
Abstract
Machine learning has transformed many fields and has recently found applications in chemistry and materials science. The small datasets commonly found in chemistry sparked the development of sophisticated machine learning approaches that incorporate chemical knowledge for each application and, therefore, require specialized expertise to develop. Here we show that GPT-3, a large language model trained on vast amounts of text extracted from the Internet, can easily be adapted to solve various tasks in chemistry and materials science by fine-tuning it to answer chemical questions in natural language with the correct answer. We compared this approach with dedicated machine learning models for many applications spanning the properties of molecules and materials to the yield of chemical reactions. Surprisingly, our fine-tuned version of GPT-3 can perform comparably to or even outperform conventional machine learning techniques, in particular in the low-data limit. In addition, we can perform inverse design by simply inverting the questions. The ease of use and high performance, especially for small datasets, can impact the fundamental approach to using machine learning in the chemical and material sciences. In addition to a literature search, querying a pre-trained large language model might become a routine way to bootstrap a project by leveraging the collective knowledge encoded in these foundation models, or to provide a baseline for predictive tasks.
Similar content being viewed by others


Main
One of the fascinating advances in machine learning has been the development of large language models (LLMs), so-called foundation models1,2,3,4,5,6. These models are appealing because of their simplicity; given a phrase, they return text that completes phrases in natural language such that, in many instances, one cannot tell that a machine wrote it.
From a scientific point of view, the most striking examples are that these foundation models can write sensible abstracts for scientific articles or even code for particular programming tasks7,8,9,10,11,12. Recently, it has been shown that these models can also solve relatively simple tabular regression and classification tasks13. However, as these models were not explicitly trained on these tasks, it is a remarkable result5.
That these models can solve simple tasks they are not trained for made us wonder whether they can also answer scientific questions for which we do not have an answer. As most chemistry problems can be represented in text form, we should be able to train these models to answer questions that chemists have. For example, ‘If I change the metal in my metal–organic framework, will it be stable in water?’ Such questions are often impossible to answer using theory or require highly sophisticated simulations or experiments.
We will always have very little (experimental) data for chemistry and material science applications. Hence, it is important that meaningful results can already be obtained with tens to hundreds of data points. We know from previous work on applications on text classification or generation that this works particularly well using models from the Generative Pre-trained Transformer 3 (GPT-3) family5, which were trained by the artificial intelligence company OpenAI. In this work, we show that these models—when provided with example data—perform surprisingly well for various chemistry questions, even outperforming the state-of-the-art machine learning models specifically developed for these tasks. It is important to realize that while language models have been used in chemistry before to predict properties14,15,16,17 or design molecules18,19,20, they have conventionally been pre-trained on chemistry-specific tasks. In contrast, the models we investigate here have been trained on text corpi compiled mainly from the Internet but still can adapt to various tasks. Although ref. 8 has probed the inherent chemistry knowledge of LLMs, we focus on how those models perform when they are fine-tuned—that is, the weights are updated—on some task-specific dataset. Note that this task-specific fine-tuning makes the models less dependent on the prompt structure than in-context learning21,22.
We benchmark our model on various datasets and applications to illustrate that these models can answer a wide range of scientific questions—ranging from the properties of materials, to how to synthesize materials and how to design materials (Fig. 1). In selecting these questions, we included some that have been addressed with machine learning. This allowed us to benchmark against state-of-the-art machine learning approaches specifically developed for these applications.

In this work, we benchmark GPT-3 on datasets spanning the chemical space from molecules over materials to reactions (Supplementary Note 1). On these datasets, we investigate different tasks ranging from classification, that is, predicting a class (for example, ‘high’, ‘low’) given a text representation of a molecule, material or reaction, to regression, that is, prediction of floating point numbers, to inverse design—the prediction of molecules. Metal–organic framework rendering created with iRASPA60.
Language-interfaced fine-tuning for classification and regression
Approach
Before discussing the different applications in detail, let us first discuss how we fine-tune23 the GPT-3 model in practice for a simple but highly non-trivial example. High-entropy alloys have attracted much interest as a novel class of structural metals. Interestingly, one has a sheer infinite number of possible combinations of metals. From a practical point of view, it is important to know whether a given combination of metals will form a solid solution or multiple phases. Hence, the question we would like to ask is: ‘What is the phase of <composition of the high-entropy alloy>?’ and our model should give a text completion from the set of possible answers {single phase, multi-phase}.
In Extended Data Table 1, we provide the set of questions and answers we used to fine-tune the GPT-3 model. These are questions and answers on high-entropy alloys for which the phase has been experimentally determined. The model tuning via the OpenAI API typically takes a few minutes and gives us a new model, which takes as input ‘Sm0.75Y0.25’ and gives as text completion ‘1’, which corresponds to single phase. This simple example already gives some remarkable results. We selected this example to directly compare its performance with the current state-the-art machine learning models with descriptors specially developed to mimic the relevant chemistry for this application24. In Fig. 2, we show that with only around 50 data points, we get a similar performance to the model of ref. 24, which was trained on more than 1,000 data points.
