
Abstract
We designed and prepared a novel series of urea derivatives with/without sulfonyl group in their structures to investigate the impact of the sulfonyl group on the biological activity of the evaluated compounds. Antibacterial investigations indicated that derivatives 7, 8, 9, and 11 had the most antibacterial property of all the compounds examined, their minimum inhibitory concentrations (MICs) determined against B. mycoides, E. coli, and C. albicans, with compound 8 being the most active at a MIC value of 4.88 µg/mL. Anti-cancer activity has been tested against eight human cancer cell lines; A549, HCT116, PC3, A431, HePG2, HOS, PACA2 and BJ1. Compounds 7, 8 and 9 emerged IC50 values better than Doxorubicin as a reference drug. Compounds 7 and 8 showed IC50 = 44.4 and 22.4 μM respectively against PACA2 compared to Doxorubicin (IC50 = 52.1 μM). Compound 9 showed IC50 = 17.8, 12.4, and 17.6 μM against HCT116, HePG2, and HOS, respectively. qRT-PCR revealed the down-regulation of PALB2 in compounds 7 and 15 treated PACA2 cells. Also, the down-regulation of BRCA1 and BRCA2 was shown in compound 7 treated PC3 cells. As regard A549 cells, compound 8 decreased the expression level of EGFR and KRAS genes. While compounds 7 and 9 down-regulated TP53 and FASN in HCT116 cells. Molecular docking was done against Escherichia coli enoyl reductase and human Son of sevenless homolog 1 (SOS1) and the results showed the promising inhibition of the studied proteins.
Similar content being viewed by others
Introduction
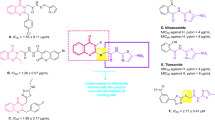
The trifluoromethyl group (abbreviated as CF3), which is more bulky than the methyl group, is one of the most common lipophilic functional groups1,2,3. Due to the impact of the trifluoromethyl substituents on the electronic characteristics of the aromatic rings, the best-reported drugs containing aromatic trifluoromethyl substitution in their chemical structure. For example (as shown in Fig. 1), Prozac (anti-depressant fluoxetine), Emend (or Aprepitant as antiemetic drug), Celecoxib (arthritis medication and COX-2 inhibitor celecoxib), Casodex (or Bicalutamide as anticancer drug) and Januvia (used in the treatment of diabetes symptoms) having aromatic trifluoromethyl substitution4,5,6,7,8. Surprisingly, the presence of the trifluoromethyl substituent is common in increasing the potency by the formation of multipolar bindings with the carbonyl groups in the targeted protein7. On the other hand, the aryl-urea or sulfonyl-urea derivatives are of great interest in various areas of organic chemistry, coordination chemistry, and medicinal chemistry (Fig. 1) 9,10,11,12,13,14,15,16. It was reported the anticancer activity of several urea derivatives including N-nitroso-urea which is an alkylating agent drug17,18. Also, glibenclamide which is sulfonylurea proved to be a tumor growth inhibitor by inducing reactive oxygen species (ROS) followed by apoptosis of cancer cells19. Indeed, the combination of aromatic trifluoromethyl substitutions with urea moiety in one compound will increase the biological activity of the final product.

Marketed drugs containing trifluoromethyl group and/or aryl-urea derivatives.
Antibiotic resistance (which refers to microorganisms' resistance to antimicrobial medicines) has made treating life-threatening bacterial infections much more difficult20,21. If the current rate of antimicrobial resistance continues, it is postulated that if no steps were taken, drug-resistant infections will be responsible for 10 million deaths worldwide per year by 2050, exceeding cancer deaths22. Antibiotic use is a major contributor to antibiotic resistance. In communities, primary care facilities, nursing homes, and particular hospitals, as well as across nations, the link between antibiotic usage and resistance has been widely documented23,24. There are various difficulties in quantifying the illness burden associated with infections caused by antibiotic-resistant bacteria. For example, sampling and microbiological protocols for testing isolates, data-gathering processes, and surveillance system architecture may differ between and within nations. Furthermore, understanding of the clinical and public health repercussions of antibiotic-resistant bacterial infections in people is relatively limited. In particular, the scientific debate is raging over the best epidemiological research design and statistical inference methodologies for making realistic estimates of adverse clinical outcomes caused by antibiotic-resistant bacterial infections21,25,26. National and international agencies have devised and are implementing action plans to slow the spread of antimicrobial resistance. Research and innovation, infection control practices, stewardship, and surveillance are the four basic principles of these action plans22,23,27.
Antibiotic resistance frequently develops as a result of extended usage, and it typically promotes the growth of resistant bacterial isolates while inhibiting the growth of susceptible germs. Most of the time, continual antibiotic pressure leads to resistance not only to the antibiotic in question but also to other antibiotics in the same class20,28. Bacterial antibiotic resistance mechanisms are mainly classified into three basic pathways: inhibition of antibiotic binding to the target site through exclusion techniques, modification or destruction of the antibiotic molecule, and mutation of the antibiotic target site. Furthermore, bacteria typically acquire resistance genes by transformation through getting resistant genes from their surroundings, transduction through transferring resistant genes from bacteriophages, and bacterial conjugation via resistant gene transfer between resistant bacterial strains29,30. As antibiotic-resistant bacteria become more common, replacements for antibiotics should indeed be recognized. Some of the proposed alternatives include antibiotic framework alteration, combinational administration of drugs, antibiotic-adjuvant hybrids, as well as the use of biopharmaceuticals20.
Consequently and in continuation of our efforts to design and develop biologically active organic compounds31,32,33,34,35,36, in the current study we decided to design and synthesize a new series of aryl-urea derivatives containing trifluoromethyl substitutions under mild and metal-free conditions to increase the biological profile of the final products as antimicrobial and anti-cancer agents.
Materials and methods
Chemistry
All reactions were carried out in aerobic conditions at room temperature. Acetonitrile was distilled and kept under an inert atmosphere. All glassware was oven-dried at 120 °C for at least 24 h before use. The starting materials of the primary amines 1, 2, 3, 4 and 5 and the isocyanate derivatives, 4-tolyl sulfonyl isocyanate (6) and 4-tolyl isocyanate (12) were purchased from Aldrich and used as received. All melting points are uncorrected and measured using Electro‐Thermal IA 9100 apparatus (Shimadzu, Japan). The Infrared spectra were recorded as potassium bromide pellets on a JASCO spectrophotometer between 4000 cm−1 and 400 cm−1. 1H NMR and 13C NMR spectra were recorded in deuterated dimethyl sulfoxide (DMSO-d6) on a Brucker spectrometer (400 MHz) at 25 °C. The chemical shifts were expressed as part per million (δ values, ppm) against TMS as an internal reference. Microanalyses were operated using Mario Elmentar apparatus, Organic Microanalysis Unit, National Research Centre (NRC), Cairo, Egypt.
