
Abstract
Immune checkpoint inhibitors (ICIs) have changed perspectives for patients with cancer, but come with severe immune-related adverse events (irAEs). To prevent fatality or chronicity, these irAEs are often promptly treated with high-dose immunosuppressants. Until recently, evidence on the effects of irAE management on ICI efficacy has been sparse. As a result, algorithms for irAE management are mostly expert-opinion based and barely consider possible detrimental effects of immunosuppressants on ICI efficacy. However, recent growing evidence suggests that vigorous immunosuppressive management of irAEs comes with unfavourable effects on ICI efficacy and survival. With expansion of the indications of ICIs, evidence-based treatment of irAEs without hampering tumour control becomes more and more important. In this review, we discuss novel evidence from pre-clinical and clinical studies on the effects of different irAE management regimens including corticosteroids, TNF inhibition and tocilizumab on cancer control and survival. We provide recommendations for pre-clinical research, cohort studies and clinical trials that can help clinicians in tailored irAE management, minimising patients’ burden while maintaining ICI efficacy.
Similar content being viewed by others

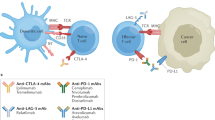
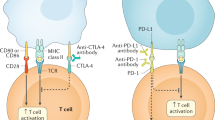
Introduction
Immunotherapy is currently widely used in cancer patients. By blocking downregulators of the immune response, immune checkpoint inhibitors (ICI) can induce long-term tumour control and survival that may last for years after treatment discontinuation in some, but not all patients with cancer. Most commonly used immune checkpoint inhibitors target cytotoxic T lymphocyte antigen 4 (CTLA-4) or programmed cell death 1 (PD-1), whose mechanisms of action are extensively reviewed elsewhere1,2.
While augmenting immune responses, ICI also induce immune-related adverse events (irAEs). irAEs are graded according to the Common Terminology Criteria for Adverse Events (CTCAE) and range from mild (grade 1–2) to severe (grade 3–4) and are sporadically lethal (grade 5)3,4. Frequency of irAEs and most often affected organs differ according to ICI regimen and (to a lesser extend) cancer type (for example more vitiligo with melanoma). In patients treated with combined checkpoint inhibition (cICI) with CTLA-4 and PD-1 blockade (typically ipilimumab plus nivolumab), irAEs of any grade occur in more than 95% of patients with severe irAEs observed in ~60%5. With blockade of the PD-1/PD-L1 interaction, irAEs are less frequent, with ~65% of patients experiencing irAEs of any grade and 15% having severe irAEs6. Whilst resembling autoimmune diseases, irAEs generally have a more acute onset, are generally more severe and chronicity can mostly be averted if irAEs are promptly and adequately managed.
irAE management guidelines generally advise ICI interruption and sometimes corticosteroid initiation with grade 2 irAEs and definite ICI discontinuation with prompt initiation of immunosuppressants for grade 3 or higher irAEs to prevent fatality and chronicity7,8,9. Therapeutic corticosteroids (also referred to as glucocorticoids) such as prednisone and methylprednisolone are propagated as the first-line immunosuppressive treatment for almost all severe irAEs. When severe irAEs do not improve within 72 hours after corticosteroid administration, escalation of immunosuppression by increasing corticosteroid dosage and/or addition of other immunosuppressive therapeutics is indicated. The choice of second-line immunosuppressant is often based on experience in autoimmune diseases with close resemblance to the particular irAE, like inflammatory bowel disease (IBD) for ICI-induced colitis and rheumatoid arthritis (RA) for ICI-induced arthritis. In accordance with the vast experience in IBD, inhibition of tumour necrosis factor (TNF) is often considered as second-line irAE treatment, for example with the monoclonal antibody infliximab.
Due to the limited evidence on irAE management, guidelines are mainly expert opinion based, aiming at fast resolution of irAEs. Since evidence of possible detrimental effects of immunosuppressants on ICI efficacy has been limited until recently, these downsides of aggressive immunosuppression have generally not been considered in irAE management.
In this review, we discuss relevant pre-clinical and clinical data on immunosuppressants in the context of irAEs and ICI efficacy (Fig. 1). To ensure a complete overview of current literature, a thorough systematic literature search was conducted with synonyms for “immune checkpoint inhibition”, “immunosuppressive agents”, specific drug names and appropriate Mesh terms. By combining theoretical and empirical evidence, we provide guidance for clinical practice and future research.

ICI immune checkpoint inhibition, irAE immune-related adverse event, TNF tumour necrosis factor, IL-6(R) interleukin-6(receptor), JAKi Janus kinase inhibitor, mTORi mammalian target of rapamycin inhibitor. Created with Biorender.com.
Immune-related adverse events and survival
Numerous reports have correlated irAE occurrence with increased overall survival10,11,12,13,14,15. These findings are prone to immortal-time bias (Box 1)11. Patients who develop an irAE after ICI initiation have guaranteed survival until irAE onset (therefore also called guarantee time bias or survivor bias), while patients who died without having an irAE might have developed one if they would not have died16. This may result in a biased estimation of survival differences in favour of the irAE group. However, several reports have shown that a correlation between irAEs and survival persisted when immortal-time bias was taken into account11,13. Interestingly, several studies have reported a survival benefit with low-grade, but not with high-grade irAEs13,17,18,19,20. This might be attributed to differences in ICI treatment duration, as ICI is often discontinued during high-grade, but not during low-grade irAEs. However, recent studies on neoadjuvant ICI demonstrate that responses occur early, with immune responses observed as early as 2 weeks after cICI initiation and pathological responses observed at 4–6 weeks21,22,23. Furthermore, ICI has been shown to induce long-term tumour control, even after discontinuation24,25. Since only 0.3–1.3% of ICI-treated patients have fatal irAEs4, it is highly unlikely that survival differences between low-grade and high-grade irAEs are due to fatal irAEs themselves. High-grade irAEs often require immunosuppression whilst low-grade irAEs do not. Therefore, survival differences between high-grade and low-grade irAEs could be an indication that immunosuppression impairs the initial survival benefit.
Corticosteroids
Corticosteroids have pleiotropic effects on various cells of the immune system26,27,28. Corticosteroids augment Treg production and activity, inhibit T cell receptor (TCR) signalling, dampen T cell effector function, and induce a less pro-inflammatory cytokine profile29,30,31,32,33. Moreover, corticosteroids upregulate immune checkpoints such as CTLA-4, PD-1, TIM-3 and LAG-3 on T cells29,30,31,34.
Several mouse studies evaluating the effect of concurrent corticosteroid and ICI treatment on tumour growth and survival have demonstrated mixed results (Supplementary table 1)30,31,32,35,36,37,38. Four studies demonstrated a significantly increased tumour growth rate and/or impaired survival in mice treated with ICI plus dexamethasone compared to ICI therapy alone30,32,35,38, and one study showed non-significant trends in the same direction36. Conversely, Xiang et al. demonstrated better tumour control in mice treated with anti-PD-1 plus low-dose dexamethasone (0.1 mg/kg every 3 days) compared to anti-PD-1 alone in two tumour types37. Importantly, only two studies investigated corticosteroid administration after ICI initiation rather than concurrently, resembling the clinical situation of corticosteroid treatment for ICI-induced irAEs35,38. In a colon adenocarcinoma mouse model, Maxwell et al. observed that only 1 out of 8 anti-PD-1 plus dexamethasone treated mice had a complete response compared to 4/8 mice treated with anti-PD-1 alone with an increased tumour growth rate in the anti-PD-1 plus dexamethasone group35. Notably, two anti-PD-1 plus dexamethasone treated mice responded before dexamethasone initiation but progressed afterwards. Opposing results were observed by Tokunaga et al. who observed slightly but significantly impaired tumour control with high dose (2000 μg) but not low-dose (20 μg) concomitant dexamethasone, while no difference in tumour control was observed when dexamethasone was started after tumour regression38. Timing and dosing of corticosteroids may be crucial when assessing their impact on ICI efficacy and notably, inconsistencies exist between animal models and clinical practice. Overall, no strong conclusions can be drawn from animal studies with respect to the impact of corticosteroids for irAEs on ICI efficacy.
