of 5 μl per test PI, and further screened by flow cytometry (BD Biosciences, San Diego, CA, USA). Annexin V-positive PI-positive cells correspond to apoptotic cells and are represented as a percent of apoptotic cells against DMSO-treated cells.
Electrophoretic mobility shift and ChIP assay
Gel shifts were performed as described (Mudduluru and Allgayer, 2008), using 5 μg of nuclear extract of DMSO- or Enz-treated (IC50, 24 h) A549 cells. Chromatin immunoprecipitation was performed according to the manufacturer's protocol (Upstate, Lake Placid, NY, USA) as described (Leupold et al, 2007; Mudduluru and Allgayer, 2008), with 2 μg specific (Sp1, Sp3, and p-c-Jun) and nonspecific IgG overnight, using aliquots of precleared lysates. DNA was purified and eluted with 100 μl elution buffer (Qiagen reaction purification kit). PCR was performed according to Leupold et al (2007), for region −190/−171 (u-PAR-AP1). Amplification of region −152/−135 (u-PAR-Sp1/Sp3/AP-2α-like) was performed by SYBR Green Master Mix, with the specific primers: For 5′-AGGCAATCTGGGGACAGAG-3′ and Rev 5′-GGACTCCTCCCAGACGTTTT-3′.
Chorionallantoic membrane assay
The chorionallantoic membrane (CAM) assay was performed as described (Leupold et al, 2007), with few modifications. Briefly, 2 × 106 cells were inoculated on the CAM of 10-day-old chicken embryos. On days 12 and 14, chicken embryos were treated intravenously with different concentrations of Enz (2 or 4 μ M) or DMSO, respectively. Untreated embryos served as a control. On day 16, the embryos were killed. Metastasis was determined by harvesting lungs and liver, and processing the tissue for genomic human DNA by quantitative alu-PCR.
Statistical analysis
Statistical significance of differences between Enz- vs DMSO-treated samples were calculated with the Student's t-test (P⩽0.05 was considered as statistically significant, P⩽0.1 as a trend).
Results
Enzastaurin inhibits the growth of human NSCLC cells
Six human NSCLC cell lines were tested for their sensitivity to Enz regarding proliferation (Figure 1A). Large cell-type H460 and adenocarcinoma LXF289 cells showed the highest sensitivity. A moderate response was observed in H1395 (adenocarcinoma) and A427 (squamous carcinoma) cells, whereas A549 (squamous carcinoma) and H1299 (adenocarcinoma) cells showed comparatively more resistance to Enz. Mean IC50 values of the cells are presented in the box in Figure 1A. Similarly, viable cells were counted after Enz treatment, confirming the IC50 concentrations determined by MTT assays (data not shown). Next, to determine if the calculated IC50 concentrations of Enz were able to induce apoptosis and inhibit the known target molecules of Enz, apoptotic assays and western blot analysis were performed. H460, A549, and H1299 cells were treated for 48 h with their respective IC50 concentrations. Enzastaurin (IC50 concentrations) induced apoptosis of H460, A549, and H1299 cells compared with the DMSO-treated samples was shown in Figure 1B. Because EGFR-induced signalling represents a major pathway driving NSCLC progression, we in addition sought to investigate the ability of Enz to inhibit essential target molecules following EGF stimulation. H460, A549, and H1299 cells were pretreated either with DMSO, or with Enz, for 24 h, and stimulated, or unstimulated, with EGF (100 nmol l−1) for 15 min before harvesting the cells. As results, we found that Enz treatment can inhibit the phosphorylation of PKC, AKT, p42/44, and GSK3β both in the presence and in the absence of EGF (Figure 1C, Supplementary Figure 1).

Enzastaurin inhibits NSCLC proliferation, migration, and invasion. (A) Cell proliferation of NSCLC after Enz treatment. Data are expressed as the percentage of control (treated with DMSO) cells. Points, mean±s.d. of three experiments. Resulting IC50 values for Enz for all NSCLC cell lines investigated are represented in the box. (B) Apoptosis analysis of H460, A549, H1299 after Enz treatment. After 48 h of Enz treatment, cells were screened by flow cytometry and percent of apoptotic cells was calculated against to DMSO-treated cells. (C) Western blot analysis of cells treated with EGF, Enz, or with the combination of both agents. Specific antibodies against the indicated molecules were used, together with anti-beta-Actin as a loading control. (C and D) Percentage of migrating cells (would-healing assay) and invading cells (Matrigel assay). Cells were treated as described in the Materials and Methods section. DMSO-treated samples served as a control (100%), and other treated samples were calculated and plotted as a percentage of this value. *P<0.05.
Enzastaurin affects cell migration and invasion
To determine whether Enz affects the migration and invasion of NSCLC cells in vitro, we evaluated the migratory (wound-healing assay) and invasive (Transwell in vitro invasion assay) capacity of H460, A549, and H1299 cells, after treatment. Enzastaurin- or Enz/EGF-treated cells displayed a significantly reduced migration and invasion as compared to DMSO- and EGF-stimulated control cells; respectively (P<0.02; Figure 1D), showing that Enz inhibits EGF-induced cell migration and invasion in addition to spontaneous invasion in NSCLC. This prompted further experiments to study changes in gene expression upon Enz treatment, to delineate major target molecules mediating Enz-inhibited invasion.