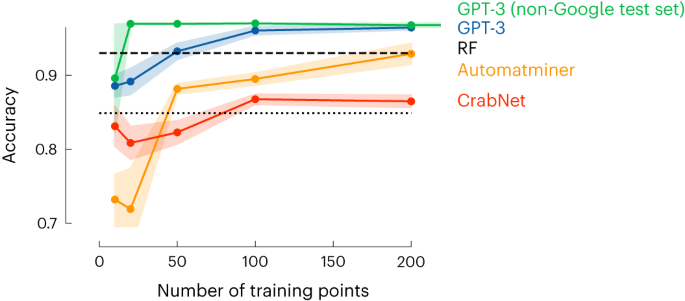
The figure compares the model’s accuracy as a function of the number of training points. The dashed horizontal line indicates the performance reported in ref. 24 using random forest (RF) with a dataset of 1,252 points and 10-fold cross-validation, that is, corresponding to a training set size of around 1,126 points. The dotted line shows the performance of a simple rule-based baseline ‘if present in the composition, classify as single phase, else multi-phase’. The yellow line we obtained using the Automatminer25, which uses as input the chemical composition. The Automatminer then returns the best featurization and model among those that are implemented using automated machine learning with genetic programming (as implemented in the TPOT package61). We additionally tested a neural network, CrabNet (red line, default settings)62, that performs well using compositions as input. The blue line is the performance of our GPT-3 model (with error bands showing s.e.m.). This figure shows that we reach similar accuracy to the model of ref. 24 with as little as around 50 data points. In addition, we also investigated a separate training and test set, for which the learning curve is shown in green. In this case, we tested on only compounds for which we could not find an exact match with a Google search. The learning curves for other metrics can be found in Supplementary Note 6.13.
Classification
These results made us wonder whether similar results can be obtained for other properties. Hence, we looked at a range of very different properties of molecules, materials and chemical reactions. We focused on those applications for which conventional machine learning methods have been developed and generally accepted as benchmarks in their field. In addition, we also compared our model with the top-performing ones on tasks from the Matbench25 suite of benchmarks (Supplementary Note 6.15).
Extended Data Table 2 compares the performance of a fine-tuned GPT-3 model with baselines (which can be found in Supplementary Note 6). For doing so, we fit the learning curves for the GPT-3 models and for the baselines and measure where they intersect, that is, we determine the factor of how much more (or fewer) data we would need to make the best baseline perform equal to the GPT-3 models in the low-data regime of the learning curves. The full learning curves for all models can be found in Supplementary Information (Supplementary Note 6).
For molecules, we investigated properties ranging from gaps between highest occupied (HOMO) and lowed unoccupied (LUMO) molecular orbitals and solubility in water to the performance in organic photovoltaics. For materials, we focused on the properties of alloys, metal–organic frameworks and polymers. Finally, for reactions, we considered two key cross-coupling reactions in organic chemistry. Extended Data Table 2 shows that in the low-data regime, our GPT-3 model is typically at least as good as the conventional machine learning model and often needs fewer data. In the high-data regime, the conventional machine learning models often catch up with the GPT-3 model. This makes sense, as for a given size of the dataset, the need for additional data and correlations (inductive biases)26 captured by GPT-3 might be less needed.
We have to mention that we did not optimize the fine-tuning of the GPT-3 model, that is, we did not try to optimize how a sentence is presented to the model; one can envision that specific tokenization can have better results for chemical sentences9,16,27,28. Also, we did not tune the number of times we show an example to a model (that is, the number of epochs or the learning rate).
Beyond fine-tuning of OpenAI models
Importantly, we are also not limited to fine-tuning; in Supplementary Note 5, we show that we can even achieve good performance without fine-tuning by incorporating examples directly into the prompt (so-called in-context learning5,29, that is, learning during inference time). This works particularly well with the largest GPT-3 models and GPT-4. We are also not limited to using models from OpenAI. In Supplementary Notes 7 and 8, we also show that we could obtain good results by fine-tuning the open-source LLM’s parameter-efficient fine-tuning techniques on consumer hardware and provide a Python package that makes it easy to apply this approach to new problems.
Representation sensitivity
An interesting question is how to represent a molecule or material. Most of the literature reports use International Union of Pure and Applied Chemistry (IUPAC) names. For machine learning applications, there has been a lot of effort to represent a chemical with unique line encodings (for example, simplified molecular-input line-entry system (SMILES) 30 or self-referencing embedded strings (SELFIES)31,32). As the GPT-3 model has been trained on natural text, one might expect that chemical names are preferred over line representations such as SMILES or SELFIES. Therefore, we investigated different representations for our molecular property prediction tasks (see also Supplementary Note 4). Interestingly, our results (Supplementary Note 6) show that good results are obtained irrespective of the representation. The fact that we often get the best performance using the IUPAC name of the molecule makes fine-tuning GPT-3 for a particular application relatively simple for non-specialists.
Regression
A more challenging task than classification is to make a regression model, which would allow us to predict the value of a continuous property such as the Henry coefficient for the adsorption of a gas in a porous material. As we are using a pre-trained language model, performing actual regression that predicts real numbers (\(\in {\mathbb{R}}\)) is impossible (without changes to the model architecture and training procedure). However, in most, if not all, practical applications, the accuracy for which we can make predictions is always limited. For example, for the Henry coefficient of a material, an accuracy of 1% (or a certain number of decimal points) is sufficient for most applications (see Supplementary Note 10 for discussion on this error source). Hence, we use molecules with Henry coefficients rounded to this accuracy as a training set and assume that the GPT-3 model can interpolate these numbers. Of course, one could also convert this into a classification problem by making tiny bins. For this more challenging regression task, we need more data for tuning the GPT-3 model, and we still get a performance that can approach the state of the art, but as this approach requires much more data, the advantage, except for the ease of training, is less. We obtain a similar conclusion for other regression problems (see Supplementary Note 10) and imbalanced classification cases (Supplementary Note 6.8).
