
Abstract
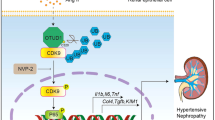
It is known that angiotensin (Ang)-converting enzyme (ACE) 2 catalyzes Ang II to Ang 1–7 to prevent the detrimental effect of Ang II on blood pressure, renal fibrosis, and inflammation. However, mechanisms of renoprotective role of Ace2 remain largely unclear. The present study tested the hypothesis that deficiency of Ace2 may accelerate intrarenal Ang II-mediated fibrosis and inflammation independent of blood pressure in a model of unilateral ureteral obstructive (UUO) nephropathy induced in Ace2+/y and Ace2−/y mice. Results showed that both Ace2+/y and Ace2−/y mice had normal levels of blood pressure and plasma Ang II/Ang 1–7. In contrast, deletion of ACE2 resulted in a fourfold increase in the ratio of intrarenal Ang II/Ang 1–7 in the UUO nephropathy. These changes were associated with the development of more intensive tubulointerstitial fibrosis (α-SMA, collagen I) and inflammation (TNF-α, IL-1β, MCP-1, F4/80+ cells, and CD3+T cells) in Ace2−/y mice at day 3 (all P<0.05) after UUO, becoming more profound at day 7 (all P<0.01). Enhanced renal fibrosis and inflammation in the UUO kidney of Ace2−/y mice were largely attributed to a marked increase in the intrarenal Ang II signaling (AT1-ERK1/2 mitogen-activated protein kinase), TGF-β/Smad2/3, and NF-κB signaling pathways. Further studies revealed that enhanced TGF-β/Smad and NF-κB signaling in the UUO kidney of Ace2−/y mice was associated with upregulation of an E3 ligase Smurf2 and a loss of renal Smad7. In conclusion, enhanced Ang II-mediated TGF-β/Smad and NF-κB signaling may be the mechanisms by which loss of Ace2 enhances renal fibrosis and inflammation. Smad7 ubiquitin degradation mediated by Smurf2 may be a central mechanism by which Ace2−/y mice promote TGF-β/Smad2/3-mediated renal fibrosis and NF-κB-driven renal inflammation in a mouse model of UUO nephropathy.
Similar content being viewed by others


Main
The renin–angiotensin system has been recognized for many years as a critical pathway leading to chronic kidney disease. Renin promotes the production of angiotensin I (Ang I), which is converted to Ang II by Ang I-converting enzyme (ACE) and also possibly by other enzymes.1 Ang II, as an important active peptide, exerts a variety of biological and pathological effects on several target tissues and organs, including blood vessels, kidney, and heart under normal and disease conditions. ACE 2 is the homolog of ACE but counterbalances the ACE activity via promoting Ang II degradation to the vasodilator peptide Ang 1–7. Ang 1–7 is a biologically active peptide and acts on the Mas receptor to exert the opposite effect on Ang II.1 Ace2 is highly expressed in the normal kidney, largely by tubular epithelial cells.2 Increasing evidence shows that Ace2 has an essential role in the cardiovascular and kidney diseases.1, 3, 4 Ace2 inhibition also leads to the development of albuminuria in a mouse model of diabetes.5, 6
Direct evidence for ACE2 in the development of hypertensive kidney disease comes from the Ace2 gene knockout mice. Loss of Ace2 leads to the late development of glomerulosclerosis by 12 months of the age, and accelerates kidney injury in mouse models of diabetes and Ang II infusion.7, 8, 9 Recent finding that administration of human recombinant ACE2 inhibits progression of diabetic kidney disease clearly demonstrated a renoprotective role for ACE2 in the progression of chronic kidney diseases.10 All these studies suggest a negative regulatory role for ACE2 in blood pressure and Ang II-mediated hypertensive and diabetic nephropathy. The function of ACE2 in non-hypertension and diabetic condition was not yet clear. Furthermore, the mechanisms of ACE2 in protection against Ang II-mediated renal injury remain largely unclear. Thus, the present study aimed to investigate the potential role of Ace2 in chronic kidney disease independent of hypertension and diabetes and explored the renoprotective mechanisms of Ace2 in progressive renal fibrosis and inflammation.
As a mouse model of UUO is a well-established chronic kidney disease model without underlying hypertensive and diabetic conditions, we thus induced the UUO nephropathy in Ace2−/y mice and examined the hypothesis that loss of Ace2 may promote intrarenal Ang II-mediated renal fibrosis and inflammation independent of blood pressure. In addition, the mechanisms by which loss of Ace2 enhances renal fibrosis and inflammation were investigated.
MATERIALS AND METHODS
Obstructive Kidney Disease Model
Ace2−/y mice (C57BL/6 background) were generated as described previously.11 A mouse model of UUO nephropathy was induced in the littermate male Ace2+/y and Ace2–/y mice (20 g body weight, 8 weeks of age) by the left ureteral ligation as described previously.12 To investigate the role of Ace2 in the early and late stages of UUO, groups of eight Ace2+/y or Ace2–/y mice were killed at day 3 and day 7 after UUO. In addition, six age-matched Ace2+/y or Ace2–/y mice were received sham operation as control. The experimental procedures were approved by the Animal Experimental Committee at the Chinese University of Hong Kong. Kidney tissue samples were collected at day 3 and day 7 after UUO for histology, immunohistochemistry, western blot, and real-time PCR analyses.