Analysis of gene expression changes after Enz treatment
To analyse changes in gene expression after Enz treatment, we applied Illumina Human Sentrix-8 BeadChip assays. A549 cells were either treated with DMSO, or with Enz, for 24 or 48 h at the same concentration as used for inhibiting invasion. Total RNA was isolated, labelled, and hybridised to the arrays. After 24 h, 1255 genes (616 up, 639 down), and after 48 h, 2711 genes (1477 up, 1234 down) were significantly (⩾1.5-fold) deregulated after Enz treatment (Supplementary Tables 2 and 3), respectively. Comparing our microarray results with data present in the literature, we found that a considerable number of genes that have been described to be either up- or downregulated specifically in NSCLC in previous publications were significantly altered upon Enz treatment (Table 1). These genes include genes regulating cell proliferation, structure, adhesion, migration, invasion, or angiogenesis, or are cell-cycle regulators. We found that all of these genes as described in Table 1 were inversely regulated upon Enz treatment in A549 cells as compared to their gene expression status reported in previous publications on NSCLC for the untreated situation. Interestingly, three tumour suppressors (FHIT, RASSF1, and VHL), which have been reported to be downregulated in NSCLC (Wikman et al, 2002; Hayes et al, 2006), were upregulated upon Enz treatment, which was validated by RT–PCR (Figure 2A–C) at the mRNA level. Also, HIF1α, VEGFC, and u-PAR (Chen et al, 2001; Werle et al, 2004; Hayes et al, 2006), genes that are known to be key regulators of various aspects of carcinogenesis, invasion, and metastasis, were significantly downregulated after Enz treatment. The expression of these genes was validated in NSCLC cell lines through real-time PCR, which confirmed their cell line-independent regulation upon Enz treatment (Figure 3A–C). Taken together, Enz negatively regulates essential genes that are reportedly relevant for NSCLC, induces the expression of three NSCLC-related tumour-suppressors, and inhibits three essential genes of invasion and metastasis at the mRNA level.

Enzastaurin induces the expression of tumour-suppressor genes. Real-time PCR quantification of (A) VHL, (B) RASSF1, and (C) FHIT mRNA in H460 and A549 cells after 24–48 h of DMSO or Enz (IC50) treatment. Relative gene expression was normalised against β-Actin mRNA. *P<0.05.

Effect of enzastaurin on the expression of pro-invasive genes. Real-time PCR quantification of (A) HIF1α, (B) VEGFC, and (C) u-PAR mRNA in H460, A549, and H1299 cells after 24–48 h of DMSO or Enz (IC50) treatment. Relative gene expression was normalised against β-Actin mRNA. *P<0.05.
Enzastaurin inhibits u-PAR gene expression in NSCLC cells
The u-PAR has been reported to be one of the major metastasis-related genes being overexpressed in many cancer types, and also in NSCLC (Beer et al, 2002; Werle et al, 2004). Previous reports including our own have shown that an overexpression of this receptor is associated with a high invasive and metastatic capacity of NSCLC, and also EGF-induced, NSCLC cells, among other cancer types (Nikolova et al, 2009). Therefore, we decided to specifically analyse a putative function of the u-PAR in Enz-regulated invasion, and molecular mediators addressed by Enz to regulate u-PAR in NSCLC. ELISA showed a significant reduction of u-PAR protein after Enz treatment in H460 (P=0.0021), A549 (P=0.02), and H1299 (P=0.0022) cells as compared to the DMSO-treated samples (Figure 4A), in addition to the downregulation of u-PAR mRNA observed in the same cell lines in the previous section. It has been well reported that u-PAR gene expression in malignant cells is largely because of the transcriptional regulation of the gene (Allgayer et al, 1999; Laufs et al, 2006; Leupold et al, 2007). Specifically, it has been reported that the u-PAR promoter, to a major part, is being regulated by transcription factors of the AP-1 and Sp family (Allgayer et al, 1999; Laufs et al, 2006; Leupold et al, 2007). Correspondingly, we found that Enz treatment in A549 and H1299 cells significantly downregulated the expression of Sp1-, Sp3-, and c-Jun-transcription factors (P<0.05; Supplementary Figure 2), which have been reported previously to be major regulators of u-PAR promoter activity. In luciferase assays of H460, A549, and H1299 cells transfected with a reporter plasmid driven by the −398/+51 upstream region of the u-PAR gene, we found a significant reduction in u-PAR wild-type promoter activity after Enz treatment (P<0.008) (Figure 4B). Moreover, site-directed mutagenesis abolishing the binding of either Sp- or AP-1-family members to their respective binding sites within the u-PAR promoter abolished the ability of Enz to induce a further reduction of promoter activity of the respective construct (Figure 4C). This was paralleled by a specific reduction of the binding of either Sp1, Sp3, or AP-1-family members to their specific cis elements within the u-PAR promoter in EMSA analysis (Supplementary Figure 3b and c), which could also be observed for the situation of EGF-induced A549 cells (Supplementary Figure 3a). Finally, to show that Enz is able to regulate transcription factor binding to the natural u-PAR promoter, we performed ChIP analysis for the endogenous u-PAR upstream region. Chromatin immunoprecipitation revealed significantly less binding of especially Sp1, and phospho-c-Jun, to the endogenous u-PAR promoter after Enz treatment (Figure 4D, Supplementary Figure 3d). Taken together, data suggest that Enz inhibits the expression of the u-PAR, at least to a relevant part by suppressing u-PAR promoter activity, by downregulation of the binding of especially Sp1, and c-Jun out of the AP-1 family.