General procedure for the synthesis of urea derivatives (7–11 and 13–17)
4-Tolyl sulfonyl isocyanate (6) (0.01 mol) or 4-tolyl isocyanate (12) (0.01 mol) was added to a solution of the primary amines (1, 2, 3, 4, or 5) (0.01 mol) in acetonitrile while stirring at room temperature. The reaction mixture was stirred to the desired time. The completion of reactions was monitored by TLC on silica gel-coated aluminum sheets. The obtained precipitate was filtered off, washed with cold acetonitrile and dried well, then recrystallized from ethyl acetate/acetonitrile (3:1) to give:
4-Methyl-N-[(4-{trifluoromethyl}phenyl)carbamoyl]benzenesulfonamide (7)
Compound 7 precipitated after 15 min as a white solid; m.p. 194–195 °C. IR (KBr): ν (cm-1) 3318, 3286 (NH), 1682 (C=O), 1341, 1167 (SO2). 1H NMR (DMSO-d6): δ (ppm) 2.35 (s, 3H, CH3), 7.26–7.85 (m, 9H, Ar + NH), 9.23 (br, 1H, NH). 13C NMR (DMSO-d6): δ (ppm) 21.4, 113.5, 119.3, 126.2, 126.6, 126.7, 128.1, 129.8, 137.4, 142.0, 142.4, 144.5, 150.0, 152.7. Anal. Calcd. for C15H13F3N2O3S (358.34): C, 50.28; H, 3.66; N, 7.82. Found: C, 50.36; H, 3.58; N, 7.77.
4-Methyl-N-[(4-{trifluoromethoxy}phenyl)carbamoyl]benzenesulfonamide (8)
Compound 8 precipitated after 7 min as a white solid; m.p. 183–184 °C. IR (KBr): ν (cm−1) 3322, 3284 (NH), 1681 (C=O), 1343, 1165 (SO2). 1H NMR (DMSO-d6): δ (ppm) 2.33 (s, 3H, CH3), 7.20–7.86 (m, 9H, Ar + NH), 9.03 (br, 1H, NH). 13C NMR (DMSO-d6): δ (ppm) 21.5, 115.0, 121.0, 122.0, 122.4, 126.2, 127.2, 129.4, 130.0, 137.8, 144.1, 144.4, 148.2, 150.0. Anal. Calcd. for C15H13F3N2O4S (374.33): C, 48.13; H, 3.50; N, 7.48. Found: C, 48.26; H, 3.59; N, 7.54.
N-[(3,5-bis{Trifluoromethyl}phenyl)carbamoyl]-4-methylbenzenesulfonamide (9)
Compound 9 precipitated after 20 min as a white solid; m.p. 172–174 °C. IR (KBr): ν (cm−1) 3323, 3281 (NH), 1685 (C=O), 1344, 1166 (SO2). 1H NMR (DMSO-d6): δ (ppm) 2.34 (s, 3H, CH3), 7.01–8.0 (m, 8H, Ar + NH), 9.50 (br, 1H, NH). 13C NMR (DMSO-d6): δ (ppm) 21.4, 100.0, 107.6, 113.4, 122.3, 125.6, 128.0, 129.5, 130.1, 131.6, 136.4, 140.9, 142.4, 144.5, 150.4, 151.0. Anal. Calcd. for C16H12F6N2O3S (426.33): C, 45.08; H, 2.84; N, 6.57. Found: C, 45.16; H, 2.99; N, 6.74.
N-[(3,5-Dimethoxyphenyl)carbamoyl]-4-methylbenzenesulfonamide (10)
Compound 10 precipitated after 35 min as a white solid; m.p. 160–161 °C. IR (KBr): ν (cm−1) 3328, 3285 (NH), 1688 (C=O), 1349, 1168 (SO2). 1H NMR (DMSO-d6): δ (ppm) 2.36 (s, 3H, CH3), 3.48 (s, 6H, OCH3), 6.75–6.79 (m, 2H, Ar), 6.98 (s, 1H, Ar), 7.38–7.40 (m, 2H, Ar), 7.78–7.80 (m, 2H, Ar), 8.60 (br, 1H, NH), 10.51 (br, 1H, NH). 13C NMR (DMSO-d6): δ (ppm) 21.6, 56.0, 105.0, 111.5, 112.7, 126.1, 128.2, 130.0, 131.9, 137.8, 141.0, 142.1, 144.3, 145.5, 149.0, 149.8. Anal. Calcd. for C16H18N2O5S (350.39): C, 54.85; H, 5.18; N, 8.00. Found: C, 54.92; H, 5.79; N, 8.14.
4-Methyl-N-[(2-{trifluoromethyl}phenyl)carbamoyl]benzenesulfonamide (11)
Compound 11 precipitated after 20 min as a white solid; m.p. 140–141 °C. IR (KBr): ν (cm−1) 3326, 3281 (NH), 1683 (C=O), 1340, 1164 (SO2). 1H NMR (DMSO-d6): δ (ppm) 2.33 (s, 3H, CH3), 7.24–7.82 (m, 8H, Ar), 8.23 (br, 1H, NH), 11.31 (br, 1H, NH). 13C NMR (DMSO-d6): δ (ppm) 21.3, 115.5, 117.3, 125.8, 126.2, 126.4, 127.9, 129.8, 130.1, 133.5, 137.3, 142.4, 144.6, 146.7, 150.2. Anal. Calcd. for C15H13F3N2O3S (358.34): C, 50.28; H, 3.66; N, 7.82. Found: C, 50.42; H, 3.74; N, 7.94.
1-(p-Tolyl)-3-[4-(trifluoromethyl)phenyl]urea (13)
Compound 13 precipitated after 13 min as a white solid; m.p. 263–265 °C. IR (KBr): ν (cm−1) 3320, 3279 (NH), 1681 (C=O). 1H NMR (DMSO-d6): δ (ppm) 2.21 (s, 3H, CH3), 7.06 (d, 2H, J = 8 Hz, Ar), 7.31 (d, 2H, J = 8 Hz, Ar), 7.60–7.63 (m, 4H, Ar), 8.65 (br, 1H, NH), 9.01 (br, 1H, NH). 13C NMR (DMSO-d6): δ (ppm) 20.8, 118.1, 118.3, 119.2, 125.0, 123.4, 126.6, 129.7, 129.6, 131.7, 137.2, 138.1, 144.1, 152.8. Anal. Calcd. for C15H13F3N2O (294.28): C, 61.22; H, 4.45; N, 9.52. Found: C, 61.32; H, 4.51; N, 9.64.