Histology and Immunohistochemistry
Changes in renal morphology were examined in methyl Carnoy's fixed, paraffin-embedded tissue sections (4 μm) using the Masson trichrome staining. Immunohistochemistry was performed on paraffin sections using a microwave-based antigen retrieval technique.12, 13 Antibodies used in this study included rabbit antibodies recognized mouse phospho-Smad2/3, MCP-1, IL-1β, TNF-α, TGF-β1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), collagen I (Southern Tech, Birmingham, AL, USA), α-SMA (Sigma, St Louis, MO, USA), and rat anti-mouse monoclonal antibody to macrophages (F4/80) (Serotec, Oxford, UK) and rabbit polyclonal antibodies to CD3+ T cells (Abcam, Cambridge, UK). All slides (except α-SMA and phospho-Smad2/3 stained sections) were counterstained with hematoxylin.
Quantitation of immunostaining was carried on coded slides as previously described.12, 13, 14 IL-1β, TNF-α, and MCP-1 in the entire cortical tubulointerstitium (a cross-section of the renal cortex) were determined by the quantitative Image-Pro plus software (Media Cybernetics, Bethesda, MD, USA) as previously described.12, 13, 14 The number of F4/80+ cells in the tubulointerstitium was counted under high-power fields ( × 40 objective) by means of a 0.0625-mm2 graticule fitted in the eyepiece of the microscope and expressed as cells per millimeters squared (mm2).
Measurement of Blood Pressure and Ang II and Ang 1–7
Blood pressure was measured by tail-cuff method using the CODA non-invasive blood pressure system (Kent Scientific, Torrington, CT, USA) in conscious mice according to the manufacturer’s instruction. Both plasma and intrarenal Ang II and Ang 1–7 were measured using commercially available enzyme immunoassay kits (Peninsula Laboratories, San Carlos, CA, USA). Briefly, the kidney tissue was weighed, homogenized in 1 ml methanol on ice. Then, the samples were centrifuged at 12 000 g at 4°C for 10 min and the supernatant was collected and dried by evaporation. The dried samples were then reconstituted with EIA buffer. Blood plasma was collected from mice, and an equal amount of buffer A was added. The mixture was centrifuged at 17 000 g for 20 min at 4°C and the supernatant was passed through the pre-treated C-18 SEP-COLUMN. The elute peptide was freezer dried and was dissolved in EIA buffer. Concentrations of Ang II and Ang 1–7 were measured by the ELISA kits following the manufacturer's instructions.
Western Blot Analysis
Renal tissues were collected by carefully removing the renal pelvis and medullar tissues, and were frozen at −80°C freezer for western blot analysis as previously described.12, 13, 14 Briefly, after the protein was transferred onto a nitrocellulose membrane, the membrane was incubated overnight with primary antibodies against phospho-Smad3, phospho-IκBα (ser32), phospho-NF-κB/p65 (ser276), ERK1/2 (Cell Signaling Technology, MA, USA); IκBα, NF-κB/p65, phospho-ERK1/2, Smad2/3, Smad7, ACE2, Smurf2, Ang II receptor-1 (AT1) (Santa Cruz Biotech); α-SMA (Sigma), collagen I (Southern Tech), and GAPDH (Chemicon, Temecula, CA, USA) followed by the LI-COR IRDye 800-labeled secondary antibodies (Rockland Immunochemicals, Gilbertsville, PA, USA) in dark for 1 h at room temperature. Signals were scanned and visualized by Odyssey Infrared Imaging System (LiCor, Lincoln, NE, USA). The ratio of the protein interested was subjected to GAPDH and was densitometrically analyzed by the Image J software (NIH, Bethesda, MD, USA).
Real-Time PCR
Renal mRNA expression was quantitatively analyzed by real-time PCR with primers against mouse mRNA of IL-1β, TNF-α, MCP-1, TGF-β1, Smad7, collagen I, α-SMA, and GAPDH as described previously,12, 13, 14 whereas the primers for AT1 and Ace2 were described below: AT1: forward 5′-TGACTTTGGCCACCAGCAT-3′, reverse 5′-CCATTGTCCACCCGATGAA-3′; Ace2: forward 5′-ACCCTTCTTACATCAGCCCTACTG -3′, reverse 5′-TGTCCAAAACCTACCCCACATAT-3′. The reaction specificity was confirmed by melting curve analysis. The housekeeping gene GAPDH was used as an internal standard and the ratio of the mRNA examined to the GAPDH was calculated.
Statistical Analyses
Data obtained from this study were expressed as the mean±s.e.m. Statistical analyses were performed using one-way ANOVA, followed by Newman–Keuls Post Test from Prism 5.0 GraphPad Software (San Diego, CA, USA).