Enzastaurin inhibits u-PAR promoter activity and u-PAR protein expression. (A) Total amounts of u-PAR protein as evaluated by ELISA as described in the Materials and Methods section. Each value represents the average of triplicate measurements. (B) Luciferase activity assay for H460, A549, and H1299 cells. Data are presented as the mean±s.d. of three independent experiments performed in quadruplicate. *P<0.05. (C) Luciferase activity assay of A549 cells transfected with wild-type (wt) u-PAR promoter, and Sp/AP1 binding site mutated reporter constructs. Values represent the mean of three independent experiments performed in triplicate; bars, s.d. (D) Real-time PCR quantification of u-PAR promoter binding after chromatin immunoprecipitation (ChIP) with Sp1, Sp3, p-c-Jun, or IgG antibodies in the presence, or absence, of Enz (24 h). (E and F) Cell viability (MTT) and apoptosis assay. At 24 h after transfection with si-u-PAR/sh-u-PAR, cells were treated with Enz for 48 h and the results compared to DMSO-treated controls for MTT assay, represented as percentage and for apoptosis cell were screened by flow cytometry and percent of apoptotic cells were calculated against to DMSO-treated cells.
u-PAR knockdown re-sensitises NSCLC cells and induces apoptosis to Enz
A427, A459, and H1299 are relatively resistant to Enz treatment as compared to other NSCLC cell lines. This observation supported the notion that endogenous u-PAR gene expression might be associated with resistance to Enz. Therefore, we conducted specific knockdown experiments with an si-RNA/sh-RNA against endogenous u-PAR, the efficient knockdown of endogenous u-PAR being confirmed with u-PAR-ELISA (Supplementary Figure 4a and b). We observed that A549 and H1299 cells treated with si-u-PAR/sh-u-PAR were significantly re-sensitised to Enz (A549/IC50⩽7.0 μ M; H1299/IC50⩽9.0 μ M) when compared to scrambled control-transfected cells (A549/IC50=10 μ M; H1299/IC50=12 μ M) (Figure 4E). Similarly, viable cells were counted after Enz treatment either with scrambled/sh-RNA or with si-u-PAR/sh-u-PAR knockdown, which confirmed the IC50 concentrations determined by MTT assay (data not shown). Furthermore, u-PAR knockdown cells significantly induced apoptosis when compared with control cells after 48 h of Enz treatment (Figure 4F), and also showed a further decrease in u-PAR protein levels after Enz treatment (Supplementary Figure 4a and b). These data suggest that u-PAR might be one molecular parameter indicating Enz resistance of NSCLC cells.
Enzastaurin reduces distant metastasis of NSCLC cells in vivo
Our previous results suggested that Enz reduces migration and invasion in vitro. To strengthen these observations, we used the chicken embryo metastasis assay (CAM) (Leupold et al, 2007) to check the ability of Enz to inhibit the formation of distant metastasis in vivo. Towards this end, H460, A549, and H1299 cells were inoculated on the upper CAM of 10-day-old chicken embryos. On day 12 and 14, the embryos were treated intravenously with 2 or 4 μ M of Enz, or equal concentrations of DMSO, respectively. On day 16, the embryos were killed, lungs and livers harvested for DNA, and the number of cells metastasised into livers and lungs were measured with a TaqMan-based PCR amplifying human alu-sequences on the chicken background (Leupold et al, 2007). As a result, we found that Enz was able to reduce the formation of distant metastasis significantly in H460 (liver P=0.001, lungs P=0.003), A549 (liver P=0.037, lungs P⩽0.01), and H1299 (liver P=0.02, lungs P=0.05) cells when compared with their respective DMSO controls (Figure 5A–C). These data suggest that Enz, besides migration and invasion, inhibits in vivo metastasis of three NSCLC cell lines.

Enzastaurin inhibits NSCLC metastasis in vivo. (A–C) Number of metastasising cells in the livers and lungs of chicken embryos treated intravenously with different concentrations of Enz (2 or 4 μ M) or DMSO, respectively. Human Alu sequences were amplified and quantified by real-time PCR. **P<0.001; *P⩽0.05.
Discussion
This is the study to show that Enz inhibits migration, invasion, and in vivo metastasis in NSCLC, and to explore first mechanisms contributing to Enz-induced inhibition of migration, invasion, and metastasis-related processes in NSCLC. We show that Enz transcriptionally inhibits u-PAR gene expression, downregulates the tumour progression-related molecules HIF1α and VEGFC, and also upregulates the tumour-suppressor genes VHL, RASSF1, and FHIT. Furthermore, our findings suggest that an si-u-PAR/sh-u-PAR knockdown sensitises resistant NSCLC cells to Enz and induces apoptosis. These findings suggest that Enz treatment in NSCLC patients might contribute to enhancing their survival by inhibiting genes of tumour progression and by inducing tumour-suppressor genes.