Inverse design
One can argue that the ultimate goal of machine learning in chemistry is to create a model that can generate molecules with a desired set of properties. This is also known as inverse design33. Broadly speaking, there are two approaches. If we have large datasets, we can train generative models such as variational autoencoders34,35 or generative adversarial neural networks36,37. Without large datasets, evolutionary techniques such as genetic algorithms can generate novel, potentially interesting molecules38,39,40,41. Those evolutionary methods work best if one can limit the underlying chemistry; for example, finding the optimal functional group on a material with a well-defined backbone42.
Given that the GPT-3 model can predict the properties of molecules and materials with a small dataset, trying an inverse design strategy is tempting. This would be particularly important in the early stages of research; one often has a small set of experimental data points and a limited understanding. Yet, we could leverage a fine-tuned GPT-3 model to generate suggestions for novel materials with similar or even better performance. This would be an important step forward. Particularly as the tuning of such a natural language model is much more accessible than the training of conventional machine learning models. Here we investigate this setting: Can a fine-tuned GPT-3 propose valid molecules that satisfy the constraints or desired properties specified in a prompt in natural language? Again, we are illustrating the potential for a few case studies.
Molecular photoswitches are organic molecules with extended aromatic systems that make them responsive to light. Upon radiation, they switch reversibly between different isomers (which changes some properties, such as dipole moments). This reversible switching makes them interesting molecules for applications ranging from sensing to drug discovery. These molecules are complex, making sufficiently accurate predictions using first-principles theory very expensive. Yet, it is important to have some guidance to identify promising molecules, and machine learning models have been developed for this. One of the important properties of these photoswitches is the wavelength at which there is a maximum in the adsorption spectrum for the E and Z isomers. Hence, we fine-tuned GPT-3 with the same data used by ref. 43. As we have shown above, we can fine-tune GPT-3 to accurately answer questions like ‘What is the pi–pi* transition wavelength of CN1C(/N=N/C2=CC=CC=C2)=C(C)C=C1C?’.
For GPT-3, inverse design is as simple as training the model with question and completion reversed. That is, answer the question ‘What is a photoswitch with transition wavelengths of 324 nm and 442 nm, respectively’ with a text completion that should be a SMILES string of a meaningful molecule. This approach should be contrasted with the approach used by ref. 43, in which a library of molecules is generated, and their machine learning model (a Gaussian process regression) is used to evaluate the transition wavelengths of each material. If one has a lot of knowledge about the system, one can design large specific libraries that contain many promising molecules, including molecules with transition wavelengths of 324.0 nm and 442 nm. But, such a brute force technique is not what we understand as inverse design, as it, by definition, cannot predict a molecule that we did not include in our library.
A simple test to see whether our model can generate new structures is to ask it to generate molecules with transition wavelengths similar to those from the dataset reported by ref. 43. Extended Data Fig. 1 shows a representative sample of the molecules generated by the model. As expected, many molecules come from the training set (coloured orange in the figure). Importantly, many molecules are not in the training set, and, interestingly, some are not even in the PubChem database of known chemicals. In Fig. 3, we show that for the molecules, the transition wavelength is within a mean absolute percentage error of around 10%. Note that as the Gaussian process regression (GPR) model of ref. 43 was shown to perform comparably to, if not better than, more costly density functional theory simulations, we chose to use their model to compute the transition wavelengths for the generated molecules.

The fraction of valid SMILES indicates the fraction of generated SMILES that can successfully be parsed using RDKit (note that it does not plateau at 0, but approximately 0.1)63. We then determine the fraction of those that are already part of the training set and find that at low temperature GPT-3 tends to restate molecules from the training set. To quantitatively capture the similarity of the distribution of the generated molecules to the ones from the training set, we compute the Fréchet ChemNet distance64, which quantifies both diversity and distribution match51 and goes through a minimum at intermediate temperatures. For quantifying how well the generated molecules match the desired transition wavelengths, we use the GPR models reported by ref. 43 to predict the transition wavelengths. The dashed horizontal lines indicate those models’ mean absolute error (MAE). Across all temperatures, we found high average synthesizability (synthetic accessibility, SA, score44 smaller than 3). Error bands indicate s.e.m.
It is interesting to quantify how novel our newly generated molecules are. We compare these molecules to those collected in ref. 43. We quantify the similarity by computing the distance between molecular fingerprints. Figure 4 visualizes this by laying out the resulting approximate nearest-neighbour graph in two dimensions. The orange and green spheres represent molecules from the ref. 43 dataset, the blue spheres show the novel ones, and the pink ones are not part of the PubChem database. As expected, we find many new structures that are derivatives of molecules in the ref. 43 database. However, we also find branches that are not part of the library of ref. 43, indicating that the model generated novel kinds of compounds.

The tree map (TMAP) algorithm builds a nearest-neighbour graph, which is then embedded in two dimensions. Therefore, similar molecules are connected with an edge. We colour the points depending on whether they are part of the original dataset of ref. 43 but not generated (green) or part of the dataset and generated by our model (orange). Our models can also generate molecules that have not been part of the photoswitch dataset (note that the model was only trained on 92 molecules from this database). In some cases, those molecules have been reported before and are part of the PubChem database (blue) or are not part of the PubChem database (pink). From this figure, we see that the generated molecules sometimes substitutions for molecules in the dataset. In other cases, newly generated molecules introduce a completely new scaffold. For this visualization, we used the TMAP65 algorithm on photoswitch molecules described using MinHash fingerprint with 2,048 permutations66.
In generating these molecules, we adjusted the so-called softmax temperature in the sampling step of GPT-3 models. This temperature is conventionally used to generate more natural text. If we set this temperature to zero, we will generate text with the most frequently used words. We can increase the temperature to make the text more natural, making it more likely that less commonly used synonyms are chosen. For chemistry, if we aim to complete a SMILES starting with carbon, the zero-temperature solution would always complete the symbol that most commonly follows carbon (‘(’ in the QMugs dataset). In contrast, too-high temperatures would randomly choose any element.
