
Abstract
Epigenetic ‘clocks’ based on DNA methylation have emerged as the most robust and widely used aging biomarkers, but conventional methods for applying them are expensive and laborious. Here we develop tagmentation-based indexing for methylation sequencing (TIME-seq), a highly multiplexed and scalable method for low-cost epigenetic clocks. Using TIME-seq, we applied multi-tissue and tissue-specific epigenetic clocks in over 1,800 mouse DNA samples from eight tissue and cell types. We show that TIME-seq clocks are accurate and robust, enriched for polycomb repressive complex 2-regulated loci, and benchmark favorably against conventional methods despite being up to 100-fold less expensive. Using dietary treatments and gene therapy, we find that TIME-seq clocks reflect diverse interventions in multiple tissues. Finally, we develop an economical human blood clock (R > 0.96, median error = 3.39 years) in 1,056 demographically representative individuals. These methods will enable more efficient epigenetic clock measurement in larger-scale human and animal studies.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

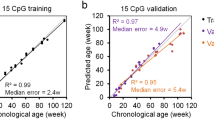
Data availability
Raw and processed sequencing data are available at the NCBI Gene Expression Omnibus under accession number GSE232346. The microarray data used for benchmarking are listed in the SuperSeries with the sequencing data under accession number GSE245630. Source data are provided with this paper.
Code availability
The code for demultiplexing and read processing, and clock analysis, is provided on GitHub at https://github.com/patricktgriffin/TIME-Seq.
References
Sprott, R. L. Biomarkers of aging and disease: introduction and definitions. Exp. Gerontol. 45, 2–4 (2010).
Bell, C. G. et al. DNA methylation aging clocks: challenges and recommendations. Genome Biol. 20, 249 (2019).
Horvath, S. & Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 19, 371–384 (2018).
Schultz, M. B. et al. Age and life expectancy clocks based on machine learning analysis of mouse frailty. Nat. Commun. 11, 4618 (2020).
Bobrov, E. et al. PhotoAgeClock: deep learning algorithms for development of non-invasive visual biomarkers of aging. Aging 10, 3249–3259 (2018).
Lehallier, B. et al. Undulating changes in human plasma proteome profiles across the lifespan. Nat. Med. 25, 1843–1850 (2019).
Fleischer, J. G. et al. Predicting age from the transcriptome of human dermal fibroblasts. Genome Biol. 19, 221 (2018).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013).
Hannum, G. et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49, 359–367 (2013).
Levine, M. E. et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 10, 573–591 (2018).
Petkovich, D. A. et al. Using DNA methylation profiling to evaluate biological age and longevity interventions. Cell Metab. 25, 954–960 (2017).
Meer, M. V., Podolskiy, D. I., Tyshkovskiy, A. & Gladyshev, V. N. A whole lifespan mouse multi-tissue DNA methylation clock. eLife 7, e40675 (2018).
Thompson, M. J. et al. A multi-tissue full lifespan epigenetic clock for mice. Aging 10, 2832–2854 (2018).
Levine, M. et al. A rat epigenetic clock recapitulates phenotypic aging and co-localizes with heterochromatin. eLife 9, e59201 (2020).
Robeck, T. R. et al. Multi-species and multi-tissue methylation clocks for age estimation in toothed whales and dolphins. Commun. Biol. 4, 642 (2021).
Wilkinson, G. S. et al. DNA methylation predicts age and provides insight into exceptional longevity of bats. Nat. Commun. 12, 1615 (2021).
Wang, T. et al. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol. 18, 57 (2017).
Horvath, S. et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging 10, 1758–1775 (2018).
Lu, Y. et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature 588, 124–129 (2020).
Kerepesi, C., Zhang, B., Lee, S.-G., Trapp, A. & Gladyshev, V. N. Epigenetic clocks reveal a rejuvenation event during embryogenesis followed by aging. Sci. Adv. 7, eabg6082 (2021).
Lu, A. T. et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 11, 303–327 (2019).
Pidsley, R. et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 17, 208 (2016).
Meissner, A. et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 33, 5868–5877 (2005).