Enzastaurin was initially developed as a specific inhibitor of PKCβ, also inhibiting the other PKC isoforms (δ, α, ɛ, and γ), PI3K/AKT, GSK3β, and S6K (Faul et al, 2003; Graff et al, 2005; Tekle et al, 2008; Faoro et al, 2009; Willey et al, 2009; Giovannetti et al, 2010). In agreement with previous studies, our initial observations in six NSCLC cell lines have shown that Enz inhibits cell growth and activation of PKC, p-42/44, AKT, and GSK3 β (Faul et al, 2003; Graff et al, 2005). Furthermore, we in addition show that it inhibits the EGF-stimulated activation of these kinases. In addition to initial findings in cultured hepatocellular carcinoma, we now show that Enz inhibits the in vitro migration and invasion of NSCLC cells (Guo et al, 2009), and that EGF-induced invasion is countered by Enz, which is a major pathway in NSCLC progression (Nikolova et al, 2009). Correspondingly, PKC isoforms have been shown to be activated in NSCLC cells, and to correlate with tumour progression in different cancers (Shipp et al, 2002; Clark et al, 2003; Ali et al, 2009; Gokmen-Polar et al, 2001). As further fundamental pathways, the MAPK/ERK and PI3K/AKT signalling axis have been reported to be activated upon PKC activation, which leads to cell proliferation and anti-apoptosis (Balendran et al, 2000; Goekjian and Jirousek, 2001). By inhibiting the activation PKC/other Ser/Thr kinases such as AKT, p-42/44, and GSK3β in NSCLC cells, in our gene expression array we now found that Enz indeed significantly downregulates a number of genes that are involved in different cellular functions such as cell proliferation, anti-apoptosis, but also cell adhesion, migration, invasion, and angiogenesis, which are known to be upregulated in the NSCLC (Bhattacharjee et al, 2001; Chen et al, 2001; Hellmann et al, 2001; Beer et al, 2002; Wikman et al, 2002; Borczuk et al, 2003; Diederichs et al, 2004; Werle et al, 2004; Hayes et al, 2006). Moreover, remarkably, it significantly upregulates the tumour-suppressor genes (FHIT, RASSF1, and VHL) that are known to be downregulated in NSCLC (Zochbauer-Muller et al, 2002; Hayes et al, 2006). It has been reported that the expression of RASSF1 and FHIT is inhibited in NSCLC tumours, their downregulation interestingly also having been shown to be associated with promoter hypermethylation (Zochbauer-Muller et al, 2002). These tumour-suppressor genes have been implicated as either a pivotal gatekeeper of cell-cycle progression (Dammann et al, 2000) or a molecule able to reverse the malignant phenotype and to induce tumour cell apoptosis (Ji et al, 1999), respectively.
Hypoxia-inducible factor 1α, one of the essential targets of Enz validated in this study, is getting stabilised in hypoxic conditions, inactivates tumour-suppressors (p53, PTEN), activates several oncogenic pathways (Src, HER/2, H-ras) (Zhang et al, 2007), and transactivates VEGF through hypoxia response elements, this being central to the initiation of pro-angiogenic signalling and neovascular formation (Forsythe et al, 1996). Intracellular HIF1α concentrations are tightly regulated by the von Hippel–Lindau tumour-suppressor protein, a product of the third tumour-suppressor gene upregulated by Enz in our study, and a component of the E3 ubiquitin ligase system (Karhausen et al, 2005). It is an interesting observation of this study that VHL is getting upregulated, and its targeted molecule HIF1α, in addition to VEGF that is transactivated by HIF1α, is getting downregulated upon Enz treatment. Correspondingly, previous reports have suggested these genes to be inversely regulated in NSCLC (Forsythe et al, 1996; Wang et al, 2009). In addition, u-PA and u-PAR, which are upregulated in NSCLC (Werle et al, 2004; Beyer et al, 2006), are getting downregulated upon Enz treatment in our study. Interestingly, all three of these tumour progression-related genes, u-PAR, HIF1α, and VEGFC, are either activated by the downstream axis of, or transcriptionally regulated at least in part, by the MAPK/ERK and/or PI3K/AKT pathways (Jo et al, 2003; Tsai et al, 2003; Phillips et al, 2005; Skurk et al, 2005), which are inhibited by Enz treatment (Faul et al, 2003; Graff et al, 2005).
The u-PAR is an essential molecule in the context of proteolysis, migration, invasion, and metastasis (Jo et al, 2003; Dass et al, 2008), and its function and transcriptional regulation are well studied (Laufs et al, 2006). Interestingly, in our study, Enz inhibited u-PAR promoter activity by inhibiting the gene expression and binding of Sp1, Sp3, and c-Jun(AP-1) to the u-PAR-promoter in vitro and in vivo (ChIP). In the context of suppressing u-PAR gene expression through Enz, it was an interesting initial observation in our study that the knockdown of u-PAR might be able to resensitise relatively resistant NSCLC cells and induces apoptosis after Enz treatment (Querfeld et al, 2006; Lee et al, 2008; Morgillo et al, 2008). This finding emphasises the general importance of defining critical molecular markers that are associated with a potential resistance towards novel targeted therapy compounds, because otherwise it might lead to the treatment of patients that, by their molecular condition, are unable to respond to the novel drug. In fact, this might be one of the explanations as to why initial clinical trials with Enz that were not associated with parallel translational molecular studies at the resected tumour tissue of treated patients, led to initial contradictory results (Casey et al, 2009; Richards et al, 2009; Kreisl et al, 2010; Wick et al, 2010). However, with our results we might give an additional encouragement for the initiation of more tailored-therapy clinical studies, in which putatively essential molecular markers associated with response or resistance are being measured before the decision for a certain combination of therapies in the individual patient.