The impact of this temperature parameter is shown in Fig. 3. At low temperatures, the generated molecules often come from the training set and only show a low diversity. Across all temperatures, the generated molecules seem synthesizable, as judged by a low synthetic accessibility (SA) score44. Increasing the temperature gives us more diverse and novel structures, but one can also expect more structures that make no chemical sense, that is, are invalid.
Stretching the limits
The results on the photoswitches illustrate the potential of LLMs for chemistry. To obtain more insight into whether we can trust these GPT-3 predictions, we carried out some experiments where we tried to stretch the limits.
We have already seen that we can obtain good results independent of how we represent a molecule (IUPAC names, SMILES or SELFIES), but can GPT-3 interpret an abstract representation of molecules we invented? A previous study45 developed a machine learning approach to design dispersants using a coarse-grained approach. This dispersant was a linear copolymer with four monomer types and a chain length between 16 and 48 units, giving a chemical design space of 58 million different dispersants. One important goal in this work was to find dispersants with the right binding free energy, that is, which polymer length and which monomer sequence is optimal. As there is no way the GPT-3 model knows about the properties or representations of the coarse-grained polymers, it is interesting to see if we can get any sensible result if we ask the question ‘What is the adsorption free energy of coarse-grained dispersant AAAABBBBDDDDAAAACCCC’ or as inverse design, ‘Give me a structure of a coarse-grained dispersant with a free energy of 17’. Interestingly, for the prediction of the adsorption free energy, the GPT-3 model outperforms the models developed by ref. 45. In addition, it can also successfully carry out the inverse design and generate monomer sequences that give the desired composition and, with a mean percentage error of around 22%, the desired adsorption free energy (the approximation of the ground truth we use already has a mean percentage error of around 9%, see Supplementary Note 11.1 for details).
In the case of the photoswitches, we have seen that the GPT-3 model can generate new molecules that are quite different from the training set. To explore in detail how far we can stretch the limits of what new molecules we can generate, we choose an application for which quantum calculations are known to predict the experimental values sufficiently accurately. The HOMO–LUMO gap is such an application. The HOMO–LUMO gap is relevant, for instance, in electronic applications that aim to excite a molecule at a specific energy. This HOMO–LUMO gap can be predicted accurately using semi-empirical quantum mechanics (GFN2-xTB46), which is computationally affordable enough for us to compute for all generated molecules (Supplementary Note 77). Moreover, the QMugs dataset47,48 has listed these HOMO–LUMO calculations for 665,000 molecules.
In Supplementary Note 11.3, we show that with the training of only 500 samples, we can get a reasonable estimate of the HOMO–LUMO gap of the molecules in the QMugs dataset. Also, by reverting the question, we have our model trained for inverse design. In Supplementary Note 11.3, we show that by asking the model ‘What is a molecule with a HOMO–LUMO gap of 3.5 eV’, we get similar to the photoswitches—a set of novel molecules. These novel molecules are not part of our training set and not even part of the QMugs dataset.
We now conduct some experiments on a dummy task to test how well the GPT-3 model can extrapolate to HOMO–LUMO gaps for which it has not received any training. To mimic this situation, we retrained our inverse design model using a dataset that has only molecules with HOMO–LUMO gaps smaller than 3.5 eV, and subsequently query the model with a question that requires the GPT-3 model to extrapolate (and, for example, to find that very small molecules are associated with large HOMO–LUMO gaps; a task we selected for only demonstration purposes and that can be exploited by generating small molecules). We do this by asking more than 1,000 times the question: ‘What is a molecule with a HOMO–LUMO gap of <XX>’, where each time we slightly change the value of the HOMO–LUMO gap, that is, we sample XX from a Gaussian centred at 4 eV. Interestingly, the GPT-3 model does provide structures with a distribution of which our quantum calculations confirm that a meaningful fraction has a HOMO–LUMO gap >4.0 eV. Again, this is a remarkable result. In our training set, there was not a single molecule with a bandgap >3.5 eV, which shows that the GPT-3 model can make extrapolations. We can do a similar experiment for the photoswitches, for which we might have a library of photoswitches whose transition wavelengths are all below 350 nm. For practical applications, however, it can often be essential to have adsorption at larger wavelengths. In this case, we can successfully use a fine-tuned GPT-3 model to generate photoswitch molecules that adsorb at lower energy (Supplementary Fig. 75, which we also validated with time-dependent density functional theory in Supplementary Note 11.2.2).
These findings inspired us to do an inverse design experiment to design molecules with properties that take us far from the training set49. We are interested in molecules that have a HOMO–LUMO gap >5 eV. From the distribution of HOMO–LUMO gaps in the QMugs database (Fig. 5), we see that the average bandgap is around 2.58 eV. Only a handful of molecules in this database have a HOMO–LUMO gap above 5 eV.

We start by fine-tuning GPT-3 on a sample of the QMugs dataset and use this model to query for around 1,000 gaps from a normal distribution with shifted mean (mean 4.0 eV, s.d. 0.2 eV). We then iteratively select the high-gap samples of the generated molecules and fine-tune the model on these data (that is, starting from the second generation, the model is fine-tuned on molecules it itself generated). Smooth curves show kernel-density estimates; the plot is truncated at 10 eV, but the models also generate some molecules with larger HOMO–LUMO gaps. We chose a comparatively large number of evaluations for this figure to increase the clarity of the visualization. For the initialization, we evaluated 2,162 compounds using xTB, followed by 1,670, 250 and 1,572. If we limit the number of quantum chemistry evaluations to or lower than 100, we can still successfully shift the distribution, as shown in Supplementary Fig. 83.