1-(p-Tolyl)-3-[4-(trifluoromethoxy)phenyl]urea (14)
Compound 14 precipitated after 5 min as a white solid; m.p. 240–241 °C. IR (KBr): ν (cm−1) 3322, 3282 (NH), 1686 (C=O). 1H NMR (DMSO-d6): δ (ppm) 2.20 (s, 3H, CH3), 7.06 (d, 2H, J = 8 Hz, Ar), 7.25–7.30 (m, 4H, Ar), 7.52 (d, 2H, J = 8 Hz, Ar), 8.57 (br, 1H, NH), 8.80 (br, 1H, NH). 13C NMR (DMSO-d6): δ (ppm) 20.8, 109.1, 118.9, 119.0, 120.1, 121.5, 122.2, 125.1, 129.7, 131.4, 137.4, 140.2, 143.0, 153.0. Anal. Calcd. for C15H13F3N2O2 (310.28): C, 58.07; H, 4.22; N, 9.03. Found: C, 58.12; H, 4.35; N, 9.14.
1-[3,5-Bis(trifluoromethyl)phenyl]-3-(p-tolyl)urea (15)
Compound 15 precipitated after 3 h as a white solid; m.p. 204–205 °C. IR (KBr): ν (cm−1) 3328, 3291 (NH), 1685 (C=O). 1H NMR (DMSO-d6): δ (ppm) 2.20 (s, 3H, CH3), 7.03–7.05 (m, 2H, Ar), 7.29–7.31 (m, 3H, Ar), 8.09 (s, 1H, Ar), 8.45 (s, 1H, Ar), 8.83 (br, 1H, NH), 9.30 (br, 1H, NH). 13C NMR (DMSO-d6): δ (ppm) 20.8, 115.1, 118.4, 118.8, 119.6, 122.3, 125.3, 129.6, 129.7, 131.0, 132.0, 136.9, 137.7, 142.5, 153.1, 153.2. Anal. Calcd. for C16H12F6N2O (362.28): C, 53.05; H, 3.34; N, 7.73. Found: C, 53.06; H, 3.55; N, 7.84.
1-[3,5-Dimethoxyphenyl]-3-(p-tolyl)urea (16)
Compound 16 precipitated after 30 s as a white solid; m.p. 188–190 °C. IR (KBr): ν (cm−1) 3325, 3280 (NH), 1687 (C = O). 1H NMR (DMSO-d6): δ (ppm) 2.20 (s, 3H, CH3), 3.67–3.72 (m, 6H, OCH3), 6.82 (s, 2H, Ar), 7.06 (d, 2H, J = 8 Hz, Ar), 7.16 (s, 1H, NH), 7.30 (d, 2H, J = 8 Hz, Ar), 8.41–8.43 (m, 2H, Ar + NH). 13C NMR (DMSO-d6): δ (ppm) 20.8, 52.5, 55.3, 55.9, 104.4, 110.6, 113.0, 118.8, 129.7, 131.0, 134.0, 137.8, 144.5, 149.3, 153.3. Anal. Calcd. for C16H18N2O3 (286.33): C, 67.12; H, 6.34; N, 9.78. Found: C, 67.19; H, 6.39; N, 9.84.
1-(p-Tolyl)-3-[2-(trifluoromethyl)phenyl]urea (17)
Compound 17 precipitated after 3 h as a white solid; m.p. 262–263 °C. IR (KBr): ν (cm−1) 3325, 3287 (NH), 1684 (C=O). 1H NMR (DMSO-d6): δ (ppm) 2.20 (s, 3H, CH3), 7.04–7.07 (m, 3H, Ar), 7.29–7.32 (m, 3H, Ar), 7.60–7.63 (m, 1H, Ar), 7.98–8.00 (m, 1H, Ar), 8.46 (s, 1H, NH), 9.25 (br, 1H, NH). 13C NMR (DMSO-d6): δ (ppm) 20.8, 118.7, 118.8, 124.5, 126.0, 126.1, 129.0, 129.5, 129.7, 131.5, 133.2, 137.4, 137.7, 153.0, 153.2. Anal. Calcd. for C15H13F3N2O (294.28): C, 61.22; H, 4.45; N, 9.52. Found: C, 61.34; H, 4.56; N, 9.61.
Antimicrobial activity evaluation
To test the antimicrobial property of prepared substances, Bacillus mycoides (Gram-positive) bacterium, Escherichia coli (Gram-negative), and Candida albicans (non-filamentous fungus) have been used as model microbes. Microorganisms were cultivated and kept at pH 7.0 in a nutritional agar medium (70148 Nutrient agar, Fluka, Spain). The antimicrobial activity of the produced substances was evaluated by using the agar well diffusion procedure as regards: Every hardened nutrient agar plate received 100 µL (4 × 107 CFU) from every 24 h re-activated culture. The infected hardened plates were punctured with 15 mm holes to accommodate a quantity of 200 µL per each dissolved component (10 mg/mL) in DMSO. The seeded cultivation dishes with test solutions were incubated for 24 h at 37 °C, and the resulting clear zones have been reported. The minimum inhibitory concentration (MIC) of constituents 7, 8, 9, and 11 was distinguished by a serial dilution method employing DMSO as a solvent. A range of doses from 10 to 0.00488 mg/mL were used. The MIC of every component along each microbe was specified as the lowest concentration of that component capable of inhibiting the development of the given microbe14,37.