Observational clinical studies on the association of corticosteroids with PFS and OS in ICI-treated cancer patients demonstrate contradictory results, originating from differences in indication and comparison group. Two meta-analyses concluded that patients receiving corticosteroids for cancer-related indications have worse survival, which might be a reflection of a worse prognosis due to bias by indication39,40. Conversely, both meta-analyses conclude that corticosteroids as irAE management do not seem to abrogate ICI efficacy. Most of the included studies compared patients whose irAEs were managed with corticosteroids to all other patients, including those without irAEs. This has two major limitations: First, results are prone to immortal-time bias, which may have resulted in an underestimation of a potential negative association between corticosteroid treatment and survival. Second, patients who develop irAEs are generally considered to have prolonged survival compared to patients who do not develop irAEs. Comparing survival of patients whose irAEs are managed with corticosteroids to all patients irrespective of irAE occurrence may obscure possible detrimental effects of corticosteroids for irAE management (Fig. 2). Indeed, all studies comparing corticosteroid-treated patients with irAEs to all other patients irrespective of irAE occurrence report no difference or a trend towards a favourable effect of corticosteroids on ICI efficacy (Supplementary table 2)41,42,43,44,45,46,47,48,49,50,51,52,53,54,55.

At baseline, it is unknown whether a patient will be treated with IS for irAEs. Comparing patients treated with IS for irAEs to all other patients (also those without irAEs) is likely incorrect. Under the assumption that patients with irAEs have a longer survival than patients without irAEs (lower left), survival of patients with irAEs treated with IS might appear similar to survival of all other patients (including those without irAEs; upper right), while it may actually be worse than survival of patients with irAEs which were not treated with IS (lower right). This is irrespective of whether the survival difference between patients with irAEs and those without is (partially) due to immortal-time bias.
Several studies making comparisons within the group of patients with irAEs, demonstrate a detrimental effect of high dose corticosteroids on ICI efficacy, while no unfavourable effect was observed in other studies (Fig. 3 and Supplementary table 3)13,41,46,47,56,57,58,59,60,61,62,63,64,65,66. Most of the latter studies reported on small groups or low-dose corticosteroid use during ICI irrespective of indication, or included multiple types of cancer or ICI which have different survival and toxicity profiles and are often unequally distributed among groups. In another analysis of randomised controlled trial data, Robert et al. reported no significant differences in PFS or OS among anti-PD-1-treated melanoma patients with irAEs treated with systemic corticosteroids compared to patients with irAEs without corticosteroid need61. The authors acknowledge the importance of immortal-time bias and the considerations regarding a landmark analysis when comparing survival of patients with irAEs to those without. However, applying this same landmark analysis in a context of two highly associated time-dependent variables (irAE onset and corticosteroid initiation) may have led to high dependency of survival outcomes on time of irAE onset relative to the landmark, possibly leading to obscured results.

Overall survival (a) and progression-free survival (b) in patients with irAEs with or without corticosteroid treatment. Corticosteroid dose cut-offs represent prednisone equivalent dose. For immune checkpoint inhibitor (ICI) type and, tumour heterogeneity type, green (+) indicates restriction to one ICI regimen/tumour type, yellow (−) indicates adjustment for ICI regimen/tumour type and red (X) indicates that heterogeneity was not accounted for. In studies by Li et al. and Faje et al., corticosteroids irrespective of their indication (irAE or non-irAE) were included, which may have induced bias. In well-intended efforts to account for immortal-time bias, Robert et al. may have introduced bias rather than corrected for time to irAE onset and Lafayolle et al. have potentially overestimated the negative effect of corticosteroids on survival. Other studies accounted for immortal-time by analysing post-irAE survival (green +), did not account for difference in time-to-irAE onset (yellow −) or did not clearly describe start-point of survival analyses (blue?). *Thompson et al. compared two dosages of corticosteroids against the lowest dose of <7.5 mg/d for at least 2 weeks prednisone equivalent. HR hazard ratio for death, CI confidence interval, n number of participants in analysis, IS immunosuppressant, N/A not applicable.
In a retrospective study of anti-PD-1-treated melanoma patients with irAEs in two independent cohorts, Bai et al. examined the correlation between both PFS and OS with early high-dose corticosteroid exposure defined as 1-day peak dosage of ≥60 mg prednisone equivalent46. They assessed post-irAE PFS and OS in a landmark analysis in which only corticosteroid use within 8 weeks after anti-PD-1 initiation was considered and patients who had progressive disease or died within 8 weeks were excluded. Early high-dose corticosteroid exposure was strongly associated with worse post-irAE PFS and post-irAE OS in both cohorts. Similarly, in a cohort of infliximab-treated colitis patients with various tumour types and ICI regimens, patients with high peak dose of corticosteroids (≥75 mg prednisolone equivalent on 1 day) had a significantly worse overall survival than patient with lower dosages corticosteroids when corrected for tumour type and age, but not for type of ICI56.
Together, these data suggest that patients who received high-dose corticosteroids for irAEs have impaired survival. It is important to note that those patients who received the highest dosages of corticosteroids are likely also the ones who were treated with second-line immunosuppressants. Therefore, uncertainty remains whether the survival disadvantage is attributable to high-dose corticosteroids specifically, or merely reflects aggressive immunosuppression as a whole.
TNF inhibitors
TNF is a molecule with both pro- and anti-inflammatory properties which are context-dependent67,68,69,70. In an effort to deduce these versatile immunological effects of TNF to a model summarising the effects of TNF inhibition in the context of ICI, Chen et al. inferred that short course TNF inhibition to treat irAEs presumably does not diminish ICI anticancer efficacy67, but this hypothesis has been questioned71,72. The complexity of TNF in the context of cancer is reflected by the opposing strategies to target TNF as a cancer therapy since its discovery. Early clinical trials using recombinant TNF in patients with advanced cancer demonstrated limited efficacy, accompanied with severe side effects73,74,75,76. Similarly, in early-phase clinical trials of TNF inhibitors in patients with advanced cancer, responses were rarely observed77,78,79,80.
Two mouse studies have shown that concomitant administration of TNF inhibition with ICI as upfront anti-cancer treatment not only resulted in less toxicity, but also in increased tumour control (Supplementary table 4)81,82. Bertrand et al. reported that simultaneous administration of TNF inhibition and anti-PD-1 6 days after tumour inoculation in mice led to tumour regression more often than anti-PD-1 alone with increased survival81. Similarly, Perez-Ruiz et al. demonstrated increased tumour control in mice treated with cICI and TNF inhibition 9 days after tumour inoculation compared to cICI alone82. This beneficial effect could not be confirmed in immunodeficient mice infused with human peripheral blood mononuclear cells, inoculated with a colon adenocarcinoma cell line82. Although no good animal models of spontaneous ICI toxicity currently exist, dextran sulphate sodium (DSS)-induced colitis severity was lower in mice treated with concomitant TNF inhibition and cICI than in those treated with cICI alone82. These results have led to the TICIMEL phase IB clinical trial in which advanced melanoma patients received a combination of upfront TNF inhibition (either infliximab or certolizumab) with cICI (NCT03293784)83. In total, ten out of 14 patients had a complete or partial response as best overall response, of whom five had an ongoing response after 1 year. Whether these data of upfront TNF inhibition are translatable to the clinical practice of irAE management after ICI initiation is uncertain.