RESULTS
Ace2–/y Mice Promote Renal Fibrosis and Inflammation Independent of Blood Pressure in the UUO Nephropathy
Western blot analysis showed that Ace2 protein was not detectable in Ace2−/y mice, but was increased in the diseased kidney of Ace2+/y mice (Figure 1a and c). Both Ace2+/y and Ace2−/y mice had similar levels of blood pressure under sham-control (126±0.5 mm Hg in Ace2+/y mice vs 122±5.8 mm Hg in Ace2−/y mice) or UUO disease conditions over 7 days (125.8±2 mm Hg in Ace2+/y mice vs 121±5.5 mm Hg in Ace2−/y mice). Histologically, Masson trichrome staining showed no detectable abnormalities in normal or sham-operation Ace2−/y mice compared with Ace2+/y mice (Figure 1b and d). However, 3 days after UUO, Ace2−/y mice developed more intensive tubulointerstitial damage, including tubular atrophy, interstitial extracellular matrix accumulation, which became much more profound at day 7 after UUO (Figure 1b and d). Immunohistochemically, compared to the Ace2+/y mice, tubulointerstitial fibrosis was enhanced in Ace2−/y mice as demonstrated by more abundant collagen I (Figure 1e) and α-SMA (Figure 1i) accumulation in the fibrotic area of tubulointerstitium. Quantitative real-time PCR and western blot analyses further confirmed these findings that disrupted Ace2 largely enhanced collagen I and α-SMA mRNA and protein expression in the UUO kidney when compared with Ace2+/y mice (Figure 1f–h and j–l).

Mice lacking Ace2 (KO) are promoted histological damage and renal fibrosis in the UUO nephropathy. (a) Western blot analysis of Ace2 expression in the kidney. (b) Masson's trichrome staining. (c) Semi-quantitative analysis of Ace2 protein expression detected by western blotting. (d) Semi-quantitative analysis of matrix protein accumulation detected by Masson's trichrome staining. (e) Immunohistochemical staining of collagen I. (f) Renal collagen I expression detected by western blot. (g) Semi-quantitative analysis of collagen I detected by western blotting. (h) Collagen I mRNA expression detected by real-time PCR. (i) Immunohistochemical staining of α-SMA expression. (j) α-SMA expression detected by western blot. (k) Semi-quantitative analysis of α-SMA protein expression by western blotting. (l) α-SMA mRNA expression detected by real-time PCR. Each bar represents mean±s.e.m. for at least six mice. *P<0.05, ***P<0.001 compared with sham-operation mice. #P<0.05, ##P<0.01, ###P<0.001 when compared with time-matched Ace2+/y (WT) mice with UUO. Magnification × 200.
As inflammation is a critical process in the development of UUO, we next examined whether disruption of Ace2 gene influences renal inflammation in the UUO kidney. As shown in Figure 2, immunohistochemistry and real-time PCR revealed that compared with Ace2+/y mice, mice deficient for Ace2 exhibited a substantial increase in renal inflammation as demonstrated by a remarkable upregulation of pro-inflammatory cytokines (TNF-α, IL-1β) and chemokine MCP-1. Enhanced expression of renal TNF-α, IL-1β, and MCP-1 in Ace2–/y mice was accompanied by a significant increase in CD3+ T cells and F4/80+ cells infiltrating the tubulointerstitium (Figure 3).


Mice lacking Ace2 (KO) are promoted renal inflammation in the UUO nephropathy. (a) Immunohistochemical staining of TNF-α expression. (b) Semi-quantitative analysis of TNF-α immunostaining. (c) TNF-α mRNA expression detected by real-time PCR. (d) Immunohistochemical staining of IL-1β. (e) Semi-quantitative analysis of IL-1β immunostaining. (f) IL-1β mRNA expression detected by real-time PCR. (g) Immunohistochemical staining of MCP-1 expression; (h) Semi-quantitative analysis of MCP-1 immunostaining. (i) MCP-1 mRNA expression detected by real-time PCR. Each bar represents mean±s.e.m. for at least six mice. **P<0.01, ***P<0.001 compared with sham-operation mice. #P<0.05, ##P<0.01, when compared with time-matched Ace2+/y (WT) mice with UUO. Magnification × 200.

Mice lacking Ace2 (KO) are promoted CD3+ T-cell and F4/80+ cell infiltration in the UUO nephropathy. (a) Immunohistochemical staining of CD3+ T cells. (b) Immunohistochemical staining of F4/80+ cell. (c) Quantitative analysis of CD3+ T cells. (d) Immunohistochemical staining of F4/80+ cell. CD3+ T-cell and F4/80+ cell infiltration in the tubulointerstitium are significantly increased in Ace2 −/y (KO) mice at day 3 after UUO, which becomes maximal at day 7. Each bar represents mean±s.e.m. for at least six mice. ***P<0.001 compared with sham-operation mice. ###P<0.001 when compared with time-matched Ace2+/y (WT) mice with UUO. Magnification × 200.