Evaluation of cell proliferation by MTT assay
The human lung carcinoma (A549), colon cancer (HCT116), prostate cancer (PC3), skin cancer (A431), hepatocellular carcinoma cell line (HePG2), osteosarcoma cell line (HOS), pancreatic cancer cell line (PACA2) and normal skin fibroblast cell line (BJ1) were purchased from American Tissue Culture Collection (Rockville, MD, USA). HCT116, A549, HePG2, A431, PACA2 and MCF7 were maintained in DMEM media, while PC3 was in RPMI-1640 media (Lonza, Biowahittkar, Belgium). The human normal BJ1 was maintained in DMEM-F12 media. The used media were supplemented with a 1% antibiotic–antimycotic mixture (10,000 µg/mL streptomycin sulfate, 10,000 U/mL potassium penicillin, 1% l-glutamine, 25 µg/mL amphotericin B and 10% fetal bovine serum (FBS) (Biowest, USA). The percentage of viable cells was determined using MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (Bio Basic Canada Inc., Canada). In brief, about 104 cells/ well were seeded into a 96-well plate and incubated at 37 °C for 24 h to be allowed to adhere. Then, the prepared compounds were added at different concentrations of 100, 50, 25 and 12.5 µg/mL to the cell monolayer in triplicate and incubated at 37 °C for 48 h. After that, the medium was replaced with another fresh medium, and 40 µL MTT (2.5 µg/mL) was added to each well and kept for a further 4 h. At last, 200 µL of 10% sodium dodecyl sulfate (SDS) was added to each well and incubated at 37 °C overnight to allow the reaction to be stopped and break up the formed formazan crystals. Subsequently, the quantity of formazan product was detected at a wavelength of 595 nm with a reference wavelength of 620 nm using a microplate reader (Bio-Rad Laboratories, model 3360, USA). Doxorubicin (Adriamycin) was used as a positive control. The vehicle was dimethyl sulfoxide (DMSO), which is used for dissolving the prepared compounds and the final concentration of it was less than 0.2%. IC50 was calculated using the Prism software program (Graph Pad software incorporated, version 3).
Gene expression analysis
Quantitative real-time PCR method
RNA was extracted from pancreas, prostate, lung and liver cell lines using a total RNA purification kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. An aliquot of RNA was diluted in RNase-free water to estimate RNA quantity. Aliquots were used immediately for reverse transcription (RT). cDNA synthesis was performed on extracted RNA, which was treated with DNase (Invitrogen, Germany) to remove any possible DNA contamination. The DNase-treated RNA was reverse transcribed into first-strand cDNA using the RevertAid First Strand cDNA Synthesis kit (Fermantas) according to the manufacturer's instructions. Determination of the pancreas, prostate, lung and liver cell line cDNA copy number was carried out using StepOne™ Real-Time PCR System from Applied Biosystems (Thermo Fisher Scientific, Waltham, MA USA). Gene expressions were detected by real-time PCR, which was performed using the Rotor-Gene Q system (Qiagen Company). A 25 µL reaction mixture consisted of 12.5 µL SYBR® Premix Ex TaqTM (TaKaRa, Biotech. Co. Ltd.), 0.5 µL of each primer (10 PMole) (Table 1), 1 µL cDNA (50 ng) and 10.5 µL RNase free water. The optimum amplification conditions were chosen empirically according to each tested gene. Generally, the amplification conditions include initial incubation (95.0 °C for 3 min), then 40 cycles of amplification with denaturation (95.0 °C for 15 s), annealing (55.0 °C for 30 s) and extension (72.0 °C for 30 s) steps then 71 cycles which started at 60.0 °C and then increased about 0.5 °C every 10 s up to 95.0 °C. Mean cycle threshold (Ct) values of triplicate samples are used for analysis. The Ct value indicates the fractional cycle number at which the amount of amplified target reaches a fixed threshold. Data from real-time PCR were analyzed using 2−∆∆Ct method38,39,40. Data were represented as the fold change in target gene expression normalized to a House-Keeping gene (HKG) and relative to the control. β-actin was used as a HKG to normalize input RNA amount, RNA quality and reverse transcription efficiency.
DNA damage using the comet assay
According to the method of Olive et al.41 after the trypsin treatment of pancreas, prostate, lung and liver cancer cell lines to produce a single cell suspension. This suspension was stirred for 5 min and filtered. 100 μL of cell suspension was mixed with 600 μL of low-melting agarose (0.8% in PBS). 100 μL of this mixture was spread on pre-coated slides which immersed in lysis buffer (0.045 M TBE, pH 8.4, containing 2.5% SDS) for 15 min. The slides were placed in an electrophoresis chamber containing the same TBE buffer, but devoid of SDS. The electrophoresis conditions were 2 V/cm for 2 min and 100 mA. Staining with ethidium bromide 20 μg/mL. at 4 °C.
Comet capture and analysis
A total of 100 randomly captured comets from each slide were examined at 400 × magnification using a fluorescence microscope connected to a CCD camera to an image analysis system [comet 5 image analysis software developed by Kinetic Imaging, Ltd. (Liverpool, UK)]. A computerized image analysis system acquires images, computes the integrated intensity profiles for each cell, estimates the comet cell components and then evaluates the range of derived parameters. To quantify the DNA damage tail length (TL), the percentage of migrated DNA (Tail DNA%) and tail moment (TM) were evaluated. The non-overlapping cells were randomly selected and were visually assigned a score on an arbitrary scale of 0–3 (i.e., class 0 = no detectable DNA damage and no tail; class 1 = tail with a length less than the diameter of the nucleus; class 2 = tail with length between 1 × and 2 × the nuclear diameter; and class 3 = tail longer than 2 × the diameter of the nucleus) based on perceived comet tail length migration and relative proportion of DNA in the nucleus42.
DNA fragmentation assay
DNA fragmentation in the pancreas, prostate, lung and liver cancer cell lines was carried out according to Yawata43 with some modifications. Briefly, after 24 h of exposure of pancreas, prostate, lung and liver cancer cell lines to the evaluated compounds in different Petri dishes (60 × 15 mm, Greiner), the cells were trypsinized, suspended and homogenized in 1 ml of medium. Then, the cells were centrifuged for 10 min at 800 rpm. After the centrifugation step, the low molecular weight genomic DNA was extracted as described in Yawata43. Approximately, 1 × 106 cells were seeded and treated with the IC50 concentration of evaluated compounds. All the cells (including floating cells) were collected by the treatment with trypsin and washed with Dulbecco`s Phosphate Buffered Saline (PBS). On ice, the cells were lysed with the lysis buffer containing 150 mM NaCl, 10 mM Tris (pH 7.4), 5 mM ethylenediaminetetraacetic acid (EDTA), and 0.5% Triton X-100 for 30 min. The lysates were mixed using a vortex and then centrifuged for 20 min at 10,000×g. The damaged DNA in the supernatant was extracted with an equal volume of neutral phenol: chloroform: isoamyl alcohol mixture (25:24:1). The percentage of fragmented DNA was then analyzed using gel electrophoresis on 2% agarose gel including 0.1 µg/mL ethidium bromide.