Several cohort studies have reported on the association between the TNF inhibitor infliximab as second-line immunosuppressant for steroid-refractory irAEs and survival (Fig. 4 and Supplementary table 5)14,59,65,84,85,86,87,88,89,90,91,92,93,94. An early report on patients with anti-CTLA-4 induced colitis showed a non-significant trend towards improved survival in 7 patients who received infliximab compared to 22 who did not88. A similar trend was observed in PFS by Favara et al. in a group of 56 advanced melanoma patients with ICI induced colitis91. However, relatively more patients in the anti-TNF-treated group were treated with cICI. Other, larger studies did not find an improved survival with TNF inhibition or demonstrated diminished survival with TNF inhibition14,65,87,92. In a study of 222 first-line ICI-treated melanoma patients with grade 3 or higher irAEs, we found a shorter overall survival and cancer-related survival in patients treated with TNF inhibition as second-line immunosuppressant compared to patients who only required corticosteroid treatment. These results remained unchanged when adjusting for ICI regimen amongst other factors. Since ICIs were reintroduced as often in both groups, the survival difference was not explained by hesitance to reintroduce ICIs after steroid-refractory irAEs14. In a second study, in a more homogeneous cohort of 350 first-line cICI-treated melanoma patients with grade 3 or higher irAEs, we demonstrated that patients receiving second-line immunosuppression had worse OS, PFS and cancer specific survival compared with patients who only received corticosteroids87. Similar trends were observed for patients receiving second-line TNF inhibition and other second-line immunosuppressants87.

Overall survival (a) and progression-free survival (b) in patients with irAEs with or without TNF inhibition. For immune checkpoint inhibitor (ICI) type and tumour, heterogeneity type, green (+) indicates restriction to one ICI regimen/tumour type, yellow (−) indicates adjustment for ICI regimen/tumour type and red (X) indicates that heterogeneity was not accounted for. Zou et al. analysed post-irAE survival which is theoretically less susceptible for difference in time-to-irAE (therefore green +), than survival from ICI initiation (yellow −, as in Fig. 2). Favara et al. did not clearly describe start-point of survival analyses (blue?). HR hazard ratio for death, CI confidence interval, n number of participants in analysis, IS immunosuppressant, GC glucocorticoid, VEDO vedolizumab, N/A not applicable.
Most studies on escalated immunosuppression to treat steroid-refractory irAEs include patients with ICI-colitis or diarrhoea only. In these patients, vedolizumab proved an alternative with approximately similar effectiveness in terms of irAE resolution85,95. By blocking the interaction of α4β7-integrin with MadCAM-1, vedolizumab is considered to prevent T-cell homing in gastrointestinal tissue specifically, with the theoretical advantage of preventing local inflammation without diminishing systemic antitumor immunity. In a retrospective comparison of cancer patients who received infliximab, vedolizumab, or both as second-line immunosuppressants for ICI-induced diarrhoea or colitis, Zou and colleagues reported inferior response and post-irAE survival of infliximab-treated patients compared to vedolizumab-treated patients85. As the authors acknowledge, ICI efficacy was not the primary endpoint of this study, and the results could have been influenced by selection bias and confounding. Infliximab-treated patients received corticosteroids for a longer time period and more often had progressive disease before onset of toxicity (41% vs 24%). Interestingly, in the subgroup of patients without progressive disease at onset of colitis, proportionally more of the infliximab-treated patients had progressed at last follow-up compared to vedolizumab-treated patients (28% vs 13%) with similar follow-up duration.
Altogether, mouse models and clinical data on the effects of TNF inhibitors on tumours in the context of ICI are conflicting. Timing of TNF inhibition in relation to start of ICI and relative to corticosteroid initiation may be of major importance, reflecting the versatile role of TNF in different stages of cancer and immune activation. No definite conclusion can yet be drawn with respect to the initiation of TNF inhibitors in the context of irAEs. However, in patients with ICI-induced colitis, vedolizumab may be preferred over TNF inhibition when considering ICI efficacy.
Interleukin-6 blockade
Interleukin (IL)-6 is a cytokine with a broad range of effects on immune and cancer cells96,97,98,99. High baseline IL-6 levels in serum and tissue and increase of serum IL-6 levels early on-treatment have been correlated to poor response and survival in ICI-treated cancer patients in several studies100,101,102,103,104. IL-6 can activate the Janus kinase (JAK) signal transducer and activator of transcription 3 (STAT3). IL-6 may induce angiogenesis and vessel permeability and lead to Th17 skewing. Activation of STAT3 has tumour promoting effects via suppression of apoptosis and tumour suppressors and induction of proliferation. Besides, STAT3 has immunosuppressive effects in the tumour microenvironment where its activation leads to inhibition of neutrophil, natural killer cell and effector T cell function, reduced DC maturation and expansion of Treg and myeloid derived suppressor cells.
Several mouse studies have shown significantly (17 experiments) or non-significantly (4 experiments) improved tumour control or survival with upfront anti-IL-6 plus ICI compared to ICI alone (Supplementary table 6)102,104,105,106,107,108,109,110. This improved tumour control is presumably due to promotion of Th1-associated immunity104,109. Conversely, no improved tumour control or survival was observed in two mouse experiments104,108 and a slightly reduced anti-tumour effect was observed in the only two models of cICI82. Although upfront IL-6 blockade has been shown to mitigate experimental autoimmune encephalomyelitis symptoms109, no pre-clinical models have been reported that mimic the timing of immunosuppression in the context of irAEs in which IL-6 blockade is initiated after ICI.
Very limited clinical reports on the use of IL-6 blockade for irAEs do not show impaired ICI efficacy111,112,113,114. Stroud et al. reported on 87 nivolumab treated patients, of whom 34 patients received tocilizumab (anti-IL-6R) for severe irAEs111. When comparing tocilizumab treated patients with patients who did not receive tocilizumab, no difference in overall survival was observed. However, the exceptionally high number of tocilizumab treated patients (39%), and the inclusion of patients without irAEs in the comparison warrant caution when interpreting these results. Similarly, Dimitriou et al. report a median PFS of 6 months in 22 advanced melanoma patients receiving tocilizumab113, which is difficult to compare given the variety of ICI regimens patients received. The COLAR study (NCT03601611) demonstrated that 6 out of 20 patients treated with tocilizumab alone for ICI colitis or arthritis had progressive disease114. Although a 14 day period without any immunosuppression was required prior to tocilizumab initiation, 9 of the 20 patients had received systemic corticosteroids, including 3 who had also received infliximab for any irAEs prior to tocilizumab administration114. Preliminary results of an ongoing phase II trial of upfront tocilizumab plus cICI in advanced melanoma patients (NCT03999749) demonstrated a favourable overall response rate (ORR) within 12 weeks of 58% in 29 evaluable patients115. Another phase II trial of upfront tocilizumab plus cICI is currently underway (NCT04940299).
Taken together, in vivo data suggest that IL-6 blockade is a promising strategy in the context of ICI with suggested synergistic rather than detrimental effects on anti-tumour efficacy when given concomitantly. However, clinical data in the context of irAEs are too limited to draw conclusions on the effects of IL-6 blockade as irAE treatment on ICI efficacy.
Other immune modulators
Other options for immunosuppression involve classical corticoid sparing disease modifying antirheumatic drugs (csDMARDs) such as methotrexate, mycophenolic acid, hydroxychloroquine and tacrolimus or kinase inhibitors such as JAK inhibitors and mammalian target of rapamycin (mTOR) inhibitors. Alternatively, intravenous immunoglobulins (IVIg), biologicals targeting other specific cytokines such as IL-1, IL-17 and IL-23 or targeting B-cells (such as rituximab) might be used to treat irAEs. Rationale for these therapeutics is reviewed elsewhere116,117,118.