Enhanced Ang II, TGF-β/Smad, and NF-κB Signaling Pathways Are Key Mechanisms by which Ace2−/y Mice Promote Renal Fibrosis and Inflammation in the UUO Nephropathy
It has been shown that Ang II is capable of activating the Smads to mediate fibrosis via both AT1-MAP kinase-crosstalk and TGF-β1-dependent pathways.15, 16, 17 We thus investigated whether enhanced renal fibrosis in the UUO kidney of Ace2−/y mice is associated with an increase in Ang II-mediated TGF-β/Smad pathway. As shown in Figure 4a and b, significant higher levels of intrarenal Ang II and Ang 1–7 were detected in Ace2+/y mice after UUO. Deletion of Ace2 resulted in doubling the intrarenal Ang II in the UUO kidney, while levels of intrarenal Ang 1–7 remained low (Figure 4a and b). Interestingly, the imbalance between Ang II generation and degradation occurred only locally in the UUO kidney, as plasma levels of Ang II and Ang 1–7 remained normal in Ace2+/y and Ace2−/y mice throughout the 7-day disease course (Figure 4c and d). Western blot and real-time PCR analyses detected that increased intrarenal Ang II in Ace2−/y mice was associated with a marked activation of the intrarenal Ang II signaling pathway, including upregulation of AT1 and activation of ERK1/2 mitogen-activated protein kinase (Figure 4e–h). Further study revealed that enhanced Ang II signaling in the UUO kidney of Ace2−/y mice was accompanied by a marked increase in renal TGF-β1 expression (Figure 5a, c and d) and higher levels of Smad2/3 phosphorylation and phospho-Smad2/3 nuclear translocation when compared with Ace2+/y mice (Figure 5b and e–g).

Mice lacking Ace2 (KO) are promoted intrarenal Ang II and Ang II signaling in the UUO nephropathy. (a, b) Intrarenal Ang II and Ang 1–7 concentrations measured by ELISA. (c, d) Plasma levels of Ang II and Ang 1–7 measured by ELISA. (e) Western blot analysis of AT1 and phospho-ERK1/2. (f, g) Semi-quantitative analysis of AT1 protein and phospho-ERK1/2 protein by western blots. (h) AT1 mRNA expression detected by real-time PCR. Results show that deletion of Ace2 largely enhances intrarenal, but not circulating, Ang II concentrations, which results in the enhancement of AT1-dependent intrarenal Ang II signaling. Each bar represents mean±s.e.m. for at least six mice. *P<0.05, ***P<0.001 compared with sham-operation mice. #P<0.05, ##P<0.01, ###P<0.001 when compared with time-matched Ace2+/y (WT) mice with UUO.

Disrupted Ace2 enhances TGF-β/Smad signaling in the UUO nephropathy. (a) Immunohistochemical staining of TGF-β1 expression. (b) Immunohistochemical staining of phospho-Smad2/3 nuclear translocation. (c) Semi-quantitative analysis of TGF-β1 immunostaining. (d) TGF-β1 mRNA expression detected by real-time PCR. (e) Semi-quantitative analysis of phospho-Smad2/3 nuclear translocation. (f) Phosphorylation of Smad2/3 (P-Smad2/3) detected by western blots. (g) Semi-quantitative analysis of phosphorylation of Smad2/3 (P-Smad2/3) by western blotting. Results show that, compared to Ace2+/y (WT) mice, disrupted Ace2 (KO) largely enhances TGF-β1 mRNA expression and protein, resulting in a further increase in Smad2/3 signaling as demonstrated by its nuclear translocation and phosphorylation. Each bar represents mean±s.e.m. for at least six mice. ***P<0.001 compared with sham-operation mice. #P<0.05, ##P<0.01 when compared with time-matched Ace2+/y (WT) mice with UUO. Magnification × 200.
We then determined the mechanism by which deletion of Ace2 promotes renal inflammation in the UUO kidney by examining the NF-κB signaling pathway. As shown in Figure 6, western blot analysis revealed that compared to Ace2+/y mice, enhanced activation of the NF-κB pathway such as an increase in the levels of phospho-IκBα and phospho-NF-κB/p65 was accompanied by degradation of IκBα and NF-κB/p65 in the UUO kidney of Ace2−/y mice (Figure 6).

Disrupted Ace2 enhances NF-κB signaling in the UUO nephropathy. (a) Representative western blots. (b) Semi-quantitative analysis of phospho-IκBα. (c) Semi-quantitative analysis of phospho-NF-κB/p65. Results show that compared to Ace2+/y (WT) mice, deletion of Ace2 (KO) largely enhances phosphorylation of both IκBα and NF-κB/p65. Each bar represents mean±s.e.m. for at least six mice. ***P<0.001 compared with sham-operation mice. #P<0.05, ##P<0.01, ###P<0.001 when compared with time-matched Ace2+/y (WT) mice with UUO.