Han, Y. et al. Epigenetic age-predictor for mice based on three CpG sites. eLife 7, e37462 (2018).
Han, Y. et al. New targeted approaches for epigenetic age predictions. BMC Biol. 18, 71 (2020).
Han, Y. et al. Targeted methods for epigenetic age predictions in mice. Sci. Rep. 10, 22439 (2020).
Wang, T. et al. Quantitative translation of dog-to-human aging by conserved remodeling of the DNA methylome. Cell Syst. 11, 176–185 (2020).
Wendt, J., Rosenbaum, H., Richmond, T. A., Jeddeloh, J. A. & Burgess, D. L. Targeted bisulfite sequencing using the SeqCap Epi enrichment system. Methods Mol. Biol. 1708, 383–405 (2018).
Mulqueen, R. M. et al. Highly scalable generation of DNA methylation profiles in single cells. Nat. Biotechnol. 36, 428–431 (2018).
Rohland, N. & Reich, D. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 22, 939–946 (2012).
Wang, M. & Lemos, B. Ribosomal DNA harbors an evolutionarily conserved clock of biological aging. Genome Res. 29, 325–333 (2019).
Zullo, J. M. et al. Regulation of lifespan by neural excitation and REST. Nature 574, 359–364 (2019).
Whitehead, J. C. et al. A clinical frailty index in aging mice: comparisons with frailty index data in humans. J Gerontol. A 69, 621–632 (2014).
Watada, E. et al. Age-dependent ribosomal DNA variations in mice. Mol. Cell. Biol. 40, e00368-20 (2020).
Rodriguez-Algarra, F. et al. Genetic variation at mouse and human ribosomal DNA influences associated epigenetic states. Genome Biol. 23, 54 (2022).
Seligman, B. J., Berry, S. D., Lipsitz, L. A., Travison, T. G. & Kiel, D. P. Epigenetic age acceleration and change in frailty in MOBILIZE Boston. J. Gerontol. A 77, 1760–1765 (2022).
Zhou, W. et al. DNA methylation dynamics and dysregulation delineated by high-throughput profiling in the mouse. Cell Genom. 2, 100144 (2022).
Lee, M. B., Hill, C. M., Bitto, A. & Kaeberlein, M. Antiaging diets: separating fact from fiction. Science 374, eabe7365 (2021).
Baur, J. A. et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444, 337–342 (2006).
Ocampo, A. et al. In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell 167, 1719–1733 (2016).
Gill, D. et al. Multi-omic rejuvenation of human cells by maturation phase transient reprogramming. eLife 11, e71624 (2022).
Matsuyama, M., WuWong, D. J., Horvath, S. & Matsuyama, S. Epigenetic clock analysis of human fibroblasts in vitro: effects of hypoxia, donor age, and expression of hTERT and SV40 largeT. Aging 11, 3012–3022 (2019).
Matsuyama, M. et al. Analysis of epigenetic aging in vivo and in vitro: factors controlling the speed and direction. Exp. Biol. Med. 245, 1543–1551 (2020).
Liu, Z. et al. Underlying features of epigenetic aging clocks in vivo and in vitro. Aging Cell 19, e13229 (2020).
Gu, H. et al. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat. Protoc. 6, 468–481 (2011).
Lehallier, B., Shokhirev, M. N., Wyss-Coray, T. & Johnson, A. A. Data mining of human plasma proteins generates a multitude of highly predictive aging clocks that reflect different aspects of aging. Aging Cell 19, e13256 (2020).
Bruinsma, S. et al. Bead-linked transposomes enable a normalization-free workflow for NGS library preparation. BMC Genomics 19, 722 (2018).
Mulqueen, R. M. et al. High-content single-cell combinatorial indexing. Nat. Biotechnol. 39, 1574–1580 (2021).
Nguyen Ba, A. N. et al. Barcoded bulk QTL mapping reveals highly polygenic and epistatic architecture of complex traits in yeast. eLife 11, e73983 (2022).
Krueger, F. & Andrews, S. R. Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics 27, 1571–1572 (2011).
Suzuki, M. et al. Whole-genome bisulfite sequencing with improved accuracy and cost. Genome Res. 28, 1364–1371 (2018).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 (2010).