In our paper, we show that Enz inhibits in vivo distant metastasis to liver and lungs in the CAM assay. Apart from its pro-apoptotic, growth inhibitory, and anti-angiogenic properties (Keyes et al, 2002; Graff et al, 2005; Querfeld et al, 2006; Willey et al, 2009), this could be, at least in part, due to downregulating tumour progression and invasion-related genes (u-PAR, HIF1α, and VEGFC) and upregulating NSCLC tumour-related suppressor genes (FHIT, RASSF1, and VHL) by Enz. We consider it unlikely that a main mechanism of metastasis inhibition by Enz can be explained by anti-angiogenesis, because we observed that DMSO- or Enz-dipped plugs did not show any significant difference in their effect on the formation of embryonic vasculature of the upper CAM (data not shown). However, certainly, an anti-angiogenic component of Enz in the context of anti-metastatic action in our model cannot be ruled out completely.
Taken together, this is the first comprehensive analysis to investigate differentially regulated genes in NSCLC cells upon Enz treatment, and to implicate novel Enz targets in the context of migration, invasion, and distant metastasis in NSCLC. Our results suggest that Enz has a major role in downregulating the u-PAR through Sp1/Sp3 and c-Jun(AP-1), besides HIF1α and VEGFC that are implicated in tumour growth and metastasis, in parallel to upregulating tumour-suppressors relevant for NSCLC. Furthermore, the knockdown of u-PAR re-sensitises NSCLC cells to Enz treatment. These findings give important information for ongoing clinical trials with Enz in NSCLC patients, and further studies investigating resected tumour tissue or blood samples of Enz treated patients for, for example, u-PAR or serum-soluble u-PAR (Henic et al, 2008), as a potentially clinically relevant marker of therapy resistance are clearly encouraged.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ali S, Al-Sukhun S, El-Rayes BF, Sarkar FH, Heilbrun LK, Philip PA (2009) Protein kinases C isozymes are differentially expressed in human breast carcinomas. Life Sci 84: 766–771
Allgayer H, Wang H, Gallick GE, Crabtree A, Mazar A, Jones T, Kraker AJ, Boyd DD (1999) Transcriptional induction of the urokinase receptor gene by a constitutively active Src. Requirement of an upstream motif (-152/-135) bound with Sp1. J Biol Chem 274: 18428–18437
Balendran A, Hare GR, Kieloch A, Williams MR, Alessi DR (2000) Further evidence that 3-phosphoinositidedependent protein kinase-1 (PDK1) is required for the stability and phosphorylation of protein kinase C (PKC) isoforms. FEBS Lett 484: 217–223
Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, Lizyness ML, Kuick R, Hayasaka S, Taylor IM, Iannettoni MD, Orringer MB, Hanash S (2002) Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med 8: 816–824
Beyer BC, Heiss MM, Simon EH, Gruetzner KU, Babic R, Jauch KW, Schildberg FW, Allgayer H (2006) Urokinase system expression in gastric carcinoma: prognostic impact in an independent patient series and first evidence of predictive value in preoperative biopsy and intestinal metaplasia specimens. Cancer 106: 1026–1035
Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, Loda M, Weber G, Mark EJ, Lander ES, Wong W, Johnson BE, Golub TR, Sugarbaker DJ, Meyerson M (2001) Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci USA 98: 13790–13795
Borczuk AC, Gorenstein L, Walter KL, Assaad AA, Wang L, Powell CA (2003) Non-small-cell lung cancer molecular signatures recapitulate lung developmental pathways. Am J Pathol 163: 1949–1960
Carducci MA, Musib L, Kies MS, Pili R, Truong M, Brahmer JR, Cole P, Sullivan R, Riddle J, Schmidt J, Enas N, Sinha V, Thornton DE, Herbst RS (2006) Phase I dose escalation and pharmacokinetic study of enzastaurin, an oral protein kinase C beta inhibitor, in patients with advanced cancer. J Clin Oncol 24: 4092–4099
Casey EM, Harb W, Bradford D, Bufill J, Nattam S, Patel J, Fisher W, Latz JE, Wu J, Hanna N (2009) Randomized, double blind, multicenter, phase II study of pemetrexed (PEM), carboplatin (CARBO), bevacizumab (BEV) with enzastaurin (ENZ) or placebo (PBO) in chemotherapy-naive patients with stage IIIB/IV non-small cell lung cancer (NSCLC) (Abstract no. 8035). J Clin Oncol 27: 15s
Chen JJ, Peck K, Hong TM, Yang SC, Sher YP, Shih JY, Wu R, Cheng JL, Roffler SR, Wu CW, Yang PC (2001) Global analysis of gene expression in invasion by a lung cancer model. Cancer Res 61: 5223–5230