In absence of clinical data, mouse studies entail the best available evidence for impact of JAK inhibitors119,120,121, mTOR inhibitors122,123,124,125, and hydroxychloroquine126,127,128 on ICI efficacy (Supplementary table 7). JAK1/2 loss-of-function mutations are associated with primary129 and secondary130 resistance to anti-PD-1. In contrast, multiple mouse models have demonstrated better tumour growth control with concomitant ICI and ruxolitinib (a JAK1/2 inhibitor) than with ICI alone119,120,121. Of note, one of these studies showed that only delayed administration of ruxolitinib (3 days after first anti-CTLA-4) provided better tumour control, which was not observed with simultaneous initiation120. A similar beneficial effect was found in mice treated with concomitant ICI with vistusertib (mTORC1/2 inhibitor) or sirolimus (mTORC1 inhibitor) compared ICI alone122,123. Interestingly, only addition of low-dose everolimus (mTORC1 inhibitor) to anti-PD-1 led to better bladder or cervical cancer control124,125, while addition of high-dose everolimus to anti-PD-1 showed a trend towards worse tumour control124. Three studies of hydroxychloroquine combined with anti-PD-1 in mice yielded contradictory results126,127,128. Sharma et al. observed better tumour control and survival in two melanoma models of simultaneous administration of anti-PD-1 and hydroxychloroquine126, while in a comparable model, Krueger et al. observed impaired tumour control when hydroxychloroquine was initiated 5 days after first anti-PD-1 initiation, again underlining the importance of timing128.
Clinical studies evaluating these agents upfront in combination with ICI are currently underway (NCT03681561, NCT03012230, NCT02423954, and NCT02890069). Although these agents have been studied in several pre-clinical models, these models lack translatability given difference in timing relative to ICI and dosage of immunosuppressants. Therefore, data are currently far too limited to draw any conclusions on the effects of these immune modulators on ICI efficacy in the context of irAEs.
Discussion and future directions
Adequate immunosuppressive treatment of irAEs is crucial to prevent fatality and chronicity and to preserve quality of life. Since evidence on the association of immunosuppressants with ICI efficacy has been limited until recently, possible unfavourable effects on ICI efficacy are generally not addressed in current irAE management guidelines. This review indicates that aggressive immunosuppression to treat irAEs with high dose corticosteroids and escalated immunosuppression with other immunosuppressants may impair survival. Whether this effect is due to specific drugs or due to an overall effect of aggressive immunosuppression has yet to be determined. Nevertheless, we defined clinical recommendations based on pre-clinical and clinical data (Box 2).
Pre-clinical studies have currently mainly focussed on upfront combination of immunosuppressants with ICI, which hampers the translatability to the clinical setting of irAE management. Experimental models have clear limitations as models for human disease, including cancer and irAE, such as differences between immune systems131 and inoculation with allogenic cell lines instead of spontaneous tumour growth. Challenges with current irAE models have been critically reviewed elsewhere132,133,134,135. Some autoimmunity, like myocarditis, can reproducibly be induced in transgenic CTLA4−/− or PDCD1−/− mice135. In contrast to observations in humans, spontaneous irAEs, which are solely induced by immune checkpoint targeted monoclonal antibodies, are sparsely observed in rodents133,136,137. Therefore, inflammation is often induced by additional means in irAE experiments, either by using mice with a predisposing genetic background (such as non-obese diabetic mice), immunisation (as with experimental autoimmune encephalitis or myocarditis), irradiation or biochemical initiation of inflammation (as with DSS-induced colitis)82,109,135,138. In general, duration of most pre-clinical irAE research may be insufficient for genuine irAEs to develop132. This was illustrated by a study that induced overt colitis in Balb/c mice by prolonged (50 days) administration of anti-CTLA-4+/− anti-PD-1139. In addition, the relatively young age of mice used for experimental models is likely pivotal to why current models poorly reflect human irAEs. A recent study showed that anti-PD-1 treatment inflicted irAE-like infiltrates in various organs along with evidence for end-organ dysfunction in aged (>18 months) but not young (<3 months) tumour-bearing C57BL/6 and Balb/c mice140. Lastly, specific pathogen-free breeding conditions of laboratory mice limit translatability, since host microbiome composition has been associated with irAE development141. Some have attempted to address this limitation by skin commensal colonisation or faecal microbiota transfer followed by ICI administration to induce skin and gut irAEs141,142. A step further towards a diverse microbiome while maintaining the specific genetic background would be to exploit wildling mice, bred by transferring laboratory mouse embryos to wild female mice143.
Apart from the above inherent shortcomings in pre-clinical models, differences in timing of immunosuppressant administration relative to ICI initiation and dosage of immunosuppression may have led to inconsistent results across pre-clinical studies. Together, these differences may account for conflicting results observed in mice concurrently treated with ICI plus immunosuppression versus patients sequentially receiving immunosuppression for irAEs upon ICI. In Box 3 we present various considerations for pre-clinical irAE research and suggestions to improve current animal experiments. Such improvements to experimental models would indispensably come with rising costs, while essential deviations between mice and man will ultimately remain. Simultaneously, generation of human multi-omics data has become increasingly affordable and data availability through open repositories facilitates reuse by a wide public. These changes will further accelerate the era of ‘human immunology’. Herein, pre-clinical models will remain important, though no longer the go-to for investigating complex immunological disease such as irAEs and their interactions with environmental, tumour and host factors.
Previous clinical studies assessing the effects of immunosuppression for irAEs were mainly observational in heterogeneous cohorts with respect to cancer type and ICI regimen, often using a suboptimal control group. Therefore, most current clinical data are prone to confounding (by indication) and immortal-time bias. Randomised controlled trials (RCTs) comparing different irAE management regimens would provide clear answers to crucial questions. Although some clinical trials investigating irAE management are ongoing, none of them is sufficiently powered to assess ICI efficacy (Supplementary table 8). Given the vast heterogeneity in terms of cancer type, type and severity of irAEs and numerous possible immunosuppressants to test, RCTs powered on survival outcomes would demand tremendous multicentre efforts. Moreover, large well-structured collaborative observational studies can result in homogeneous and relevant cohorts, facilitating assessment of specific immunosuppressive regimens and validate results in different settings (Box 4).
Numerous questions remain to be answered in further well-designed pre-clinical and clinical studies. To guide clinical practice, data are needed to decipher whether impaired survival in patients treated with second-line immunosuppressants is due to specific therapeutics, reflects high dose corticosteroid usage or is a result of escalated immunosuppression as a whole. This could also provide clues for early versus late initiation of second-line immunosuppression and might be the steppingstone for skipping corticosteroids as first line and moving directly to more targeted immunosuppressants. Unravelling whether short course of high dose corticosteroids or lower dosages for a longer duration are most harmful is key to provide guidance for corticosteroid escalation and tapering strategies. Furthermore, whether associations between irAE treatment and ICI efficacy are similar for both early-onset and late-onset irAEs deserves further attention.
Taken together, clinicians should be aware of possible detrimental effects of immunosuppressants on ICI efficacy. Pre-clinical studies should consider timing and co-administration of several immunosuppressants at several dosages. Large multicentre randomised controlled trials and observational cohort studies are warranted to provide clinicians with more leads to effectively treat irAEs while maintaining ICI anti-tumour response.