Hence, this is a challenging inverse design problem, as only a few materials in the database have the desired properties. Here our experiment is the quantum calculation, and we typically assume that we can evaluate hundreds to thousands of materials in a reasonable time. From a machine learning point of view, a set of thousands of materials is in a very low-data regime. However, from an experimental point of view, this is a large but sometimes doable effort. Of course, this is a somewhat arbitrary limit, and in Supplementary Fig. 83, we also give data for fewer experiments.
We start with the training using a set of hundreds of molecules randomly selected from the QMugs dataset (blue distribution in Fig. 5). These selected molecules will have bandgap distribution similar to the QMugs dataset. We then query for HOMO–LUMO gaps, now around 1,000 times requesting a molecule with a bandgap taken from a normal distribution with shifted mean (mean 4.0 eV, s.d. 0.2 eV). We evaluated these new molecules (green curve in Fig. 5), which indeed shows a shift of the distribution to higher HOMO–LUMO gaps. In the next iteration, we retrain the model with the new data and query again higher HOMO–LUMO gaps. Figure 5 shows that we have achieved our aim after four iterations.
Concluding remarks
Our results raise a very important question: how can a natural language model with no prior training in chemistry outperform dedicated machine learning models, as we were able to show in the case of high-entropy alloys in Fig. 2 and for various molecule, material and chemical reaction properties in Extended Data Table 2? To our knowledge, this fundamental question has no rigorous answer. The fact that we get good results independent of the chemical representation illustrates that these language models are very apt at extracting correlations from any text15. For example, we found promising results using both conventional chemical names and entirely hypothetical representations. In both cases, the model could quantitatively correlate the pattern of repeating units correctly to different kinds of properties.
Of course, if we say that the GPT-3 model is successful, it implies only that we have established that the GPT-3 model has identified correlations in the current training data that can be successfully exploited to make predictions. However, this does not imply that the correlations are always meaningful or related to cause–effect relationships. Hence, our research does not stop here. The next step will be to use GPT-3 to identify these correlations and ultimately get a deeper understanding. In this context, we argue that GPT-3 is only a tool to make more effective use of the knowledge scientists have collected over the years. It is also important to mention that while the training corpus contains chemistry information, many, if not most, scientific articles and results (including all failed or partially successful experiments50) have not been seen by GPT-3. Hence, one can expect an even more impressive performance if these data are added to the training data.
As we show in this Article, a machine learning system built using GPT-3 works impressively well for a wide range of questions in chemistry—even for those for which we cannot use conventional line representations such as SMILES. Compared with conventional machine learning, it has many advantages. GPT-3 can be used for many different applications. Each application uses the same approach, in which the training and use of the model are based on questions formulated in natural language. This raises the bar for future machine learning studies, as any new models should at least outperform this simple approach instead.
The other important practical point is that using a GPT-3 model in a research setting is similar to a literature search. It will allow chemists to leverage the chemical knowledge we have collected. GPT-3 has been designed to discover correlations in text fragments, and the fact that these correlations are extremely relevant to chemistry opens many possibilities for chemists and material scientists alike.
Methods
For all the results shown in the main text, we used the smallest ada variant of GPT-3 available via the OpenAI API. For fine-tuning, we used the same setting for all case studies (8 epochs, learning rate multiplier of 0.02). Error bands show, if not otherwise indicated, the standard error of the mean.
Data efficiency comparison
To compare the data-efficiency of the GPT-3 models with our baselines, we fitted all learning curves to power laws (−a exp(−bx + c)). We then used these power laws to find where the best-performing baseline shows the same performance as the best GPT-3-based approach at the first learning curve point (that performs better than random, as measured using the Cohen’s kappa (κ) metric).
Validity checks
To check the validity of the generated SMILES we use the is_valid method from the Guacamol package51, which effectively considers a SMILES as valid if it can be parsed using RDKit.
GPT-J model
We also performed some of our experiments by fine-tuning the GPT-J-6B model52,53 (which has been trained on the Pile dataset54) on consumer hardware using 8-bit quantization55 and 8-bit optimizers56 in addition to the low-rank adaptation (LoRA) technique57.
Data availability
All data used in this work was obtained from public sources and can be downloaded from GitHub (https://github.com/kjappelbaum/gptchem)58.
Code availability
All code created in this work is available on GitHub. The gptchem repository (https://github.com/kjappelbaum/gptchem)58 contains all experiments with the OpenAI API. The chemlift repository (https://github.com/lamalab-org/chemlift)59 contains an implementation supporting open-source LLMs.
References
Bommasani, R. et al. On the opportunities and risks of foundation models. Preprint at https://arxiv.org/abs/2108.07258 (2021).
Vaswani, A. et al. Attention is all you need. Adv. Neural Inf. Process. Syst. https://proceedings.neurips.cc/paper/2017/file/3f5ee243547dee91fbd053c1c4a845aa-Paper.pdf (2017).
Chowdhery, A. et al. PaLM: scaling language modeling with pathways. J. Mach. Learn. Res. 24, 1–113 (2023).
Hoffmann, J. et al. An empirical analysis of compute-optimal large language model training. Adv. Neural Inf. Process. Syst. 35, 30016–30030 (2022).
Brown, T. et al. Language models are few-shot learners. Adv. Neural Inf. Process. Syst. 33, 1877–1901 (2020).