Clark AS, West KA, Blumberg PM, Dennis PA (2003) Altered protein kinase C (PKC) isoforms in non-small cell lung cancer cells: PKCdelta promotes cellular survival and chemotherapeutic resistance. Cancer Res 63: 780–786
da Rocha AB, Mans DR, Regner A, Schwartsmann G (2002) Targeting protein kinase C: new therapeutic opportunities against high-grade malignant gliomas? Oncologist 7: 17–33
Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP (2000) Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet 25: 315–319
Dass K, Ahmad A, Azmi AS, Sarkar SH, Sarkar FH (2008) Evolving role of uPA/uPAR system in human cancers. Cancer Treat Rev 34: 22–36
Diederichs S, Bulk E, Steffen B, Ji P, Tickenbrock L, Lang K, Zänker KS, Metzger R, Schneider PM, Gerke V, Thomas M, Berdel WE, Serve H, Müller-Tidow C (2004) S100 family members and trypsinogens are predictors of distant metastasis and survival in early-stage non-small cell lung cancer. Cancer Res 64: 5564–5569
Douglas H, Robert AW (2000) The hallmarks of cancer. Cell 100: 57–70
Eberwine J, Spencer C, Miyashiro K, Mackler S, Finnell R (1992) Complementary DNA synthesis in situ: methods and applications. Methods Enzymol 216: 80–100
Fang X, Yu S, Tanyi JL, Lu Y, Woodgett JR, Mills GB (2002) Convergence of multiple signaling cascades at glycogen synthase kinase 3: Edg receptor-mediated phosphorylation and inactivation by lysophosphatidic acid through a protein kinase C-dependent intracellular pathway. Mol Cell Biol 22: 2099–2110
Faoro L, Cervantes GM, Ferguson BD, Seiwert TY, Yala S, Vigneswaran WT, Westerhoff M, Tretiakova MS, Ferguson MK, Moura GL, Husain AN, Vokes EE, Salgia R (2009) MET/PKCss expression correlate with metastasis and inhibition is synergistic in lung cancer. J Carcinog 8: 15
Faul MM, Gillig JR, Jirousek MR, Ballas LM, Schotten T, Kahl A, Mohr M (2003) Acyclic N-(azacycloalkyl)bisindolylmaleimides: isozyme selective inhibitors of PKCbeta. Bioorg Med Chem Lett 13: 1857–1859
Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL (1996) Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 16: 4604–4613
Gemma A, Takenaka K, Hosoya Y, Matuda K, Seike M, Kurimoto F, Ono Y, Uematsu K, Takeda Y, Hibino S, Yoshimura A, Shibuya M, Kudoh S (2001) Altered expression of several genes in highly metastatic subpopulations of a human pulmonary adenocarcinoma cell line. Eur J Cancer 37: 1554–1561
Giovannetti E, Honeywell R, Hanauske AR, Tekle C, Kuenen B, Sigmond J, Giaccone G, Peters GJ (2010) Pharmacological aspects of the enzastaurin-pemetrexed combination in non-small cell lung cancer (NSCLC). Curr Drug Targets 11: 12–28
Goekjian PG, Jirousek MR (2001) Protein kinase C inhibitors as novel anticancer drugs. Expert Opin Investig Drugs 10: 2117–2140
Gokmen-Polar Y, Murray NR, Velasco MA, Gatalica Z, Fields AP (2001) Elevated protein kinase C betaII is an early promotive event in colon carcinogenesis. Cancer Res 61: 1375–1381
Graff JR, McNulty AM, Hanna KR, Konicek BW, Lynch RL, Bailey SN, Banks C, Capen A, Goode R, Lewis JE, Sams L, Huss KL, Campbell RM, Iversen PW, Neubauer BL, Brown TJ, Musib L, Geeganage S, Thornton D (2005) The protein kinase Cbeta-selective inhibitor, Enzastaurin (LY317615.HCl), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res 65: 7462–7469
Gridelli C, Langer C, Maione P, Rossi A, Schild SE (2007) Lung cancer in the elderly. J Clin Oncol 25: 1898–1907
Guo K, Li Y, Kang X, Sun L, Cui J, Gao D, Liu Y (2009) Role of PKCbeta in hepatocellular carcinoma cells migration and invasion in vitro: a potential therapeutic target. Clin Exp Metastasis 26: 189–195
Hanauske AR, Lahn M, Musib LC, Weigang-Köhler K, Yilmaz E, Graefe T, Kuenen B, Thornton D, McNealy P, Giaccone G (2009) Phase Ib safety and pharmacokinetic evaluation of daily and twice daily oral enzastaurin in combination with pemetrexed in advanced/metastatic cancer. Ann Onco 20: 1565–1575
Hayes DN, Monti S, Parmigiani G, Gilks CB, Naoki K, Bhattacharjee A, Socinski MA, Perou C, Meyerson M (2006) Gene expression profiling reveals reproducible human lung adenocarcinoma subtypes in multiple independent patient cohorts. J Clin Oncol 24: 5079–5090
Heighway J, Knapp T, Boyce L, Brennand S, Field JK, Betticher DC, Ratschiller D, Gugger M, Donovan M, Lasek A, Rickert P (2002) Expression profiling of primary non-small cell lung cancer for target identification. Oncogene 21: 7749–7763
Hellmann GM, Fields WR, Doolittle DJ (2001) Gene expression profiling of cultured human bronchial epithelial and lung carcinoma cells. Toxicol Sci 61: 154–163