Methods
To ensure a complete overview of current literature, a thorough systematic literature search was conducted in PubMed with synonyms for “immune checkpoint inhibition”, “immunosuppressive agents”, specific drug names and appropriate Mesh terms, resulting in 7836 records. Relevant pre-clinical studies providing insight into pathophysiology of immunosuppression in the context of ICI and pre-clinical studies using animal models with ICI ± immunosuppression as determinant, and tumour volume or any measure of survival as outcome were selected. Clinical studies reporting on tumour response or survival in relation to immunosuppressive irAE management were selected, except for case reports and case series. Four relevant reports that were encountered through reference tracking were also included. To maximise comprehensiveness of this review, for immunosuppressants that yielded peer-reviewed (pre-clinical) studies through PubMed, we complemented our search with bioRxiv/medRxiv database screening for recently published manuscripts on the combined effect of immunosuppressants and ICI. Search strategies are displayed in Supplementary methods.
Main findings of mouse and human data are summarised in tables and if a hazard ratio with 95% confidence interval were reported, these were visualised in a forest plot per type of immunosuppressant. Adjusted hazard ratios from multivariable analyses were reported whenever possible. Most relevant and rigorous studies are discussed in detail in the main text. The majority of studies suffer from the same methodological pitfalls and generalisability issues and our outcome of interest was not the main endpoint of most clinical studies. Therefore, we have chosen to discuss these limitations and their implications for clinical practice in general and refrained from conducting a formal risk of bias assessment or meta-analysis.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data referenced in this review can be accessed by following resources numbered in the Reference section.
Code availability
The code to create Figs. 2 and 3 is available via: https://github.com/rjverheijden/ICI_immunosuppression_review_2023.
References
Rowshanravan, B., Halliday, N. & Sansom, D. M. CTLA-4: a moving target in immunotherapy. Blood 131, 58–67 (2018).
Sharpe, A. H. & Pauken, K. E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 18, 153–167 (2018).
National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. U.S. Department of Health and Human Services (2017).
Wang, D. Y. et al. Fatal toxic effects associated with immune checkpoint inhibitors. JAMA Oncol. 4, 1721 (2018).
Wolchok, J. D. et al. Long-term outcomes with nivolumab plus ipilimumab or nivolumab alone versus ipilimumab in patients with advanced melanoma. J. Clin. Oncol. 40, 127–137 (2022).
Wang, Y. et al. Treatment-related adverse events of PD-1 and PD-L1 inhibitors in clinical trials: a systematic review and meta-analysis. JAMA Oncol. 5, 1008–1019 (2019).
Schneider, B. J. et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: ASCO Guideline Update. J. Clin. Oncol. 39, 4073–4126 (2021).
Haanen, J. et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 33, 1217–1238 (2022).
Brahmer, J. R. et al. Society for immunotherapy of cancer (sitc) clinical practice guideline on immune checkpoint inhibitor-related adverse events. J. Immunother. Cancer 9, e002435 (2021).
Das, S. & Johnson, D. B. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 7, 1–11 (2019).
Dall’Olio, F. G. et al. Immortal time bias in the association between toxicity and response for immune checkpoint inhibitors: a meta-analysis. Immunotherapy 13, 257–270 (2021).
Kfoury, M. et al. Analysis of the association between prospectively collected immune-related adverse events and survival in patients with solid tumor treated with immune-checkpoint blockers, taking into account immortal-time bias. Cancer Treat. Rev. 110, 102452 (2022).
Eggermont, A. M. M. M. et al. Association between immune-related adverse events and recurrence-free survival among patients with stage III melanoma randomized to receive pembrolizumab or placebo: a secondary analysis of a randomized clinical trial. JAMA Oncol. 6, 519–527 (2020).
Verheijden, R. J. et al. Association of anti-TNF with decreased survival in steroid refractory ipilimumab and anti-PD1-treated patients in the dutch melanoma treatment registry. Clin. Cancer Res. 26, 2268–2274 (2020).
Jing, Y., Yang, J., Johnson, D. B., Moslehi, J. J. & Han, L. Harnessing big data to characterize immune-related adverse events. Nat. Rev. Clin. Oncol. 19, 269–280 (2022).
Giobbie-Hurder, A., Gelber, R. D. & Regan, M. M. Challenges of guarantee-time bias. J. Clin. Oncol. 31, 2963–2969 (2013).
Zhong, L., Wu, Q., Chen, F., Liu, J. & Xie, X. Immune-related adverse events: promising predictors for efficacy of immune checkpoint inhibitors. Cancer Immunol. Immunother. 70, 2559–2576 (2021).
Li, Y. et al. Effect of immune-related adverse events and pneumonitis on prognosis in advanced non–small cell lung cancer: a comprehensive systematic review and meta-analysis. Clin. Lung Cancer 22, e889–e900 (2021).
Fan, Y. et al. Association of immune related adverse events with efficacy of immune checkpoint inhibitors and overall survival in cancers: a systemic review and meta-analysis. Front. Oncol. 11, 1081 (2021).
Tarhini, A. A. et al. Immune adverse events (irAEs) with adjuvant ipilimumab in melanoma, use of immunosuppressants and association with outcome: ECOG-ACRIN E1609 study analysis. J. Immunother. Cancer 9, e002535 (2021).
Rozeman, E. A. et al. Identification of the optimal combination dosing schedule of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma (OpACIN-neo): a multicentre, phase 2, randomised, controlled trial. Lancet Oncol. 20, 948–960 (2019).
Reijers, I. L. M. et al. Personalized response-directed surgery and adjuvant therapy after neoadjuvant ipilimumab and nivolumab in high-risk stage III melanoma: the PRADO trial. Nat. Med. 28, 1178–1188 (2022).
Luoma, A. M. et al. Tissue-resident memory and circulating T cells are early responders to pre-surgical cancer immunotherapy. Cell 185, 2918–2935.e29 (2022).
Jansen, Y. J. L. et al. Discontinuation of anti-PD-1 antibody therapy in the absence of disease progression or treatment limiting toxicity: clinical outcomes in advanced melanoma. Ann. Oncol. 30, 1154–1161 (2019).
Schadendorf, D. et al. Efficacy and safety outcomes in patients with advanced melanoma who discontinued treatment with nivolumab and ipilimumab because of adverse events: a pooled analysis of randomized phase II and III trials. J. Clin. Oncol. 35, 3807–3814 (2017).
Cain, D. W. & Cidlowski, J. A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 17, 233–247 (2017).
Kalfeist, L. et al. Impact of glucocorticoid use in oncology in the immunotherapy era. Cells 11, 1–22 (2022).
Aldea, M. et al. How to manage patients with corticosteroids in oncology in the era of immunotherapy? Eur. J. Cancer 141, 239–251 (2020).
Xing, K., Gu, B., Zhang, P. & Wu, X. Dexamethasone enhances programmed cell death 1 (PD-1) expression during T cell activation: an insight into the optimum application of glucocorticoids in anti-cancer therapy. BMC Immunol. 16, 1–9 (2015).
Acharya, N. et al. Endogenous glucocorticoid signaling regulates CD8+ T cell differentiation and development of dysfunction in the tumor microenvironment. Immunity 53, 658–671.e6 (2020).
Giles, A. J. et al. Dexamethasone-induced immunosuppression: mechanisms and implications for immunotherapy. J. Immunother. Cancer 6, 1–13 (2018).
Iorgulescu, B. J. et al. Concurrent dexamethasone limits the clinical benefit of immune checkpoint blockade in glioblastoma. Clin. Cancer Res. 27, 276–287 (2021).
Bereshchenko, O. et al. GILZ promotes production of peripherally induced treg cells and mediates the crosstalk between glucocorticoids and TGF-β signaling. Cell Rep. 7, 464–475 (2014).
Maeda, N. et al. Glucocorticoids potentiate the inhibitory capacity of programmed cell death 1 by up-regulating its expression on T cells. J. Biol. Chem. 294, 19896–19906 (2019).