Molecular docking study
Molecular operating environment (MOE 2009.10) and BIOVIA Discovery Studio programs were used to perform the molecular docking study44. The mode of interaction between the evaluated compounds 8 and 9 and the active site of the studied proteins (Escherichia coli enoyl reductase and human SOS1) was visualized in 2-dimensional and 3-dimensional states using the BIOVIA Discovery Studio program. The studied proteins were downloaded from the protein data bank (www.pdb.org), where the protein codes were (1C14 and 6SCM) respectively. The co-crystalized standard ligand complexed with the studied proteins were (Triclosan and BI-3406) respectively. The target compounds 8 and 9 were drawn using the MOE builder interface and then subjected to local and global energy minimization using the included MOPAC. The energy of the target compounds was calculated by performing the systematic conformational search where RMS gradient and RMS distance were set as default at 0.01 kcal/mole and of 0.1Ao respectively. The lowest value of energy for the target compounds was selected to be used in the next docking step. Several modifications were done on the selected proteins for the subsequent docking studies which included the following steps: (a) The hydrogen atoms were added to the selected target proteins; (b) The co-crystalized standard ligand molecule was removed from the protein active site; (c) The active site was selected using MOE alpha site finder and dummy atoms were prepared from the obtained alpha spheres; (d) The prepared model was then subjected to the docking step to predict the ligand–protein binding interactions at the active site.
Results and discussion
Chemistry
Under simple and free-metal condition reaction, the treatment of 4-tolyl sulfonyl isocyanate (6) with primary amines of 4-(trifluoromethyl)aniline (1), 4-(trifluoromethoxy)aniline (2), 3,5-bis(trifluoromethyl)aniline (3), 3,5-dimethoxy aniline (4), and 2-(trifluoromethyl)aniline (5) in acetonitrile at room temperature afforded the corresponding 4-tolyl sulfonylurea derivatives 7–11, respectively. Likewise, the reaction of 4-tolylisocyanate (12) with the same previous amines afforded 4-tolylurea derivatives 13–17, respectively.
All compounds 7–11 and 13–17 were prepared in good to excellent yield. The chemical structures of the newly synthesized compounds 7–11 and 13–17 were approved by IR, 1H-NMR, 13C-NMR, and elemental analysis. Compound 9 (Fig. 2) will be discussed in detail as a represented example of sulfonyl-urea derivatives 7–11. In the 1H-NMR spectrum (400 MHz, DMSO-d6) the protons of the aromatic rings and one NH group were assigned at range δ 7.01–8.0 ppm as multiplet signals, the other NH group which attached to the sulfonyl group was assigned at δ 9.50 ppm as a singlet signal. The protons of the methyl group were assigned at δ 2.34 ppm as a singlet signal. Likewise, all the expected number of carbon signals was observed in the 13C-NMR spectrum of 9. In the IR, the NH group was assigned at ν 3323 and 3281 cm−1, while the C=O was attributed at ν 1685 cm−1 and the sulfonyl group (SO2) was assigned at ν1344 and 1166 cm−1. On the other hand, compound 13 (Fig. 2) will be explained as a represented example of aryl-urea derivatives 13–17. The 1H-NMR spectrum of 13 showed the NH groups as singlet in the downfield zone at δ 8.65 and 9.01 ppm, while the methyl group was assigned as expected at δ 2.21 ppm. The aromatic protons of 4-tolyl ring were assigned at δ 7.06 and 7.31 ppm as a doublet with coupling constant (J = 8 Hz), while the aromatic protons of the 4-trifluoromethyl phenyl ring were observed at δ 7.61 ppm as a multiplet signal. In the 13C-NMR spectrum, all the carbons were observed at their expected chemical shifts. The IR spectrum of 13 visualized the NH group at ν 3320 and 3279 cm−1 and the carbonyl group at ν 1681 cm−1.

Synthesis of 4-tolyl sulfonylurea (7–11) and 4-tolylurea derivatives (13–17). All the reactions were carried out in acetonitrile at room temperature.
Antimicrobial activity
Antimicrobial activity testing utilizing the agar diffusion method
To assess the efficacy of the prepared formulations as antimicrobial agents, different microbial populations, B. mycoides, E. coli, and C. albicans, have been chosen as examples for Gram-positive, Gram-negative bacteria, and non-filamentous fungi, respectively. To evaluate the antimicrobial properties of the produced substances against the preceding indicated microbes, the agar well diffusion technique was used. The specific antimicrobial agent's activity, and therefore its efficacy as an antiseptic agent, is dependent mostly on the cell structure of microbial species, along with the main ingredients of the evaluated chemical14,45,46.
The antimicrobial efficacy of the generated components (compounds 7–17) was displayed in Table 2. The acquired findings demonstrated the disparity in action and responsiveness between each of the various evaluated chemicals and microbes (Table 2). Compound 9 had the maximum activity versus B. mycoides and C. albicans (41 mm), trailed by compound 8, which exhibited comparable results against B. mycoides, and C. albicans (37 and 38 mm, respectively). Compounds 7 and 11 also showed significant antimicrobial properties against investigated microbial species, with greater values in opposition to C. albicans. Other substances, on the other hand, had reduced antimicrobial properties against all evaluated microbes, with compounds 13, 14, 16, and 17 exhibiting the worst activity. As a side note, E. coli, as a typical Gram-negative bacterium, demonstrated the highest resilience amongst studied microorganisms, which might be attributed to the composition of its cell wall (Table 1). Because of their significant antibacterial properties, compounds 7, 8, 9, and 11 have been selected to determine their MIC values to stop the proliferation of tested microorganisms.
The serial dilution procedure was applied to determine the MICs of the most powerful synthesized compounds. The results provided for determining the MIC values of compounds 7, 8, 9, and 11 have been included in Table 3. The antimicrobial activity of evaluated compounds was shown to be proportionate to their dosages (Table 3 and Fig. 3). Furthermore, the MIC values of every compound against evaluated microorganisms are affected by both the cell structure of the microbes, as well as the overall constitution of the examined compound45. A compound with a lower MIC value has a greater potential to inhibit the development of the designated microbe14. Compounds 7 and 8 have a minimum MIC level (≥ 4.88 µg/mL) versus different classes of microorganisms, with a MIC value of 4.88 µg/mL in all cases except the case of compound 7 against B. mycoides (9.75 µg/mL). On the other hand, the highest MIC value was recorded with compound 11 against E. coli (Table 4).

Effect of different concentrations of compounds (a) 7 (b) 8 (c) 9 and (d) 11 on their antimicrobial activity. Each sample (150 µL) at different concentrations was dissolved in DMSO and added to a 15 mm agar well. Inoculum (4 × 107 CFU) was added to each 20 cm plate (150 mL of 70148 nutrient agar, Fluka), then the plates were incubated overnight at 37 °C.