Loss of Renal Smad7 Is an Underlying Mechanism Required for the Promotion of TGF-β/Smad-Mediated Renal Fibrosis and NF-κB-Driven Renal Inflammation in the UUO Nephropathy in Ace2−/y Mice
We next examined whether enhanced TGF-β/Smad and NF-κB signaling in Ace2−/y mice is associated with a loss of renal Smad7 via the Smurf2-ubiquitin degradation mechanism. As shown in Figure 7, western blot analysis detected that Smurf2 expression was significantly increased in the UUO kidney of Ace2+/y mice, which was further increased in the Ace2−/y mice (Figure 7a and b). Importantly, an increase in Smurf2 expression in the UUO kidney was associated with a reduction of renal Smad7 protein in Ace2+/y mice, which became more profound in mice lacking Ace2 (Figure 7a and c). Interestingly, in contrast to Smad7 protein, real-time PCR detected that Smad7 mRNA expression was significantly increased in the UUO kidney of Ace2+/y mice, which became higher in Ace2−/y mice (Figure 7d). An increase in Smad7 mRNA but loss of Smad7 protein along with upregulation of Smurf2 suggests that the Smurf2-dependent ubiquitin degradation of renal Smad7 occurs within the UUO kidney.

Disrupted Ace2 enhances Smurf2-mediated degradation of renal Smad7 protein in the UUO nephropathy. (a) Representative western blots for renal Smurf2 and Smad7 expression. (b) Semi-quantitative analysis of Smurf2 protein. (c) Semi-quantitative analysis of Smad7 protein. (d) Real-time PCR detection of renal Smad7 mRNA. Results show that compared to Ace2+/y (WT), disrupted Ace2 (KO) largely enhances Smurf2 expression, which results in renal Smad7 protein degradation. Note that renal Smad7 mRNA is upregulated in the UUO nephropathy in both Ace2+/y and Ace2−/y mice. Each bar represents mean±s.e.m. for at least six mice. **P<0.01, ***P<0.001 compared with sham-operation mice. #P<0.05, ##P<0.01, ###P<0.001 when compared with time-matched Ace2+/y (WT) mice with UUO.
DISCUSSION
The present study provided the evidence that loss of Ace2 enhanced renal fibrosis and inflammation in a mouse model of UUO nephropathy. Results from this study also delineated a critical role of Ace2 in negatively regulating endogenous Ang II-mediated progressive renal injury independent of systemic hypertension. Thus, Ace2 is an essential regulator in maintaining the balance between Ang II generation and Ang II degradation locally within the kidney regardless of systemic disease conditions. Furthermore, the present study also found that enhanced TGF-β/Smad and NF-κB signaling pathways were mechanisms by which Ace2−/y mice promote renal fibrosis and inflammation, which was attributed to a loss of renal Smad7 mediated by a mechanism of Ang II-induced Smurf2-dependent ubiquitin-degradation pathway.
Many studies have shown that the expression of Ace2 within the kidney varies with disease conditions. In hypertensive nephropathy, we found that renal ACE2 is largely reduced along with a marked upregulation of ACE and AT1 receptor in patients with hypertension.2 This is consistent with the findings in animal models of hypertension,18, 19 although renal ACE2 expression and activity is unaltered during established hypertension in adult SHRSP and TGR(mREN2)27 rats.20 Similarly, in diabetic nephropathy, while upregulation of Ace2 is noted in the diabetic kidney in mice8, 10, 21, 22 and in humans,23 downregulation of Aces is also reported.24, 25, 26 The functional importance of Ace2 is now established by recent findings that loss of Ace2 promotes but administration of recombinant human ACE2 inhibits exogenous Ang II-induced hypertensive nephropathy and streptozotocin-induced diabetic nephropathy.8, 9, 10, 11, 27 All these findings imply that Ace2 may protect against progressive renal injury in diseases associated with hypertension and diabetes. In the present study, we found that disruption of Ace2 largely enhanced intrarenal Ang II levels (a twofold increase), which was accompanied by much more severe renal fibrosis and inflammation, demonstrating a renoprotective role for Ace2 in obstructive nephropathy.
The finding of Ace2 as a critical regulator in maintaining the balance between endogenous Ang II generation and degradation locally in a mouse model of UUO nephropathy is clinically relevant. It implies that Ace2 not only functions to systemically regulate hypertensive renal injury but also acts as a local regulator for renal protection against the endogenous Ang II-mediated injury independent of hypertension. Indeed, the present study revealed that the local, but not systemic, Ang II generation and degradation was critically regulated by Ace2 as evidenced by the finding that disruption of Ace2 impaired the ACE2/Ang 1–7 axis but enhanced largely the ACE/Ang II/AT1-dependent Ang II generation in Ace2−/y mice (Ang II vs Ang 1–7=4:1). This finding was consistent with the observation in both animals and patients with diabetes or IgA nephropathy.21, 22, 23, 24, 25, 26, 28 Thus, Ace2 is an important counter-regulator for systemic hypertension and locally in determining the disease progression by balancing the intrarenal Ang II generation and degradation pathways.