Maxwell, R. et al. Contrasting impact of corticosteroids on anti-PD-1 immunotherapy efficacy for tumor histologies located within or outside the central nervous system. Oncoimmunology 7, 1–14 (2018).
Aston, W. J. et al. Dexamethasone differentially depletes tumour and peripheral blood lymphocytes and can impact the efficacy of chemotherapy/checkpoint blockade combination treatment. Oncoimmunology 8, e1641390 (2019).
Xiang, Z. et al. Dexamethasone suppresses immune evasion by inducing GR/STAT3 mediated downregulation of PD-L1 and IDO1 pathways. Oncogene 40, 5002–5012 (2021).
Tokunaga, A. et al. Selective inhibition of low-affinity memory CD8+ T cells by corticosteroids. J. Exp. Med. 216, 2701–2713 (2019).
Petrelli, F. et al. Association of steroids use with survival in patients treated with immune checkpoint inhibitors: a systematic review and meta-analysis. Cancers 12, 1–11 (2020).
Wang, Y. et al. Corticosteroid administration for cancer-related indications is an unfavorable prognostic factor in solid cancer patients receiving immune checkpoint inhibitor treatment. Int. Immunopharmacol. 99, 108031 (2021).
Riudavets, M. et al. Immune-related adverse events and corticosteroid use for cancer-related symptoms are associated with efficacy in patients with non-small cell lung cancer receiving anti-PD-(L)1 blockade agents. Front. Oncol. 10, 1–11 (2020).
Mouri, A. et al. Effect of systemic steroid use for immune-related adverse events in patients with non-small cell lung cancer receiving PD-1 blockade drugs. J. Clin. Med. 10, 3744 (2021).
Ishihara, H. et al. Prognostic impact of early treatment interruption of nivolumab plus ipilimumab due to immune-related adverse events as first-line therapy for metastatic renal cell carcinoma: a multi-institution retrospective study. Target. Oncol. 16, 493–502 (2021).
Paderi, A. et al. Association of systemic steroid treatment and outcome in patients treated with immune checkpoint inhibitors: a real-world analysis. Molecules 26, 5789 (2021).
Weber, J. et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin. Cancer Res. 15, 5591–5598 (2009).
Bai, X. et al. Early use of high-dose glucocorticoid for the management of irAE is associated with poorer survival in patients with advanced melanoma treated with anti-PD-1 monotherapy. Clin. Cancer Res. 27, 5993–6000 (2021).
Lafayolle de la Bruyère, C. et al. Investigating the impact of immune-related adverse events, glucocorticoid use and immunotherapy interruption on long-term survival outcomes. Cancers 13, 2365 (2021).
Johnson, D. B. et al. Survivorship in immune therapy: assessing chronic immune toxicities, health outcomes, and functional status among long-term ipilimumab survivors at a single referral center. Cancer Immunol. Res. 3, 464–469 (2015).
Horvat, T. Z. et al. Immune-related adverse events, need for systemic immunosuppression, and effects on survival and time to treatment failure in patients with melanoma treated with ipilimumab at memorial sloan kettering cancer center. J. Clin. Oncol. 33, 3193–3198 (2015).
Maher, V. E. et al. Analysis of the association between adverse events and outcome in patients receiving a programmed death protein 1 or programmed death ligand 1 antibody. J. Clin. Oncol. 37, 2730–2737 (2019).
Vitale, M. G. et al. Correlation between immune-related adverse event (IRAE) occurrence and clinical outcome in patients with metastatic renal cell carcinoma (mRCC) treated with nivolumab: IRAENE Trial, an Italian Multi-institutional Retrospective Study. Clin. Genitourin. Cancer 18, 477–488 (2020).
Shafqat, H., Gourdin, T. & Sion, A. Immune-related adverse events are linked with improved progression-free survival in patients receiving anti-PD-1/PD-L1 therapy. Semin. Oncol. 45, 156–163 (2018).
Arheden, A. et al. Real-world data on PD-1 inhibitor therapy in metastatic melanoma. Acta Oncol. 58, 962–966 (2019).
Skribek, M. et al. Effect of corticosteroids on the outcome of patients with advanced non–small cell lung cancer treated with immune-checkpoint inhibitors. Eur. J. Cancer 145, 245–254 (2021).
Gaucher, L. et al. Impact of the corticosteroid indication and administration route on overall survival and the tumor response after immune checkpoint inhibitor initiation. Ther. Adv. Med. Oncol. 13, 1758835921996656 (2021).
Dahl, E. K. et al. Safety and efficacy of infliximab and corticosteroid therapy in checkpoint inhibitor-induced colitis. Aliment. Pharmacol. Ther. 56, 1370–1382 (2022).
Gauci, M. L. et al. Severe immune-related hepatitis induced by immune checkpoint inhibitors: clinical features and management proposal. Clin. Res. Hepatol. Gastroenterol. 45, 101491 (2021).
Romanski, N. A., Holmstroem, R. B., Ellebaek, E. & Svane, I. M. Characterization of risk factors and efficacy of medical management of immune-related hepatotoxicity in real-world patients with metastatic melanoma treated with immune checkpoint inhibitors. Eur. J. Cancer 130, 211–218 (2020).
Dimitriou, F. et al. Frequency, treatment and outcome of immune-related toxicities in patients with immune-checkpoint inhibitors for advanced melanoma: results from an institutional database analysis. Cancers 13, 1–15 (2021).
Pan, E. Y., Merl, M. Y. & Lin, K. The impact of corticosteroid use during anti-PD1 treatment. J. Oncol. Pharm. Pract. 26, 814–822 (2020).
Robert, C. et al. Long-term safety of pembrolizumab monotherapy and relationship with clinical outcome: a landmark analysis in patients with advanced melanoma. Eur. J. Cancer 144, 182–191 (2021).
Faje, A. T. et al. High-dose glucocorticoids for the treatment of ipilimumab-induced hypophysitis is associated with reduced survival in patients with melanoma. Cancer 124, 3706–3714 (2018).
Li, M. et al. Effect of corticosteroid dosing on outcomes in high-grade immune checkpoint inhibitor hepatitis. Hepatology 75, 531–540 (2022).
Thompson, L. L. et al. Impact of systemic corticosteroids on survival outcomes in immune checkpoint inhibitor–induced gastroenterocolitis. Eur. J. Cancer 142, 143–146 (2021).
Wang, Y. et al. Immune-checkpoint inhibitor-induced diarrhea and colitis in patients with advanced malignancies: retrospective review at MD Anderson. J. Immunother. Cancer 6, 1–13 (2018).
Min, L. et al. Systemic high-dose corticosteroid treatment does not improve the outcome of ipilimumab-related hypophysitis: a retrospective cohort study. Clin. Cancer Res. 21, 749–755 (2015).
Chen, A. Y., Wolchok, J. D. & Bass, A. R. TNF in the era of immune checkpoint inhibitors: friend or foe? Nat. Rev. Rheumatol. 17, 213–223 (2021).
Salomon, B. L. Insights into the biology and therapeutic implications of TNF and regulatory T cells. Nat. Rev. Rheumatol. 17, 487–504 (2021).
Moatti, A. & Cohen, J. L. The TNF-α/TNFR2 pathway: targeting a brake to release the anti-tumor immune response. Front. Cell Dev. Biol. 9, 725473 (2021).
Benoot, T., Piccioni, E., De Ridder, K. & Goyvaerts, C. TNFα and immune checkpoint inhibition: friend or foe for lung cancer? Int. J. Mol. Sci. 22, 1–17 (2021).
Suijkerbuijk, K. P. M. & Verheijden, R. J. TNF inhibition for immune checkpoint inhibitor-induced irAEs: the jury is still out. Nat. Rev. Rheumatol. 17, 505–505 (2021).