Anti-cancer activity
Primary screening
Compounds 7–11 and 13–17 were screened against seven human cancer cell lines, lung carcinoma (A549), colon cancer (HCT116), prostate cancer (PC3), human skin cancer (A431), hepatocellular carcinoma cell line (HePG2), osteosarcoma cell line (HOS), pancreatic cancer cell line (PACA2), at 100 μg/mL. Also, the compounds were evaluated against the normal skin fibroblast cell line (BJ1) for comparison purposes. As shown in Table 5, compound 7 exhibited 100% mortality against all the evaluated cancer cell lines except A431 which showed 55.4% mortality. Compound 8 exhibited 100% mortality against A549, HCT116, PC3, HePG2 and PACA2, and more than 50% mortality against HOS. Also, 100% mortality was exhibited for compound 9 against HCT116, A431, HePG2, and HOS (Table 5). Compound 11 exhibited more than 75% mortality against A549, HCT116, PC3, and PACA2, while compound 13 showed 100% mortality against only HePG2 and about 74.3% mortality against HOS. Regarding compound 14, 100% mortality was exhibited against HOS and 65.3% mortality against HePG2. Compound 15 exhibited more than 75% mortality against PC3, HOS, and PACA2, and more than 50% mortality against A549. More than 50% mortality was exhibited for compounds 10, 16, and 17 against PACA2, (HOS and PACA2), (PC3, HOS and PACA2) respectively. So, the evaluated compounds against these selected cell lines were subjected to secondary screening to determine their IC50 and selectivity index values.
Secondary screening
As depicted in Table 6, the most promising compounds against HCT116 were compounds 8 and 9 with IC50 values 47.3 and 17.8 µM, respectively compared to doxorubicin (69.2 µM). Regarding HePG2 cell line, compound 9 showed the highest activity with an IC50 value 12.4 µM as compared to doxorubicin (39.7 µM), while compounds 7 and 8 exerted moderate activity with IC50 values of 97.4 and 74 µM respectively. However, compound 8 exerted lower activity than compound 9 against HCT116 and HePG2, it was more selective. Compounds 7 and 8 exhibited promising activity against A549 and PACA2 with IC50 values 86.5 and 44.4 µM respectively for compound 7, and 55.8 and 22.4 µM respectively for compound 8. While compounds 11 and 15 showed moderate activity compared to doxorubicin (IC50 = 52.1 µM) against PACA2 with IC50 values 80.7 and 82.5 µM respectively. Regarding PC3, compound 8 was the most active one with an IC50 value of 57.2 µM, while compounds 7 and 11 showed moderate activity with IC50 values of 91.3 and 114.7 µM respectively. The most active compound against HOS was compound 9 with an IC50 value of 17.6 µM, also compound 15 showed promising activity with an IC50 value of 37.3 µM, while compound 7 showed mild activity with an IC50 value of 102.15 µM compared to doxorubicin (IC50 = 24.3 µM). Regarding A431, only compound 9 exerted activity which was moderate with an IC50 value of 113.3 µM. From these results, it was noticed that compounds 7, 8, 9 and 15 exerted the best cytotoxic effect and compound 8 was the most selective one. So, further molecular studies were done on these selected compounds.
Gene expression analysis
Gene expression in pancreatic cell line
The gene expression result of selected gene PALB2 (Partner And Localizer Of BRCA2) and CDKN2A in PACA2 cells revealed that the treated pancreatic cell line showed a significant reduction (P < 0.01) in the expression levels of PALB2 and CDKN2A genes compared with negative untreated sample (-ve) (Fig. 4a,b). Also, compared to the negative sample of the pancreatic cancer cell line, the expression values of PALB2 and CDKN2A genes were significantly reduced (P < 0.05) in the treated (7 and 15) and positive control pancreatic cell line (doxorubicin). Moreover, the PALB2 gene expression level was significantly suppressed in treated 7 and 15 versus positive control PACA2 cells (+ ve). PALB2 gene plays an important role in double-strand break repair and its down-regulation resulted in DNA damage in the treated cells47. This was confirmed in the subsequent sections of the DNA damage analysis using comet and gel electrophoresis assays.

Alterations in the gene expression level of (a) PALB2 and (b) CDKN2A; genes in Paca2 cancer cell line treated with 7 and 15. (c) BRCA1 and (d) BRCA2; genes in PC3 cancer cell lines treated with 7. Data are presented as mean ± SEM. (a, b, c) Mean values within tissue with unlike superscript letters were significantly different (P < 0.05).
Gene expression in prostate cancer cell line
The BRCA1 (Breast cancer 1) and BRCA2 (Breast cancer 2) genes, which are associated with PC3 prostatic carcinoma, were used to perform gene expression analysis in these tissue. The results presented in Figs. 4c & d indicated that negative samples of PC3 showed significantly high expression levels of BRCA1 and BRCA2 genes (P < 0.01) compared with treated PC3 cells. While the expression values of BRCA1 and BRCA2 genes were decreased significantly (P < 0.05) in treated 7 and positive control PC3 cells compared with negative samples. Additionally, the BRCA1 and BRCA2 gene expression levels were downregulated in 7 much lower than those in positive control PC3 cells (doxorubicin). BRCA1 and BRCA2 genes are implicated in DNA repair and hence decreasing the expression level of them leads to DNA fragmentation48. Herein, the downregulation of BRCA1 and BRCA2 supported our results in the subsequent DNA damage sections.
Gene expression in lung cancer cell line
The results of selected genes EGFR (Epidermal Growth Factor Receptor) and KRAS (Kirsten rat sarcoma virus) in A549 lung cancer cell line showed a significant increase of their expression levels (P < 0.01) in negative samples of A549 cells compared with treated samples (Fig. 5a,b). While the treated 8 and doxorubicin-treated A549 cells showed a significant reduction of EGFR and KRAS gene expression levels (P < 0.05) compared with negative samples. Likewise, the expression levels of EGFR and KRAS genes were downregulated in treated 8 much lower than those in positive control A549 cells. In lung carcinoma, EGFR and KRAS are found to be tumorigenesis drivers and are implicated in invasion and metastasis processes49,50. So, compound 8 exerted anticancer activity through the down-regulation of EGFR and KRAS in A549 lung carcinoma.

Alterations in the gene expression of (a) EGFR and (b) KRAS; genes in A549 cancer cell lines. (c) TP53 and (d) FASN; genes in HCT116 cancer cells treated with 7 and 9. Data are presented as mean ± SEM. (a, b, c) Mean values within tissue with unlike superscript letters were significantly different (P < 0.05).