Edwards, C. N., Lai, T., Ros, K., Honke, G. & Ji, H. Translation between molecules and natural language. in Conference On Empirical Methods In Natural Language Processing (eds Goldberg, Y. et al.) 375–413 (Association for Computational Linguistics, 2022).
Hocky, G. M. & White, A. D. Natural language processing models that automate programming will transform chemistry research and teaching. Digit. Discov. 1, 79–83 (2022).
White, A. D. et al. Assessment of chemistry knowledge in large language models that generate. Digit. Discov. 2, 368–376 (2023).
Taylor, R. et al. Galactica: a large language model for science. Preprint at https://arxiv.org/abs/2211.09085 (2022).
Dunn, A. et al. Structured information extraction from complex scientific text with fine-tuned large language models. Adv. Neural Inf. Process. Syst. 35, 11763–11784 (2022).
Choudhary, K. & Kelley, M. L. ChemNLP: a natural language-processing-based library for materials chemistry text data. J. Phys. Chem. C 127, 17545–17555 (2023).
Jablonka, K. M. et al. 14 examples of how LLMs can transform materials science and chemistry: a reflection on a large language model hackathon. Digit. Discov. 2, 1233–1250 (2023).
Dinh, T. et al. LIFT: language-interfaced fine-tuning for non-language machine learning tasks. Adv. Neural Inf. Process. Syst. 35, 11763–11784 (2022).
Karpov, P., Godin, G. & Tetko, I. V. Transformer-CNN: Swiss knife for QSAR modeling and interpretation. J. Cheminform. 12, 17 (2020).
Tshitoyan, V. et al. Unsupervised word embeddings capture latent knowledge from materials science literature. Nature 571, 95–98 (2019).
Born, J. & Manica, M. Regression transformer enables concurrent sequence regression and generation for molecular language modelling. Nat. Mach. Intell. 5, 432–444 (2023).
Yüksel, A., Ulusoy, E., Ünlü, A. & Doğan, T. SELFormer: molecular representation learning via SELFIES language models. Mach. Learn. Sci. Technol. 4, 025035 (2023).
van Deursen, R., Ertl, P., Tetko, I. V. & Godin, G. GEN: highly efficient SMILES explorer using autodidactic generative examination networks. J. Cheminform.12, 22 (2020).
Flam-Shepherd, D., Zhu, K. & Aspuru-Guzik, A. Language models can learn complex molecular distributions. Nat. Commun. 13, 3293 (2022).
Grisoni, F. Chemical language models for de novo drug design: challenges and opportunities. Curr. Opin. Struct. Biol. 79, 102527 (2023).
Ramos, M. C., Michtavy, S. S., Porosoff, M. D. & White, A. D. Bayesian optimization of catalysts with in-context learning. Preprint at https://arxiv.org/abs/2304.05341 (2023).
Guo, T. et al. What indeed can GPT models do in chemistry? A comprehensive benchmark on eight tasks. Preprint at https://arxiv.org/abs/2305.18365 (2023).
Howard, J. & Ruder, S. Universal language model fine-tuning for text classification. In Proc. 56th Annual Meeting of the Association for Computational Linguistics (Volume 1: Long Papers) 328–339 (Association for Computational Linguistics, 2018); https://aclanthology.org/P18-1031
Pei, Z., Yin, J., Hawk, J. A., Alman, D. E. & Gao, M. C. Machine-learning informed prediction of high-entropy solid solution formation: beyond the Hume–Rothery rules. npj Comput. Mater. https://doi.org/10.1038/s41524-020-0308-7 (2020).
Dunn, A., Wang, Q., Ganose, A., Dopp, D. & Jain, A. Benchmarking materials property prediction methods: the Matbench test set and Automatminer reference algorithm. npj Comput. Mater. https://doi.org/10.1038/s41524-020-00406-3 (2020).
Goldblum, M., Finzi, M., Rowan, K. & Wilson, A. The no free lunch theorem, Kolmogorov complexity, and the role of inductive biases in machine learning. ICLR 2024 Conference, OpenReview https://openreview.net/forum?id=X7nz6ljg9Y (2023).
Schwaller, P. et al. Molecular transformer: a model for uncertainty-calibrated chemical reaction prediction. ACS Cent. Sci. 5, 1572–1583 (2019).
Winter, B., Winter, C., Schilling, J. & Bardow, A. A smile is all you need: predicting limiting activity coefficients from SMILES with natural language processing. Digit. Discov. 1, 859–869 (2022).
Dai, D. et al. Why can GPT learn in-context? Language models secretly perform gradient descent as meta-optimizers. Preprint at https://arxiv.org/abs/2212.10559 (2022).
Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28, 31–36 (1988).
Krenn, M., Häse, F., Nigam, A., Friederich, P. & Aspuru-Guzik, A. Self-referencing embedded strings (SELFIES): a 100% robust molecular string representation. Mach. Learn. Sci. Technol. 1, 045024 (2020).
Krenn, M. et al. SELFIES and the future of molecular string representations. Patterns 3, 100588 (2022).
Sanchez-Lengeling, B. & Aspuru-Guzik, A. Inverse molecular design using machine learning: generative models for matter engineering. Science 361, 360–365 (2018).
Yao, Z. et al. Inverse design of nanoporous crystalline reticular materials with deep generative models. Nat. Mach. Intell. 3, 76–86 (2021).
Gómez-Bombarelli, R. et al. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 4, 268–276 (2018).
Kim, B., Lee, S. & Kim, J. Inverse design of porous materials using artificial neural networks. Sci. Adv. 6, eaax9324 (2020).
Lee, S., Kim, B. & Kim, J. Predicting performance limits of methane gas storage in zeolites with an artificial neural network. J. Mater. Chem. A 7, 2709–2716 (2019).