Although ACE2 is a critical enzyme to degrade Ang I and Ang II to Ang 1–7, a number of alternative enzymes, including neprilysin, prolyl-endopeptidase, and thimet oligopeptidase, as well as ACE, are also capable of converting the Ang I and Ang II to Ang 1–7.4, 29, 30 Thus, the regulation of Ang 1–7 generation is under control by the multiple enzymes systems. It is possible that these Ang 1–7-generating enzymes may compensate each other to maintain the steady-state levels of Ang II and Ang 1–7 both locally and systemically, despite Ace2 as a major pathway for Ang 1–7 generation. This compensatory mechanism may explain our finding that the Ace2−/y and Ace+/y mice have similar levels of plasma Ang II and Ang 1–7 in both normal and the UUO conditions. It could be also true that because obstructive nephropathy was induced in the left kidney only, this leaves the opposite right kidney normal and enables to overcome the functional loss from the left nephropathy. In contrast, the balance between Ang II and Ang 1–7 was altered locally within the UUO kidney, which was much more profound in Ace2−/y mice, resulting in a fourfold increase in the ratio of intrarenal Ang II/Ang 1–7. This finding suggested that the compensatory mechanism locally in the UUO kidney was lost when the Ace2 pathway is disrupted. Therefore, the local vs systemically regulatory networking for both Ang II and Ang 1–7-generating pathways within the body may be attributed to the discrepancy in the levels of Ang II and Ang 1–7 between the plasma and the intrarenal tissue, although the precise mechanisms remain unknown.
A significant finding in the present study was the identification that enhanced TGF-β/Smad-mediated renal fibrosis and NF-κB-driven renal inflammation were key mechanisms by which loss of Ace2 enhanced progressive renal injury in a mouse model of UUO nephropathy. We and other investigators have previously demonstrated that Ang II is able to activate the TGF-β/Smad signaling via both TGF-β-dependent and -independent (through the ERK/p38 mitogen-activated protein kinase crosstalk pathway) mechanisms in vascular and renal cells.15, 16, 17 In the present study, we also found that loss of Ace2 largely increased intrarenal Ang II signaling via the AT1-mediated activation of ERK1/2 mitogen-activated protein kinase pathway. This was associated with enhanced TGF-β/Smad signaling (Smad2/3 phosphorylation and nuclear translocation) and progressive tubulointerstitial fibrosis in Ace2−/y mice when compared with Ace2+/y mice. Furthermore, in the context of renal inflammation, we also found that deletion of Ace2 promoted Ang II-stimulated NF-κB-dependent renal inflammation such as upregulation of IL-1β, TNF-α, MCP-1, and increased macrophage and T-cell infiltration in the UUO nephropathy. This was consistent with the known role of Ang II to activate NF-κB-mediated renal inflammation.31, 32 It is also possible that the development of intrarenal hypoxia within the UUO kidney may also be attributed to the development of renal inflammation.33 Prevention of Ang II-mediated renal oxidative stress, inflammation, and fibrosis by ACE2 supports the notion that loss of Ace2 promoted renal inflammation via the oxidative stress mechanism.9
More importantly, we also found that increased Ang II-induced Smurf2-dependent ubiquitin degradation of renal Smad7 may be the underlying mechanism required for promotion of TGF-β/Smad-mediated renal fibrosis and NF-κB-driven renal inflammation in the UUO nephropathy in Ace2−/y mice. We have previously shown that Smad7 is an integrated regulator that negatively regulates TGF-β/Smad-mediated renal fibrosis via its negative feedback-loop and NF-κB-dependent renal inflammation by the induction of IκBα, an inhibitor of NF-κB.34 Indeed, Smad7 acts as an adaptor protein that binds and recruits Smurf2, an E3 ubiquitin ligase, to the TGF-β1 receptor complex to promote its degradation through the proteasomal pathway.35 At the same time, ubiquitin-degradation of Smad7 occurs simultaneously.35 We have recently shown that Ang II can induce Smurf2 to degrade renal Smad7 in tubular epithelial cells in vitro.36 Once Smad7 is degraded, activation of Smad2/3 and renal fibrosis are enhanced. This is clearly demonstrated by the findings that Ang II activates Smad3 to induce epithelial-myofibroblast transition and renal fibrosis via Smurf2-dependent ubiquitin degradation of Smad7,36 and that upregulation of renal Smurf2 degrades renal Smad7 to promote TGF-β/Smad-dependent renal fibrosis in the UUO nephropathy.37 In addition, we also found that Smad7 is a negative regulator of NF-κB signaling.34 Overexpression of Smad7 is capable of inducing IκBα expression and preventing IκBα from phosphorylation and activation of NF-κB signaling, thereby inhibiting renal inflammation in vivo and in vitro.38, 39 In contrast, deletion of Smad7 promotes NF-κB-dependent renal inflammation in a mouse model of UUO.12 Taken together, loss of Smad7 promotes, while overexpression of Smad7 inhibits, TGF-β/Smad-mediated progressive renal fibrosis and NF-κB-driven renal inflammation as evidenced in a number of kidney disease models including UUO nephropathy,12, 37, 40 hypertension-associated remnant kidney disease,38, 41 diabetic nephropathy,14 and immunologically-mediated glomerulonephritis.42 Therefore, loss of Ace2 promoted Ang II-induced Smurf2-dependent ubiquitin degradation of renal Smad7 may be another essential mechanism by which Ace2−/y mice were promoted TGF-β/Smad-mediated renal fibrosis and NF-κB-driven renal inflammation in a mouse model of UUO nephropathy.