Ackert-Bicknell, C. L. et al. Aging research using mouse models. Curr. Protoc. Mouse Biol. 5, 95–133 (2015).
Zhou, W. et al. DNA methylation dynamics and dysregulation delineated by high-throughput profiling in the mouse. Cell Genom. 2, 100144 (2022).
Acknowledgements
We thank the following individuals for their contributions to this work: R. Rogers for help editing this paper; A. Nguyen Ba and S. Boswell for providing the Tn5 purification and reaction protocols; C. Ricci-Tam for providing the initial hybridization enrichment protocols; the Bauer Core Facility at Harvard for sequencing and DNA extraction; the Harvard Microbiology Department for allowing us access to their Illumina MiSeq; the Seidman laboratory at the Harvard Medical School for allowing us to use their Agilent TapeStation; and the Jackson Laboratory Nathan Shock Center of Excellence in the Basic Biology of Aging (National Institutes of Health (NIH) number AG038070) for providing 200 mouse blood samples. This work was supported by the following grants, research programs and organizations, and gifts: the National Science Foundation Graduate Research Fellowship (number DGE1745303 to P.T.G.); the NIH/NIA F99/K00 Fellowship (number AG073499 to P.T.G.); the Diamond/AFAR Postdoctoral Transition Award in Aging (number DIAMOND19036 to A.E.K.); the NIH/NIA K99/R00 Fellowship (number AG070102 to A.E.K.); the Intramural Research Program of the NIH/NIA (to N.N.H. and M.K.E.); several NIH/NIA research project grants (numbers R01AG019719 and R21HG011850 to D.A.S.; number P01AG055369 to S.J.M.; and numbers AG065403 and AG047200 to V.N.G.); the Glenn Foundation for Medical Research (to D.A.S.); the Milky Way Research Foundation (to D.A.S.); gifts from M. Chambers, R. Rosenkranz, T. Robbins, P. Diamandis, S. Aoki, D. and S. Hoff, D. Dalio and Dalio Philanthropies, and the Glenn Foundation for Medical Research (to D.A.S.).
Author information
Authors and Affiliations
Contributions
P.T.G. conceived the project, designed the TIME-seq library preparations and carried out most of the preliminary experiments and optimization of the TIME-seq protocol. A.E.K. organized most of the mouse cohorts and performed the frailty index and blood parameter measurements. P.T.G., A.E.K., J.L., M.A., N.H., M.S.M., A.L.M., C.C., D.L.V. and R.J.C. collected the mouse samples, and extracted and normalized the DNA. P.T.G., A.E.K. and J.L. performed the library preparations. P.T.G. developed the bioinformatics pipeline for read processing and performed most of the bioinformatics analyses. C.K. provided the coverage-filtered RRBS methylation matrix for TIME-seq rDNA (RRBS) clock training. J.R.P., K.Y. and V.N.G. obtained and deidentified the human blood DNA samples, administered the related data and provided input on the human clock analysis. A.T., M.V.M. and V.N.G. provided the JAX mice blood samples used for clock training. N.N.H. and M.K.E. consulted on the human clock analysis. M.R.M., J.R.M. and S.J.M. provided the dietary intervention samples. J.A.A. performed the treatments and provided the liver samples from the CR and HFD mouse experiments. X.T. performed the treatments and provided the mouse livers from the AAV-OSK and AAV-GFP control mice. P.T.G. completed the cell culture time course with help from J.L. P.T.G. wrote the paper with input from D.A.S. and A.E.K. All authors edited and approved the paper.
Corresponding author
Ethics declarations
Competing interests
P.T.G. and D.A.S. are named inventors on a patent application related to TIME-seq methods filed by the Harvard Medical School (patent application number PCT/US21/37069) and licensed to Tally Health. D.A.S. is a founder and equity owner of Tally Health. P.T.G. has received minor equity compensation as a consultant to Tally Health. Additional information on D.A.S. affiliations not directly related to this work can be found at https://sinclair.hms.harvard.edu/david-sinclairs-affiliations. The other authors declare no competing interests.