Nigam, A., Friederich, P., Krenn, M. & Aspuru-Guzik, A. Augmenting genetic algorithms with deep neural networks for exploring the chemical space. In ICLR (2019).
Jablonka, K. M., Mcilwaine, F., Garcia, S., Smit, B. & Yoo, B. A reproducibility study of ‘augmenting genetic algorithms with deep neural networks for exploring the chemical space’. Preprint at https://arxiv.org/abs/2102.00700 (2021).
Chung, Y. G. et al. In silico discovery of metal-organic frameworks for precombustion CO2 capture using a genetic algorithm. Sci. Adv. 2, e1600909 (2016).
Lee, S. et al. Computational screening of trillions of metal–organic frameworks for high-performance methane storage. ACS Appl. Mater. Interfaces 13, 23647–23654 (2021).
Collins, S. P., Daff, T. D., Piotrkowski, S. S. & Woo, T. K. Materials design by evolutionary optimization of functional groups in metal–organic frameworks. Sci. Adv. https://doi.org/10.1126/sciadv.1600954 (2016).
Griffiths, R.-R. et al. Data-driven discovery of molecular photoswitches with multioutput Gaussian processes. Chem. Sci. 13, 13541–13551 (2022).
Ertl, P. & Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 1, 8 (2009).
Jablonka, K. M., Jothiappan, G. M., Wang, S., Smit, B. & Yoo, B. Bias free multiobjective active learning for materials design and discovery. Nat. Commun. https://doi.org/10.1038/s41467-021-22437-0 (2021).
Bannwarth, C., Ehlert, S. & Grimme, S. GFN2-xTB—an accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J. Chem. Theory Comput. 15, 1652–1671 (2019).
Isert, C., Atz, K., Jiménez-Luna, J. & Schneider, G. QMugs: quantum mechanical properties of drug-like molecules https://doi.org/10.3929/ethz-b-000482129 (2021).
Isert, C., Atz, K., Jiménez-Luna, J. & Schneider, G. QMugs, quantum mechanical properties of drug-like molecules. Sci. Data 9, 273 (2022).
Westermayr, J., Gilkes, J., Barrett, R. & Maurer, R. J. High-throughput property-driven generative design of functional organic molecules. Nat. Comput. Sci. 3, 139–148 (2023).
Jablonka, K. M., Patiny, L. & Smit, B. Making the collective knowledge of chemistry open and machine actionable. Nat. Chem. 14, 365–376 (2022).
Brown, N., Fiscato, M., Segler, M. H. & Vaucher, A. C. GuacaMol: benchmarking models for de novo molecular design. J. Chem. Inf. Model. 59, 1096–1108 (2019).
Wang, B. Mesh-Transformer-JAX: model-parallel implementation of transformer language model with JAX. GitHub https://github.com/kingoflolz/mesh-transformer-jax (2021).
Wang, B. & Komatsuzaki, A. GPT-J-6B: a 6 billion parameter autoregressive language model. GitHub https://github.com/kingoflolz/mesh-transformer-jax (2021).
Gao, L. et al. The Pile: an 800BG dataset of diverse text for language modeling. Preprint at https://arxiv.org/abs/2101.00027 (2020).
Dettmers, T., Lewis, M., Belkada, Y. & Zettlemoyer, L. GPT3.int8(): 8-bit matrix multiplication for transformers at scale. Adv. Neural Inf. Process. Syst. 35, 30318–30332 (2022).
Dettmers, T., Lewis, M., Shleifer, S. & Zettlemoyer, L. 8-bit optimizers via block-wise quantization. in The Tenth International Conference on Learning Representations (2022).
Hu, E. J. et al. LoRA: low-rank adaptation of large language models. in International Conference On Learning Representations (2021).
Jablonka, K. M. kjappelbaum/gptchem: initial release. Zenodo https://doi.org/10.5281/zenodo.7806672 (2023).
Jablonka, K. M. chemlift. Zenodo https://doi.org/10.5281/zenodo.10233422 (2023).
Dubbeldam, D., Calero, S. & Vlugt, T. J. iRASPA: GPU-accelerated visualization software for materials scientists. Mol. Simul. 44, 653–676 (2018).
Le, T. T., Fu, W. & Moore, J. H. Scaling tree-based automated machine learning to biomedical big data with a feature set selector. Bioinformatics 36, 250–256 (2020).
Wang, A. Y.-T., Kauwe, S. K., Murdock, R. J. & Sparks, T. D. Compositionally restricted attention-based network for materials property predictions. npj Comput. Mater. 7, 77 (2021).
RDKit contributors. RDKit: Open-source Cheminformatics; (2023) http://www.rdkit.org
Preuer, K., Renz, P., Unterthiner, T., Hochreiter, S. & Klambauer, G. Fréchet ChemNet distance: a metric for generative models for molecules in drug discovery. J. Chem. Inf. Model. 58, 1736–1741 (2018).
Probst, D. & Reymond, J.-L. Visualization of very large high-dimensional data sets as minimum spanning trees. J. Cheminform. 12, 12 (2020).
Probst, D. & Reymond, J.-L. A probabilistic molecular fingerprint for big data settings. J. Cheminform. 10, 66 (2018).
Ertl, P. & Rohde, B. The Molecule Cloud—compact visualization of large collections of molecules. J. Cheminform. 4, 12 (2012).
Wang, Y., Wang, J., Cao, Z. & Farimani, A. B. Molecular contrastive learning of representations via graph neural networks. Nat. Mach. Intell. 4, 279–287 (2022).