References
Ferrario CM . ACE2: more of Ang-(1–7) or less Ang II? Curr Opin Nephrol Hypertens 2010;20:1–6.
Koka V, Huang XR, Chung AC, et al. Angiotensin II up-regulates angiotensin I-converting enzyme (ACE), but down-regulates ACE2 via the AT1-ERK/p38 MAP kinase pathway. Am J Pathol 2008;172:1174–1183.
Danilczyk U, Penninger JM . Angiotensin-converting enzyme II in the heart and the kidney. Circ Res 2006;98:463–471.
Tikellis C, Bernardi S, Burns WC . Angiotensin-converting enzyme 2 is a key modulator of the renin-angiotensin system in cardiovascular and renal disease. Curr Opin Nephrol Hypertens 2010;20:62–68.
Soler MJ, Wysocki J, Ye M, et al. ACE2 inhibition worsens glomerular injury in association with increased ACE expression in streptozotocin-induced diabetic mice. Kidney Int 2007;72:614–623.
Ye M, Wysocki J, William J, et al. Glomerular localization and expression of angiotensin-converting enzyme 2 and angiotensin-converting enzyme: implications for albuminuria in diabetes. J Am Soc Nephrol 2006;17:3067–3075.
Oudit GY, Herzenberg AM, Kassiri Z, et al. Loss of angiotensin-converting enzyme-2 leads to the late development of angiotensin II-dependent glomerulosclerosis. Am J Pathol 2006;168:1808–1820.
Wong DW, Oudit GY, Reich H, et al. Loss of angiotensin-converting enzyme-2 (Ace2) accelerates diabetic kidney injury. Am J Pathol 2007;171:438–451.
Zhong J, Guo D, Chen CB, et al. Prevention of angiotensin II-mediated renal oxidative stress, inflammation, and fibrosis by angiotensin-converting enzyme 2. Hypertension 2011;57:314–322.
Oudit GY, Liu GC, Zhong J, et al. Human recombinant ACE2 reduces the progression of diabetic nephropathy. Diabetes 2010;59:529–538.
Crackower MA, Sarao R, Oudit GY, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002;417:822–828.
Chung AC, Huang XR, Zhou L, et al. Disruption of the Smad7 gene promotes renal fibrosis and inflammation in unilateral ureteral obstruction (UUO) in mice. Nephrol Dial Transplant 2009;24:1443–1454.
Lan HY, Mu W, Nikolic-Paterson DJ, et al. A novel, simple, reliable, and sensitive method for multiple immunoenzyme staining: use of microwave oven heating to block antibody crossreactivity and retrieve antigens. J Histochem Cytochem 1995;43:97–102.
Chen HY, Huang XR, Wang W, et al. The protective role of Smad7 in diabetic kidney disease: mechanism and therapeutic potential. Diabetes 2011;60:590–601.
Rodriguez-Vita J, Sanchez-Lopez E, Esteban V, et al. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation 2005;111:2509–2517.
Wang W, Huang XR, Canlas E, et al. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ Res 2006;98:1032–1039.
Yang F, Chung AC, Huang XR, et al. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: the role of Smad3. Hypertension 2009;54:877–884.
Tikellis C, Cooper ME, Bialkowski K, et al. Developmental expression of ACE2 in the SHR kidney: a role in hypertension? Kidney Int 2006;70:34–41.
Dilauro M, Zimpelmann J, Robertson SJ, et al. Effect of ACE2 and angiotensin-(1–7) in a mouse model of early chronic kidney disease. Am J Physiol Renal Physiol 2010;298:F1523–F1532.
Kamilic J, Hamming I, Kreutz R, et al. Renal ACE2 expression and activity is unaltered during established hypertension in adult SHRSP and TGR(mREN2)27. Hypertens Res 2009;33:123–128.
Wysocki J, Ye M, Soler MJ, et al. ACE and ACE2 activity in diabetic mice. Diabetes 2006;55:2132–2139.
Ye M, Wysocki J, Naaz P, et al. Increased ACE 2 and decreased ACE protein in renal tubules from diabetic mice: a renoprotective combination? Hypertension 2004;43:1120–1125.
Lely AT, Hamming I, van Goor H, et al. Renal ACE2 expression in human kidney disease. J Pathol 2004;204:587–593.
Mizuiri S, Hemmi H, Arita M, et al. Expression of ACE and ACE2 in individuals with diabetic kidney disease and healthy controls. Am J Kidney Dis 2008;51:613–623.
Reich HN, Oudit GY, Penninger JM, et al. Decreased glomerular and tubular expression of ACE2 in patients with type 2 diabetes and kidney disease. Kidney Int 2008;74:1610–1616.
Tikellis C, Johnston CI, Forbes JM, et al. Characterization of renal angiotensin-converting enzyme 2 in diabetic nephropathy. Hypertension 2003;41:392–397.
Shiota A, Yamamoto K, Ohishi M, et al. Loss of ACE2 accelerates time-dependent glomerular and tubulointerstitial damage in streptozotocin-induced diabetic mice. Hypertens Res 2010;33:298–307.