Peer review
Peer review information
Nature Aging thanks Andrew Adey, Trey Ideker, Andrea Maier and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Biotinylated-RNA bait production and initial hybridization enrichment testing.
a, Schematic of steps involved in production of biotinylated-RNA baits from single-stranded oligo pools for target enrichment in TIME-Seq libraries. The percent of reads overlapping target RRBS mouse rDNA clock CpGs (b) and an IGV browser screenshot of mapped-read pileups (c) using version 1 rDNA baits for enrichment of a TIME-Seq pool. Reads on-target (d) and mouse RRBS blood clock (Petkovich et al., 2017) CpG coverage (e) using mouse-blood specific baits in a pilot experiment targeting non-repetitive clock loci. Dotted line represents coverage cut-off of 10. Pools in both rDNA and blood clock pilot enrichments were sequenced with approximately 1 million paired end (PE) reads each in pool of 16 samples. (f) Adaptor design schematic for comparison of TIME-Seq adaptors with longer barcoded adaptors. Comparison of on-target reads in short TIME-Seq and long cytosine-depleted adaptor designs for both mouse blood clock (g) and (h) rDNA (version 1) baits enrichments.
Extended Data Fig. 2 TIME-Seq library and sequencing schematic.
Schematic representation of final library structure (top) and Illumina sequencing (bottom) steps required to sequence TIME-Seq libraries. Index read 1 and read 2 primers are custom primers.
Extended Data Fig. 3 Small pilot-experiment sample metrics, correlation of rDNA CpG methylation, and age predictions using a reported RRBS-based rDNA clock.
a, TIME-Seq pilot experimental design using mouse blood DNA from 4 age groups and preparing 2 replicates of each sample with rDNA baits (version 1) as well as RRBS libraries to be sequenced as a fraction of an Illumina MiSeq sequencing run. b, Demultiplexed reads from TIME-Seq pools. c, Mean CpG methylation from reads mapped to the mouse ribosomal DNA meta-locus. Unmethylated lambda phage DNA control is represented as a diamond. d, Percent methylation from reads mapped to ribosomal DNA meta-locus in replicate 1 and replicate 2 in CpGs with coverage of at least 125 reads. e, Replicate correlation from different coverage cutoffs in the rDNA. f, Pileup tracks for samples from a TIME-Seq pool (replicate 1) as well as mapped reads from one sample (mouse ID 3, aged 24 months). Reads are colored by mismatch: blue for T (unmethylated) and red for C (methylated). RRBS rDNA clock coordinates are illustrated on the bottom by black rectangles. g, Percent of reads directly overlapping clock CpGs from TIME-Seq libraries (N = 12; mean from 2 replicates) and shallow-sequenced RRBS libraries (N = 10). h, RRBS rDNA clock predictions using TIME-Seq data enriched for clock loci (N = 12, both replicates) i, Coverage of each clock locus in the original RRBS rDNA clock. CpGs shown in red have a mean coverage of less than 50. Boxplot lengths (panels b, c, g, h) represent the interquartile range (IQR) with the middle line representing median values and the whiskers 1.5 times the IQR.
Extended Data Fig. 4 Additional data related to mouse multi-tissue and tissue-specific clock training and testing.
a, Baits overlapping target loci used for mouse clock CpG enrichment. b, Age predictions from the TIME-Seq Mouse Multi-tissue Clock applied to the 157 mouse muscle samples. Pearson correlation between predicted and actual age is shown. c, TIME-Seq White Adipose Clock train (N = 107) and testing set (N = 27) predictions. d, TIME-Seq Kidney Clock train (N = 156) and testing set (N = 38) predictions. For panels c and d, Pearson correlation between predicted and actual age is shown for train and test. The median absolute error is shown for the testing set.