Henic E, Borgfeldt C, Christensen IJ, Casslén B, Høyer-Hansen G (2008) Cleaved forms of the urokinase plasminogen activator receptor in plasma have diagnostic potential and predict postoperative survival in patients with ovarian cancer. Clin Cancer Res 14: 5785–5793
Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, Feuer EJ, Thun MJ (2005) Cancer statistics. CA Cancer J Clin 55: 10–30
Ji L, Fang B, Yen N, Fong K, Minna JD, Roth JA (1999) Induction of apoptosis and inhibition of tumorigenicity and tumor growth by adenovirus vector-mediated fragile histidine triad (FHIT) gene overexpression. Cancer Res 59: 3333–3339
Jo M, Thomas KS, O’Donnell M, Gonias SL (2003) Epidermal growth factor receptor-dependent and -independent cell-signaling pathways originating from the urokinase receptor. J Biol Chem 278: 1642–1646
Karhausen J, Kong T, Narravula S, Colgan SP (2005) Induction of the von Hippel-Lindau tumor suppressor gene by late hypoxia limits HIF-1 expression. J Cell Biochem 95: 1264–1275
Keyes K, Cox K, Treadway P, Mann L, Shih C, Faul MM, Teicher BA (2002) An in vitro tumor model: analysis of angiogenic factor expression after chemotherapy. Cancer Res 62: 5597–5602
Kreisl TN, Kotliarova S, Butman JA, Albert PS, Kim L, Musib L, Thornton D, Fine HA (2010) A phase I/II trial of enzastaurin in patients with recurrent high-grade gliomas. Neuro Oncol 12: 181–189
Laufs S, Schumacher J, Allgayer H (2006) Urokinase-receptor (u-PAR): an essential player in multiple games of cancer: a review on its role in tumor progression, invasion, metastasis, proliferation/dormancy, clinical outcome and minimal residual disease. Cell Cycle 5: 1760–1771
Lee KW, Kim SG, Kim HP, Kwon E, You J, Choi HJ, Park JH, Kang BC, Im SA, Kim TY, Kim WH, Bang YJ (2008) Enzastaurin, a protein kinase C beta inhibitor, suppresses signaling through the ribosomal S6 kinase and bad pathways and induces apoptosis in human gastric cancer cells. Cancer Res 68: 1916–1926
Leupold JH, Asangani I, Maurer GD, Lengyel E, Post S, Allgayer H (2007) Src induces urokinase receptor gene expression and invasion/intravasation via activator protein-1/p-c-Jun in colorectal cancer. Mol Cancer Res 5: 485–496
Marchetti A, Tinari N, Buttitta F, Chella A, Angeletti CA, Sacco R, Mucilli F, Ullrich A, Iacobelli S (2002) Expression of 90K (Mac-2 BP) correlates with distant metastasis and predicts survival in stage I non-small cell lung cancer patients. Cancer Res 62: 2535–2539
Morgillo F, Martinelli E, Troiani T, Laus G, Pepe S, Gridelli C, Ciardiello F (2008) Sequence-dependent, synergistic antiproliferative and proapoptotic effects of the combination of cytotoxic drugs and enzastaurin, a protein kinase Cbeta inhibitor, in non-small cell lung cancer cells. Mol Cancer Ther 7: 1698–1707
Mudduluru G, Allgayer H (2008) The human receptor tyrosine kinase Axl gene – promoter characterization and regulation of constitutive expression by Sp1, Sp3 and CpG methylation. Biosci Rep 28: 161–176
Müller-Tidow C, Metzger R, Kügler K, Diederichs S, Idos G, Thomas M, Dockhorn-Dworniczak B, Schneider PM, Koeffler HP, Berdel WE, Serve H (2001) Cyclin E is the only cyclin-dependent kinase 2-associated cyclin that predicts metastasis and survival in early stage nonsmall cell lung cancer. Cancer Res 61: 647–653
Nikolova DA, Asangani IA, Nelson LD, Hughes DP, Siwak DR, Mills GB, Harms A, Buchholz E, Pilz LR, Manegold C, Allgayer H (2009) Cetuximab attenuates metastasis and u-PAR expression in non-small cell lung cancer: u-PAR and E-cadherin are novel biomarkers of cetuximab sensitivity. Cancer Res 69: 2461–2470
Phillips RJ, Mestas J, Gharaee-Kermani M, Burdick MD, Sica A, Belperio JA, Keane MP, Strieter RM (2005) Epidermal growth factor and hypoxia-induced expression of CXC chemokine receptor 4 on non-small cell lung cancer cells is regulated by the phosphatidylinositol 3-kinase/PTEN/AKT/mammalian target of rapamycin signaling pathway and activation of hypoxia inducible factor-1alpha. J Biol Chem 280: 22473–22481
Querfeld C, Rizvi MA, Kuzel TM, Guitart J, Rademaker A, Sabharwal SS, Krett NL, Rosen ST (2006) The selective protein kinase C beta inhibitor enzastaurin induces apoptosis in cutaneous T-cell lymphoma cell lines through the AKT pathway. J Invest Dermatol 126: 1641–1647