Gene expression in colon cancer cell line
The results of TP53 (tumor protein p53) and FASN (Fatty Acid Synthase) genes expression analysis in HCT116 cells revealed that the expression levels of TP53 and FASN genes were improved significantly (P < 0.01) in negative samples compared with treated samples of HCT116 cells (Fig. 5c,d). Where a suppression (P < 0.05) in the expression levels of TP53 and FASN genes was recorded in 7, 9 and doxorubicin (positive control) treated HCT116 cells compared with negative samples. Also, 7 and doxorubicin-treated HCT116 cells showed a significant decrease in TP53 and FASN gene expression levels compared with 9 treated cells. The lipid metabolism provides an alternative source of energy required to afford tumor growth. FASN plays a central role in lipid metabolism and hence helps in the survival of tumor cells51. So, compounds 7 and 9 had an antisurvival effect on tumor cells via decreasing the expression level of the FASN gene.
DNA damage analysis
DNA damage in pancreatic cell line
The data in Table 7 and Fig. 6 represented the DNA damage in the PACA2 cell line. The mean values of DNA damage were significantly decreased (P < 0.05) in negative untreated samples of the PACA2 cell line compared with treated cell lines. On the other hand, the DNA damage values were increased significantly (P < 0.01) in treated 7 and 15 and doxorubicin-treated cells and the highest values of DNA damage were recorded in 7 much more than those in 15 and doxorubicin-treated cells.

The visual score of normal DNA (class 0) and damaged DNA (classes 1, 2 and 3) using the comet assay.
DNA damage in prostate cell line
As shown in Table 7 and Fig. 6, the DNA damage values in the PC3 cancer cell line were significantly decreased (P < 0.05) in negative untreated samples of the PC3 compared with the treated cells. Meanwhile, the treated 7 and doxorubicin-treated PC3 cells showed a significant increase (P < 0.01) in DNA damage values whereas the highest values were observed in 7 much more than those in doxorubicin-treated cells.
DNA damage in lung cell line
The results of DNA damage in the A549 cancer cell line showed that negative untreated samples of the A549 exhibited a significant decrease (P < 0.05) in DNA damage values compared with treated cell lines (Table 7 and Fig. 6). However, the DNA damage values were increased significantly (P < 0.01) in 8 and doxorubicin-treated A549 samples and the highest values of DNA damage were observed in 8 treated cells much more than those in the doxorubicin-treated sample.
DNA damage in the colon cell line
The current results showed a significant decrease (P < 0.05) in DNA damage values in negative untreated samples of HCT116 compared with treated (Table 7 and Fig. 6). Further, treated 7 and doxorubicin HCT116 cells showed a significant increase (P < 0.01) in DNA damage values and the highest values were observed in 7 much more than those in doxorubicin and 9 treated cells.
DNA fragmentation analysis
Measurement of DNA fragmentation in pancreatic cancer cell line
The effect of different treatments on DNA damage in PACA2 cancer cell line revealed that DNA fragmentation rates were significantly increased in treated samples 7 and 15 and doxorubicin compared with negative untreated samples as summarized in Table 8 and Fig. 4. However, the DNA fragmentation values were increased significantly (P < 0.01) in treated PACA2 samples compared with negative control. Whereas, the highest value of DNA fragmentation was observed in 7 much more than 15 and those in doxorubicin-treated PACA2 cells.
Measurement of DNA fragmentation in prostate cancer cell line
The data in Table 8 and Fig. 4 revealed that the rate of DNA fragmentation in PC3 cancer cell line was significantly suppressed (P < 0.01) in negative samples of the PC3 compared with those in treated samples 7 and doxorubicin (positive control). Although, the DNA fragmentation values were increased significantly (P < 0.01) in treated PC3 samples compared with negative control, the highest values were found in 7 treated cells as compared to the doxorubicin-treated cells.
Measurement of DNA fragmentation in lung cancer cell line
The results of DNA fragmentation in A549 determination as presented in Table 8 and Fig. 7 showed that negative samples of A549 exhibited a significant decrease (P < 0.01) in DNA fragmentation rates compared with those in 8 and doxorubicin-treated samples. However, treated A549 samples showed a significant reduction in the DNA fragmentation values (P < 0.01) compared with negative control. Moreover, the highest value of DNA fragmentation was observed in 8 much more than those in doxorubicin-treated cells.

DNA fragmentation detected with Agarose gel in (a) PACA2 (lane 1: negative control, lane 2: 7, lane3: 15, lane 4: positive control); (b) PC3 (lane 1: negative control, lane 2: 7, lane 3: positive control); (c) A549 (lane 1: negative control, lane 2: 8, lane 3: positive control); (d) HCT116 (lane 1: negative control, lane 2: 7, lane 3: 9, lane 4: positive control), M: represent DNA marker.
Measurement of DNA fragmentation in colon cancer cell line
The results of DNA fragmentation in HCT116 revealed that negative samples HCT116 showed a significant decrease (P < 0.01) in DNA fragmentation rates compared with those in treated samples 7, 9 and doxorubicin (positive control) (Table 8 and Fig. 7). However, the DNA fragmentation values were increased significantly (P < 0.01) in treated HCT116 samples compared with negative control. Additionally, the highest value of DNA fragmentation was observed in 7 much more than those in doxorubicin and 9 treated samples. DNA fragmentation into oligonucleosomal fragments is a biochemical feature of programmed cell death (apoptosis)52. It is worth to be mentioned that DNA fragmentation is a late event of the apoptosis process. So, our prepared evaluated compounds induced apoptosis in the above-mentioned cancer cell lines.