Extended Data Fig. 5 Additional Data from validation and benchmarking of TIME-Seq.
a-b, TIME-Seq Mouse rDNA Clock predictions with samples colored for (a) validation library preparation (prep) and (b) cohort of the mouse. Pearson correlation is shown for each panel. c, Correlation between age-adjusted prediction residuals in the validation sets from the different prediction approaches. d, Correlation and significance matrix between ∆Age from each approach and ∆Medage(blood), that is, the difference in median value from similar aged mice for each blood measurement. The color and size of each circle represent the correlation and p-value significance, respectively. WBC = white blood cell count, NE (%) = percent of neutrophils, LY (%) = percent of lymphocytes, MO (%) = percent of monocytes, EO (%) = percent of eosinophils, BA (%) = percent of basophils, RBC = red blood cell count, Hb = hemoglobin, HCT = hematocrit, MCV = mean corpuscular volume, MCH = mean corpuscular hemoglobin, MCHC = mean corpuscular hemoglobin concentration, RDW = red blood cell distribution width, PLT = platelets, MPV = mean platelet volume. e, Frailty indexes for each of the assayed mice along with Pearson correlation with age. f, Comparison of ∆Age and ∆Medage(FI) for mice in the validation cohort. Pearson correlation is shown without adjusting for multiple comparisons. g, Comparison between TIME-Seq CpG methylation and RRBS methylation in the same sample and CpG. Pearson correlation between CpG methylation levels is shown.
Extended Data Fig. 6 Additional Data for TIME-Seq clocks applied to intervention mice and an in vitro time course.
a, Comparison TIME-Seq Multi-Tissue Clock predictions of high-fat diet mouse liver (N = 12) with standard diet controls (N = 5). b, Comparison of TIME-Seq Liver Clock predictions in OSK-expressing, (+) OSK (N = 5), and control, (-) OSK (N = 9), mice. For panels a-b, statistical comparison between groups was performed using a two-sided Student’s t-test after assessing normality with Shapiro-Wilk’s test. c, Predictions of cell culture samples using the TIME-Seq Mouse Skin Clock. The slope from the linear models fit to data points from each cell line is shown.
Extended Data Fig. 7 Comparison of methylation levels from TIME-Seq and BeadChip on the same samples.
Pearson correlation between BeadChip and TIME-Seq DNAme values for each sample and CpG (R = 0.93, p < 2.2e − 16).
Supplementary information
Supplementary Information
Supplementary Note.
Supplementary Table 1
Estimated cost of reagents for the TIME-seq library preparation. Reagents estimated to be used in quantities costing less than US$0.01 per sample were excluded from the total cost per sample calculation.
Supplementary Table 2
Information on biotinylated RNA bait pools used for the targeted enrichment of the TIME-seq libraries. The asterisk represents the total genomic space enriched in repetitive regions, which is equal to the target area multiplied by the copy number. Mean rDNA repeats are approximately equal to 1,400 copies per haploid (C57BL/6 mice).
Supplementary Table 3
Sequencing costs for the TIME-seq experiments. Sample number, sequencing kit and estimated cost of sequencing for each experiment.
Supplementary Table 4
Estimated costs for library preparation and clock data generation.
Supplementary Table 5
Oligonucleotides used for the TIME-seq library preparation.
Supplementary Table 6
Clock CpG positions, coefficients and intercepts.
Source data
Source Data Fig. 1
Statistical source data for Fig. 1.
Source Data Fig. 2
Statistical source data for Fig. 2.
Source Data Fig. 3
Statistical source data for Fig. 3.
Source Data Fig. 4
Statistical source data for Fig. 4.
Source Data Fig. 5
Statistical source data for Fig. 5.
Source Data Extended Data Fig. 1
Statistical source data for Extended Data Fig. 1.
Source Data Extended Data Fig. 3
Statistical source data for Extended Data Fig. 3.
Source Data Extended Data Fig. 4
Statistical source data for Extended Data Fig. 4.
Source Data Extended Data Fig. 5
Statistical source data for Extended Data Fig. 5.
Source Data Extended Data Fig. 6
Statistical source data for Extended Data Fig. 6.
Source Data Extended Data Fig. 7
Statistical source data for Extended Data Fig. 7.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Griffin, P.T., Kane, A.E., Trapp, A. et al. TIME-seq reduces time and cost of DNA methylation measurement for epigenetic clock construction. Nat Aging 4, 261–274 (2024). https://doi.org/10.1038/s43587-023-00555-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43587-023-00555-2