Richards DA, Kuefler PR, Becerra C, Wilfong LS, Gersh RH, Boehm KA, Zhan F, Asmar L, Myrand SP, Hozak RR, Zhao L, Gill JF, Mullaney BP, Obasaju CK, Nicol SJ (2009) Gemcitabine plus enzastaurin or single-agent gemcitabine in locally advanced or metastatic pancreatic cancer: results of a Phase II, randomized, noncomparative study. Invest New Drugs [Epub ahead of print]
Shipp MA, Ross KN, Tamayo P, Weng AP, Kutok JL, Aguiar RC, Gaasenbeek M, Angelo M, Reich M, Pinkus GS, Ray TS, Koval MA, Last KW, Norton A, Lister TA, Mesirov J, Neuberg DS, Lander ES, Aster JC, Golub TR (2002) Diffuse large B cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med 8: 68–74
Skurk C, Maatz H, Rocnik E, Bialik A, Force T, Walsh K (2005) Glycogen-synthase kinase3beta/beta-catenin axis promotes angiogenesis through activation of vascular endothelial growth factor signaling in endothelial cells. Circ Res 96: 308–318
Tekle C, Giovannetti E, Sigmond J, Graff JR, Smid K, Peters GJ (2008) Molecular pathways involved in the synergistic interaction of the PKC beta inhibitor enzastaurin with the antifolate pemetrexed in non-small cell lung cancer cells. Br J Cancer 99: 750–759
Tsai PW, Shiah SG, Lin MT, Wu CW, Kuo ML (2003) Up-regulation of vascular endothelial growth factor C in breast cancer cells by heregulin-beta 1. A critical role of p38/nuclear factor-kappa B signaling pathway. J Biol Chem 278: 5750–5759
Wang J, Zhao W, Guo Y, Zhang B, Xie Q, Xiang D, Gao J, Wang B, Chen Z (2009) The expression of RNA-binding protein HuR in non-small cell lung cancer correlates with vascular endothelial growth factor-C expression and lymph node metastasis. Oncology 76: 420–429
Watkins V, Hong S, Lin B (2006) Enzastaurin safety review: data from phase I and phase II trials. J Clin Oncol 24: 608
Werle B, Kotzsch M, Lah TT, Kos J, Gabrijelcic-Geiger D, Spiess E, Schirren J, Ebert W, Fiehn W, Luther T, Magdolen V, Schmitt M, Harbeck N (2004) Cathepsin B, plasminogenactivator-inhibitor (PAI-1) and plasminogenactivator-receptor (uPAR) are prognostic factors for patients with non-small cell lung cancer. Anticancer Res 24: 4147–4161
Wick W, Puduvalli VK, Chamberlain MC, van den Bent MJ, Carpentier AF, Cher LM, Mason W, Weller M, Hong S, Musib L, Liepa AM, Thornton DE, Fine HA (2010) Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J Clin Oncol 28: 1168–1174
Wikman H, Kettunen E, Seppänen JK, Karjalainen A, Hollmén J, Anttila S, Knuutila S (2002) Identification of differentially expressed genes in pulmonary adenocarcinoma by using cDNA array. Oncogene 21: 5804–5813
Willey CD, Xiao D, Tu T, Woon Kim K, Moretti L, Niermann KJ, Tawtawy MN, Quarles CC, Lu B (2009) Enzastaurin (LY317615), a protein kinase C beta selective inhibitor, enhances antiangiogenic effect of radiation. Int J Radiat Oncol Biol Phys 20: 1565–1575
Zhang Q, Tang X, Zhang ZF, Velikina R, Shi S, Le AD (2007) Nicotine induces hypoxia-inducible factor-1alpha expression in human lung cancer cells via nicotinic acetylcholine receptor-mediated signaling pathways. Clin Cancer Res 13: 4686–4694
Zochbauer-Muller S, Gazdar AF, Minna JD (2002) Molecular pathogenesis of lung cancer. Annu Rev Physiol 64: 681–708
Acknowledgements
This project was partly supported by Lilly (Indianapolis, IN, USA). HA was supported by Alfried Krupp von Bohlen und Halbach Foundation (Award for Young Full Professors; Essen), Hella-Bühler-Foundation (Heidelberg), Dr Ingrid zu Solms Foundation (Frankfurt/Main), the Hector Foundation (Weinheim, Germany), the FRONTIER Excellence Initiative of the University of Heidelberg, and the Walter Schulz Foundation (Munich, Germany). This publication contains parts of the dissertation (Dr sc. hum.) of Alexandra Körner at Mannheim Faculty of Medicine, University of Heidelberg, Germany. We thank Dr Bernhard Korn for his support in microarray analysis and Erika Hillerich and Paolo Ceppi for excellent help and critical appraisal of the paper.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Körner, A., Mudduluru, G., Manegold, C. et al. Enzastaurin inhibits invasion and metastasis in lung cancer by diverse molecules. Br J Cancer 103, 802–811 (2010). https://doi.org/10.1038/sj.bjc.6605818
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6605818
Keywords
This article is cited by
-
The protein kinase C (PKC) inhibitors combined with chemotherapy in the treatment of advanced non-small cell lung cancer: meta-analysis of randomized controlled trials
Clinical and Translational Oncology (2015)
-
Enzastaurin has anti-tumour effects in lung cancers with overexpressed JAK pathway molecules
British Journal of Cancer (2012)