Bass, A. R. & Chen, A. Y. Reply to: TNF inhibition for immune checkpoint inhibitor-induced irAEs: the jury is still out. Nat. Rev. Rheumatol. 17, 505–506 (2021).
Selby, P. et al. Tumour necrosis factor in man: clinical and biological observations. Br. J. Cancer 56, 803–808 (1987).
Blick, M., Sherwin, S. A., Rosenblum, M. & Gutterman, J. Phase I study of recombinant tumor necrosis factor in cancer patients1 | Cancer Research | American Association for Cancer Research. Cancer Res. 47, 2986–2989 (1987).
Gamm, H., Lindemann, A., Mertelsmann, R. & Herrmann, F. Phase I trial of recombinant human tumour necrosis factor α in patients with advanced malignancy. Eur. J. Cancer Clin. Oncol. 27, 856–863 (1991).
Brown, T. D. et al. A phase II trial of recombinant tumor necrosis factor in patients with adenocarcinoma of the pancreas: a Southwest Oncology Group study. Immunother. 10, 376–378 (1991).
Madhusudan, S. et al. A phase II study of etanercept (enbrel), a tumor necrosis factor α inhibitor in patients with metastatic breast cancer. Clin. Cancer Res. 10, 6528–6534 (2004).
Madhusudan, S. et al. Study of etanercept, a tumor necrosis factor-alpha inhibitor, in recurrent ovarian cancer. J. Clin. Oncol. 23, 5950–5959 (2005).
Harrison, M. L. et al. Tumor necrosis factor α as a new target for renal cell carcinoma: two sequential phase II trials of infliximab at standard and high dose. J. Clin. Oncol. 25, 4542–4549 (2007).
Brown, E. R. et al. A clinical study assessing the tolerability and biological effects of infliximab, a TNF-α inhibitor, in patients with advanced cancer. Ann. Oncol. 19, 1340–1346 (2008).
Bertrand, F. et al. TNFα blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat. Commun. 8, 2256 (2017).
Perez-Ruiz, E. et al. Prophylactic TNF blockade uncouples efficacy and toxicity in dual CTLA-4 and PD-1 immunotherapy. Nature 569, 428–432 (2019).
Montfort, A. et al. Combining Nivolumab and Ipilimumab with Infliximab or Certolizumab in Patients with Advanced Melanoma: First Results of a Phase Ib Clinical Trial. Clin. Cancer Res. 27, 1037–1047 (2021).
Abu-Sbeih, H. et al. Early introduction of selective immunosuppressive therapy associated with favorable clinical outcomes in patients with immune checkpoint inhibitor-induced colitis. J. Immunother. Cancer 7, 1–11 (2019).
Zou, F. et al. Efficacy and safety of vedolizumab and infliximab treatment for immune-mediated diarrhea and colitis in patients with cancer: a two-center observational study. J. Immunother. Cancer 9, e003277 (2021).
Burdett, N. et al. Cancer outcomes in patients requiring immunosuppression in addition to corticosteroids for immune-related adverse events after immune checkpoint inhibitor therapy. Asia. Pac. J. Clin. Oncol. 16, e139–e145 (2019).
van Not, O. J. et al. Association of Immune-Related Adverse Event Management With Survival in Patients With Advanced Melanoma. JAMA Oncol. 8, 2374–2445 (2022).
Arriola, E., Wheater, M., Karydis, I., Thomas, G. & Ottensmeier, C. Infliximab for IPILIMUMAB-related colitis-letter. Clin. Cancer Res. 21, 5642–5643 (2015).
Johnson, D. H. et al. Infliximab associated with faster symptom resolution compared with corticosteroids alone for the management of immune-related enterocolitis. J. Immunother. Cancer 6, 103–108 (2018).
Lesage, C. et al. Incidence and clinical impact of anti-TNFalpha treatment of severe immune checkpoint inhibitor-induced colitis in advanced melanoma: the mecolit survey. J. Immunother. 42, 175–179 (2019).
Favara, D. M. et al. Five-year review of corticosteroid duration and complications in the management of immune checkpoint inhibitor-related diarrhoea and colitis in advanced melanoma. ESMO Open 5, e000585 (2020).
Nahar, K. J. et al. Clinicopathological characteristics and management of colitis with anti-PD1 immunotherapy alone or in combination with ipilimumab. J. Immunother. Cancer 8, e001488 (2020).
Alexander, J. L. et al. Clinical outcomes of patients with corticosteroid refractory immune checkpoint inhibitor-induced enterocolitis treated with infliximab. J. Immunother. Cancer 9, 1–9 (2021).
Araujo, D. V. et al. Real world outcomes and hepatotoxicity of infliximab in the treatment of steroid-refractory immune-related adverse events. Curr. Oncol. 28, 2173–2179 (2021).
Abu-Sbeih, H. et al. Outcomes of vedolizumab therapy in patients with immune checkpoint inhibitor–induced colitis: a multi-center study. J. Immunother. Cancer 6, 142 (2018).
Johnson, D. E., O’Keefe, R. A. & Grandis, J. R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 15, 234–248 (2018).
Kang, S., Tanaka, T., Narazaki, M. & Kishimoto, T. Targeting Interleukin-6 Signaling in Clinic. Immunity 50, 1007–1023 (2019).
Kumari, N., Dwarakanath, B. S., Das, A. & Bhatt, A. N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumor Biol. 37, 11553–11572 (2016).
Tsukamoto, H. et al. Immune-suppressive effects of interleukin-6 on T-cell-mediated anti-tumor immunity. Cancer Sci. 109, 523–530 (2018).
Kang, D. H. et al. Baseline serum interleukin-6 levels predict the response of patients with advanced non-small cell lung cancer to pd-1/pd-l1 inhibitors. Immune Netw. 20, 1–11 (2020).
Myojin, Y. et al. Interleukin-6 is a circulating prognostic biomarker for hepatocellular carcinoma patients treated with combined immunotherapy. Cancers 14, 883 (2022).
Liu, C. et al. Systematic analysis of IL-6 as a predictive biomarker and desensitizer of immunotherapy responses in patients with non-small cell lung cancer. BMC Med. 20, 1–15 (2022).
Keegan, A. et al. Plasma IL-6 changes correlate to PD-1 inhibitor responses in NSCLC. J. Immunother. Cancer 8, 1–8 (2020).
Tsukamoto, H. et al. Combined blockade of IL6 and PD-1/PD-L1 signaling abrogates mutual regulation of their immunosuppressive effects in the tumor microenvironment. Cancer Res. 78, 5011–5022 (2018).
Liu, H., Shen, J. & Lu, K. IL-6 and PD-L1 blockade combination inhibits hepatocellular carcinoma cancer development in mouse model. Biochem. Biophys. Res. Commun. 486, 239–244 (2017).
Ohno, Y. et al. Lack of interleukin-6 in the tumor microenvironment augments type-1 immunity and increases the efficacy of cancer immunotherapy. Cancer Sci. 108, 1959–1966 (2017).
Li, J. et al. Targeting interleukin-6 (IL-6) sensitizes anti-PD-L1 treatment in a colorectal cancer preclinical model. Med. Sci. Monit. 24, 5501–5508 (2018).
Mace, T. A. et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 67, 320–332 (2018).
Hailemichael, Y. et al. Interleukin-6 blockade abrogates immunotherapy toxicity and promotes tumor immunity. Cancer Cell 40, 509–523.e6 (2022).
Li, W. et al. Blockade of IL-6 inhibits tumor immune evasion and improves anti–PD-1 immunotherapy. Cytokine 158, 155976 (2022).