Breuck, P.-P. D., Evans, M. L. & Rignanese, G.-M. Robust model benchmarking and bias-imbalance in data-driven materials science: a case study on MODNet. J. Phys. Condens. Matter 33, 404002 (2021).
Hollmann, N., Müller, S., Eggensperger, K. & Hutter, F. TabPFN: a transformer that solves small tabular classification problems in a second. Preprint at https://arxiv.org/abs/2207.01848 (2022).
Griffiths, R.-R. et al. Gauche: a library for Gaussian processes in chemistry. in ICML 2022 2nd AI for Science Workshop https://openreview.net/forum?id=i9MKI7zrWal (2022)
Chen, T. & Guestrin, C. XGBoost: a scalable tree boosting system. in Proc. 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 785–794 (ACM, 2016).
Moosavi, S. M. et al. Understanding the diversity of the metal-organic framework ecosystem. Nat. Commun. 11, 4068 (2020).
Moosavi, S. M. et al. A data-science approach to predict the heat capacity of nanoporous materials. Nat. Mater. 21, 1419–1425 (2022).
Probst, D., Schwaller, P. & Reymond, J.-L. Reaction classification and yield prediction using the differential reaction fingerprint DRFP. Digit. Discov. 1, 91–97 (2022).
Raffel, C. et al. Exploring the limits of transfer learning with a unified text-to-text transformer. J. Mach. Learn. Res. 21, 5485–5551 (2020).
Radford, A. et al. Language models are unsupervised multitask learners. OpenAI blog 1, 9 (2019).
Mobley, D. L. & Guthrie, J. P. FreeSolv: a database of experimental and calculated hydration free energies, with input files. J. Comput. Aided Mol. Des. 28, 711–720 (2014).
Delaney, J. S. ESOL: estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 44, 1000–1005 (2004).
Mitchell, J. B. O. DLS-100 solubility dataset. University of St Andrews https://risweb.st-andrews.ac.uk:443/portal/en/datasets/dls100-solubility-dataset(3a3a5abc-8458-4924-8e6c-b804347605e8).html (2017).
Walters, P. Predicting aqueous solubility—it’s harder than it looks. Practical Cheminformatics https://practicalcheminformatics.blogspot.com/2018/09/predicting-aqueous-solubility-its.html (2018).
Bento, A. P. et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 42, D1083–D1090 (2014).
Gaulton, A. et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40, D1100–D1107 (2012).
Nagasawa, S., Al-Naamani, E. & Saeki, A. Computer-aided screening of conjugated polymers for organic solar cell: classification by random forest. J. Phys. Chem. Lett. 9, 2639–2646 (2018).
Kawazoe, Y., Yu, J.-Z., Tsai, A.-P. & Masumoto, T. (eds) Nonequilibrium Phase Diagrams of Ternary Amorphous Alloys Landolt-Börnstein: Numerical Data and Functional Relationships in Science and Technology—New Series (Springer, 2006).
Zhuo, Y., Tehrani, A. M. & Brgoch, J. Predicting the band gaps of inorganic solids by machine learning. J. Phys. Chem. Lett. 9, 1668–1673 (2018).
Ahneman, D. T., Estrada, J. G., Lin, S., Dreher, S. D. & Doyle, A. G. Predicting reaction performance in C–N cross-coupling using machine learning. Science 360, 186–190 (2018).
Perera, D. et al. A platform for automated nanomole-scale reaction screening and micromole-scale synthesis in flow. Science 359, 429–434 (2018).
Acknowledgements
K.M.J., A.O.-G. and B.S. were supported by the MARVEL National Centre for Competence in Research funded by the Swiss National Science Foundation (grant agreement ID 51NF40-182892). P.S. acknowledges support from NCCR Catalysis (grant number 180544), a National Centre of Competence in Research funded by the Swiss National Science Foundation. The research of K.M.J. and B.S. was also supported by the USorb-DAC Project, which is funded by a grant from The Grantham Foundation for the Protection of the Environment to RMI’s climate tech accelerator programme, Third Derivative. In addition, the work of K.M.J. was supported by the Carl-Zeiss Foundation.
Funding
Open access funding provided by EPFL Lausanne.
Author information
Authors and Affiliations
Contributions
K.M.J. developed the machine learning approach with feedback from P.S. and B.S. K.M.J. and B.S. wrote the article. A.O.-G. contributed to the density functional theory calculations.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Guillaume Godin, Glen Hocky and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Molecule Cloud for randomly generated photoswitch molecules.
Molecule Cloud generated using the tool reported by Ertl and Rohde67. Aquamarine background indicates samples from molecules in the database reported by Griffiths et al.43 that our model did not generate, coral indicates the molecules our model generated and that are part of Griffiths et al43’s database, light steel blue background indicates samples that are generated by our model and that are not part of the database of Griffiths et al.43 but part of the PubChem database. Pale violet-red background indicates molecules that our model generated but that are part neither of PubChem nor the database of Griffiths et al.43.
Supplementary information
Supplementary Information
Supplementary Figs. 1–86, Notes 1–14 and Tables 1–15.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jablonka, K.M., Schwaller, P., Ortega-Guerrero, A. et al. Leveraging large language models for predictive chemistry. Nat Mach Intell 6, 161–169 (2024). https://doi.org/10.1038/s42256-023-00788-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42256-023-00788-1
This article is cited by
-
Connecting molecular properties with plain language
Nature Machine Intelligence (2024)
-
Leveraging language representation for materials exploration and discovery
npj Computational Materials (2024)
-
AI chatbot shows surprising talent for predicting chemical properties and reactions
Nature (2024)
-
The rise of data repositories in materials chemistry
Communications Chemistry (2024)