Mizuiri S, Hemmi H, Arita M, et al. Increased ACE and decreased ACE2 expression in kidneys from patients with IgA nephropathy. Nephron Clin Pract 2010;117:c57–c66.
Zhuo JL, Li XC . New insights and perspectives on intrarenal renin-angiotensin system: focus on intracrine/intracellular angiotensin II. Peptides 2011;32:1551–1565.
Navar LG, Kobori H, Prieto MC, et al. Intratubular renin-angiotensin system in hypertension. Hypertension 2011;57:355–362.
Ruster C, Wolf G . Renin-angiotensin-aldosterone system and progression of renal disease. J Am Soc Nephrol 2006;17:2985–2991.
Zhuo JL, Li XC . Novel roles of intracrine angiotensin II and signalling mechanisms in kidney cells. J Renin Angiotensin Aldosterone Syst 2007;8:23–33.
Eltzschig HK, Carmeliet P . Hypoxia and inflammation. N Engl J Med 2011;364:656–665.
Lan HY . Smad7 as a therapeutic agent for chronic kidney diseases. Front Biosci 2008;13:4984–4992.
Kavsak P, Rasmussen RK, Causing CG, et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell 2000;6:1365–1375.
Yang F, Huang XR, Chung AC, et al. Essential role for Smad3 in angiotensin II-induced tubular epithelial-mesenchymal transition. J Pathol 2010;221:390–401.
Fukasawa H, Yamamoto T, Togawa A, et al. Down-regulation of Smad7 expression by ubiquitin-dependent degradation contributes to renal fibrosis in obstructive nephropathy in mice. Proc Natl Acad Sci USA 2004;101:8687–8692.
Ng YY, Hou CC, Wang W, et al. Blockade of NFkappaB activation and renal inflammation by ultrasound-mediated gene transfer of Smad7 in rat remnant kidney. Kidney Int Suppl 2005; S83–S91.
Wang W, Huang XR, Li AG, et al. Signaling mechanism of TGF-beta1 in prevention of renal inflammation: role of Smad7. J Am Soc Nephrol 2005;16:1371–1383.
Lan HY, Mu W, Tomita N, et al. Inhibition of renal fibrosis by gene transfer of inducible Smad7 using ultrasound-microbubble system in rat UUO model. J Am Soc Nephrol 2003;14:1535–1548.
Hou CC, Wang W, Huang XR, et al. Ultrasound-microbubble-mediated gene transfer of inducible Smad7 blocks transforming growth factor-beta signaling and fibrosis in rat remnant kidney. Am J Pathol 2005;166:761–771.
Ka SM, Huang XR, Lan HY, et al. Smad7 gene therapy ameliorates an autoimmune crescentic glomerulonephritis in mice. J Am Soc Nephrol 2007;18:1777–1788.
Acknowledgements
This work has been supported by Grants from Research Grant Council of Hong Kong SAR (GRF767508 and 768409, and CUHK5/CRF/09).
Author contributions. ZL conceived all the experiments, analyzed data and drafted the manuscript. XRH and HYC conceived the generation of Ace2+/y and Ace2−/y mice, data analysis, and manuscript editing. JMP provided Ace2−/y mice and manuscript editing. HYL was responsible for the experimental design, data interpretation, and writing up manuscript for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
The renin-angiotensin system is critical in chronic kidney disease. The protective role of Ace2 in intrarenal Ang II-mediated renal injury in obstructive nephropathy is examined. Deletion of Ace2 results in a 4-fold increase in the intrarenal Ang II/Ang 1-7 ratio, enhanced TGF-β/Smad2/3-mediated renal fibrosis, and NF-κB-driven renal inflammation via the Smurf2-dependent degradation of renal Smad7.
Rights and permissions
About this article
Cite this article
Liu, Z., Huang, X., Chen, HY. et al. Loss of angiotensin-converting enzyme 2 enhances TGF-β/Smad-mediated renal fibrosis and NF-κB-driven renal inflammation in a mouse model of obstructive nephropathy. Lab Invest 92, 650–661 (2012). https://doi.org/10.1038/labinvest.2012.2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2012.2
Keywords
This article is cited by
-
Sodium–glucose cotransporter inhibitors and kidney fibrosis: review of the current evidence and related mechanisms
Pharmacological Reports (2023)
-
Rutin protects against gamma-irradiation and malathion-induced oxidative stress and inflammation through regulation of mir-129-3p, mir-200C-3p, and mir-210 gene expressions in rats’ kidney
Environmental Science and Pollution Research (2023)
-
Curcumin analog C66 alleviates inflammatory colitis by inhibiting the activation of NF-κB
Inflammopharmacology (2022)
-
Stimulation of ACE2/ANG(1–7)/Mas Axis by Diminazene Ameliorates Alzheimer’s Disease in the D-Galactose-Ovariectomized Rat Model: Role of PI3K/Akt Pathway
Molecular Neurobiology (2018)
-
Deletion of angiotensin-converting enzyme 2 exacerbates renal inflammation and injury in apolipoprotein E-deficient mice through modulation of the nephrin and TNF-alpha-TNFRSF1A signaling
Journal of Translational Medicine (2015)