Stroud, C. R. G. et al. Tocilizumab for the management of immune mediated adverse events secondary to PD-1 blockade. J. Oncol. Pharm. Pract. 25, 551–557 (2019).
Campochiaro, C. et al. Tocilizumab for the treatment of immune-related adverse events: a systematic literature review and a multicentre case series. Eur. J. Intern. Med. 93, 87–94 (2021).
Dimitriou, F., Hogan, S., Menzies, A. M., Dummer, R. & Long, G. V. Interleukin-6 blockade for prophylaxis and management of immune-related adverse events in cancer immunotherapy. Eur. J. Cancer 157, 214–224 (2021).
Holmstroem, R. B. et al. COLAR: open-label clinical study of IL-6 blockade with tocilizumab for the treatment of immune checkpoint inhibitor-induced colitis and arthritis. J. Immunother. Cancer 10, e005111 (2022).
Weber, J. S. et al. 1040O Phase II trial of ipilimumab, nivolumab and tocilizumab for unresectable metastatic melanoma. Ann. Oncol. 32, S869 (2021).
Martins, F. et al. New therapeutic perspectives to manage refractory immune checkpoint-related toxicities. Lancet Oncol. 20, e54–e64 (2019).
Esfahani, K. et al. Moving towards personalized treatments of immune-related adverse events. Nat. Rev. Clin. Oncol. 17, 504–515 (2020).
Henderson Berg, M. H., del Rincón, S. V. & Miller, W. H. Potential therapies for immune-related adverse events associated with immune checkpoint inhibition: from monoclonal antibodies to kinase inhibition. J. Immunother. Cancer 10, 1–11 (2022).
Zak, J. et al. Myeloid reprogramming by JAK inhibition enhances checkpoint blockade therapy. bioRxiv https://doi.org/10.1101/2022.06.24.497435. (2022).
Benci, J. L. et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 167, 1540–1554.e12 (2016).
Lu, C., Talukder, A., Savage, N. M., Singh, N. & Liu, K. JAK-STAT-mediated chronic inflammation impairs cytotoxic T lymphocyte activation to decrease anti-PD-1 immunotherapy efficacy in pancreatic cancer. Oncoimmunology 6, 1–15 (2017).
Langdon, S. et al. Combination of dual mTORC1/2 inhibition and immune-checkpoint blockade potentiates anti-tumour immunity. Oncoimmunology 7, e1458810 (2018).
Bai, X. et al. Improvement of PD-1 blockade efficacy and elimination of immune-related gastrointestinal adverse effect by mTOR inhibitor. Front. Immunol. 12, 1–14 (2021).
Xia, W. et al. The combination therapy of Everolimus and anti-PD-1 improves the antitumor effect by regulating CD8+ T cells in bladder cancer. Med. Oncol. 39, 1–10 (2022).
Song, L., Liu, S. & Zhao, S. Everolimus (RAD001) combined with programmed death-1 (PD-1) blockade enhances radiosensitivity of cervical cancer and programmed death-ligand 1 (PD-L1) expression by blocking the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of. Bioengineered 13, 11240–11257 (2022).
Sharma, G. et al. PPT1 inhibition enhances the antitumor activity of anti–PD-1 antibody in melanoma. JCI Insight 5, e165688 (2020).
Wabitsch, S. et al. Hydroxychloroquine can impair tumor response to anti-PD1 in subcutaneous mouse models. iScience 24, 101990 (2021).
Krueger, J. et al. Hydroxychloroquine (HCQ) decreases the benefit of anti-PD-1 immune checkpoint blockade in tumor immunotherapy. PLoS ONE 16, 1–20 (2021).
Shin, D. S. et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 7, 188–201 (2017).
Zaretsky, J. M. et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375, 819–829 (2016).
Medetgul-Ernar, K. & Davis, M. M. Standing on the shoulders of mice. Immunity 55, 1343–1353 (2022).
Bayless, N. L. et al. Development of preclinical and clinical models for immune-related adverse events following checkpoint immunotherapy: a perspective from SITC and AACR. J. Immunother. Cancer 9, e002627 (2021).
Liu, J., Blake, S. J., Smyth, M. J. & Teng, M. W. Improved mouse models to assess tumour immunity and irAEs after combination cancer immunotherapies. Clin. Transl. Immunol. 3, e22 (2014).
Saito, R., Kobayashi, T., Kashima, S., Matsumoto, K. & Ogawa, O. Faithful preclinical mouse models for better translation to bedside in the field of immuno-oncology. Int. J. Clin. Oncol. 25, 831–841 (2020).
Wong, C.-K. et al. Immunopathogenesis of immune checkpoint inhibitor induced myocarditis: insights from experimental models and treatment implications. Biomedicines 11, 107 (2023).
Liu, S.-Y. et al. Sequential blockade of PD-1 and PD-L1 causes fulminant cardiotoxicity—from case report to mouse model validation. Cancers 11, 580 (2019).
Zhang, Y. et al. Hormonal therapies up-regulate MANF and overcome female susceptibility to immune checkpoint inhibitor myocarditis. Sci. Transl. Med. 14, eabo1981 (2022).
Tsuruoka, K. et al. Exacerbation of autoimmune myocarditis by an immune checkpoint inhibitor is dependent on its time of administration in mice. Int. J. Cardiol. 313, 67–75 (2020).
Bauché, D. et al. Antitumor efficacy of combined CTLA4/PD-1 blockade without intestinal inflammation is achieved by elimination of FcγR interactions. J. Immunother. Cancer 8, e001584 (2020).
Tsukamoto, H. et al. Aging-associated and CD4 T-cell–dependent ectopic CXCL13 activation predisposes to anti–PD-1 therapy-induced adverse events. Proc. Natl Acad. Sci. USA 119, e2205378119 (2022).
Andrews, M. C. et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat. Med. 27, 1432–1441 (2021).
Hu, Z. I. et al. Immune checkpoint inhibitors unleash pathogenic immune responses against the microbiota. Proc. Natl Acad. Sci. USA 119, e2200348119 (2022).
Rosshart, S. P. et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 365, eaaw4361 (2019).
Acknowledgements
We would like to acknowledge Frederieke van der Baan for her critical review and thoughtful suggestions during the preparation of this manuscript. This study received no funding.
Author information
Authors and Affiliations
Contributions
R.J.V. and M.J.M.v.E. have searched, critically appraised, and summarised the literature. R.J.V. has drafted the manuscript. All authors have designed, revised and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
K.P.M.S. has advisory relationships with Bristol Myers Squibb, Novartis, MSD, Pierre Fabre and AbbVie and received honoraria from Novartis, MSD and Roche. Research funding: BMS, Philips and TigaTx are All paid to the institution. F.v.W. has received advisory/speaker fees from Takeda and Johnson&Johnson, and has received research funding from BMS, Takeda, Sanofi, Pfizer, Galapagos and Leo Pharma. No potential conflicts of interest were disclosed by the other authors.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Verheijden, R.J., van Eijs, M.J.M., May, A.M. et al. Immunosuppression for immune-related adverse events during checkpoint inhibition: an intricate balance. npj Precis. Onc. 7, 41 (2023). https://doi.org/10.1038/s41698-023-00380-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-023-00380-1
This article is cited by
-
Rare, late onset of immune checkpoint inhibitor-induced type 1 diabetes mellitus in a patient with small-cell lung cancer treated with serplulimab: a case report and review of the literature
Journal of Medical Case Reports (2024)
-
Severe cutaneous adverse reactions
Nature Reviews Disease Primers (2024)
-
Clinical and translational attributes of immune-related adverse events
Nature Cancer (2024)
-
Update on Neuro-ophthalmic Manifestations of Immune Checkpoint Inhibitors
Current Neurology and Neuroscience Reports (2024)