Molecular docking study
Compounds 8 and 9 were chosen for the molecular docking study. As it was seen from the anticancer activity section, compounds 8 and 9 had the best cytotoxic effect against the majority of the evaluated cancer cell lines and had a little cytotoxic effect on the evaluated normal cell line. Also, they had promising antimicrobial activity. Compounds 8 and 9 were studied against Escherichia coli enoyl reductase and only compound 8 was studied against human Son of sevenless homolog 1 (SOS1). The proteins were downloaded from the protein database with their co-crystalized ligands which were used as a reference standard ligand. It was found that the root mean squared deviation (RMSD) of the co-crystalized ligand were (1.34, and 0.5 A0) for Escherichia coli enoyl reductase and SOS1, respectively. It was noticed that the RMSD value was ≤ 2 which confirmed the accuracy of the docking parameters. Regarding the bacterial protein (E. coli enoyl reductase), the binding energy (S) were − 23.1 and − 18.9 kcal/mol for compounds 8 and 9 respectively which were better than that of the standard (− 16.8 kcal/mol) (Table 9). While, for the human protein (SOS1), the S value of compound 8 was equaled − 18.7 kcal/mol. As compared to the S value of the standard ligand (− 23.8 kcal/mol) (Table 9), compound 8 showed lower binding energy but was still in negative charge which demonstrated the spontaneous interaction with the selected protein. As shown in Fig. 8, compound 8 interacted with the active site of E. coli enoyl reductase through seven hydrogen bonds, one between fluorine atom and PRO 191 with bond distance 4.05 A°, two hydrogen bonds between the amino acid SER 91and the oxygen of the SO2 and NH of amide group with bond distances 4.39 and 3.09 A° respectively. Two hydrogen bonds between oxygen of SO2 and two different amino acids, ALA 21 and SER 19 with bond distances 4.26 and 4.02 A° respectively. The last two hydrogen bonds were between ILE 20 and two different oxygens of the SO2 group with comparable bond distances of 4.31 and 4.35 A°. Also, there were two electrostatic interactions between the benzene ring, ALA 15 and ALA 196. Regarding compound 9, there were eight interactions with the E. coli enoyl reductase enzyme (Fig. 8). These interactions included, two hydrogen bonds between a fluorine atom and both GLY 93 and ILE 92 with bond distances of 3.9 and 4.9 A° respectively, a hydrogen bond between another fluorine atom and LYS 163 with a bond distance 4.98 A°, two hydrogen bonds between the oxygen of the SO2, ILE 200 and PHE 94 with bond distances 3.6 and 4.68 A° respectively, two electrostatic interactions between two different benzene rings and ALA 196, a third electrostatic interaction was seen between benzene and LYS 163. Regarding human SOS1 protein, it was found that compound 8 interacted through nine interactions; four interactions between fluorine atoms and PHE 890, ASP 887, and GLU 891 residues, one conventional hydrogen bond which was between fluorine atom and LYS 898 with bond distance 5.8 A°, pi-donor hydrogen bond between benzene ring and TYR 884 with bond distance 6.64 A°, another two hydrogen bonds with GLU 902 and ASP 887 with bond distances 3.7 and 3.8 A° respectively and pi-pi stacked between benzene ring and PHE 890 (Fig. 8). Enoyl reductase enzyme is responsible for the last step of the fatty acid biosynthesis in E.coli bacteria53. According to the molecular docking study, the antibacterial activity of our prepared compounds might be due to the inhibition of the enoyl reductase enzyme. The human SOS1 is a co-activator transcriptional factor for the KRAS gene54. The inhibition of the SOS1 protein leads to the down-regulation of the KRAS gene. Our result in the above-mentioned gene expression analysis of KRAS in A549 lung carcinoma confirmed our result of the molecular docking study on SOS1 protein.

The molecular interactions of compounds 8 and 9 with the active site of E. coli enoyl reductase and compound 8 with the active site of human SOS1.
Structure–activity relationship (SAR)
To explain the relationship between the chemical structure and the biological properties of the newly synthesized compounds, the Structure–Activity Relationship (SAR) of urea derivatives 7–11 and 13–17 was visualized in Fig. 9. The presence of the sulfonyl group in compounds 7–11 is essential for increasing the antimicrobial and anti-cancer activity of these compounds in comparison with compounds 13–17 as the non-containing sulfonyl group. Compounds 7, 8, 9, and 11 were found as the most antibacterial agents compared to all the examined compounds, with the most potent effect on both Bacillus mycoides (36, 37, 41, and 31 mm, respectively) and Candida albicans (37, 38, 41, and 32 mm, respectively), while the minimum inhibitory concentrations (MICs) of these compounds were determined against B. mycoides, E. coli, and C. albicans, and compound 8 is the most active at 4.88 µg/mL. On the other hand, compounds 7, 8 and 9 (with sulfonyl group) showed potent anti-cancer properties against most of the tested cell lines. For example, compound 8 has strong anti-cancer activity against A549, HCT116, PC3 and PACA2 cell lines with IC50 55.8, 47.3, 57.2 and 22.4 µM, respectively, and compound 9 gave excellent anti-cancer activity against HCT116, HePG2 and HOS cell lines with IC50 17.8, 12.4 and 17.6 µM, respectively. The presence of the trifluoromethyl group at positions 3 and 5 in compounds 9 and 15 increased the anti-cancer properties than that observed in compounds 10 and 16 which have methoxy groups at positions 3 and 5. It was worth noting that, the presence of the trifluoromethoxy group at position 4 in compound 14 increased the anti-cancer activity on the HOS cell line with IC50 61.6 µM even in the absence of the sulfonyl group. Likewise, the presence of trifluoromethyl group at positions 3 and 5 increased the anti-cancer properties on HOS cell line in the presence of sulfonyl group (as in compound 9 with IC50 17.6 µM) or absence of sulfonyl group (as in compound 15 with IC50 37.3 µM).

SAR of the newly synthesized urea compounds 7–11 and 13–17.
Conclusion
A novel series of sulfonylurea derivatives (7–11), and aryl-urea derivatives (13–17) were evaluated as antimicrobial and anticancer agents. The current study's antimicrobial results demonstrated the efficacy of the synthesized derivatives as antimicrobial agents versus diverse microbial populations. That data can aid in the development of innovative medications to combat the challenge of resistant pathogens. As anticancer candidates, compounds 7, 8, 9 and 15 showed promising activity with the lowest IC50 values as compared to doxorubicin against different cancer cell lines. The anticancer activity of these promising compounds was studied at the gene expression level which showed the down-regulation of PALB2, (BRCA1 and BRCA2), (EGFR and KRAS) and (TP53 and FASN) in PACA2, PC3, A549 and HCT116 treated cells respectively. It was concluded from the percentage of DNA damage that was studied via two different techniques that our evaluated compounds induced apoptotic cell death in the treated cancer cell lines. The molecular docking study revealed the inhibition possibility of compounds 8 and 9 against the E. coli enoyl reductase and compound 8 against human SOS1. The structure–activity relationship revealed the great impact of the added sulfonyl group on the activity of our prepared urea derivatives.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Glyn, R. J. & Pattison, G. Effects of replacing oxygenated functionality with fluorine on lipophilicity. J. Med. Chem. 64, 10246–10259. https://doi.org/10.1021/acs.jmedchem.1c00668 (2021).
Yale, H. L. The trifluoromethyl group in medical chemistry. J. Med. Pharm. Chem. 1, 121–133. https://doi.org/10.1021/jm50003a001 (2002).
Cho, E. J. et al. The palladium-catalyzed trifluoromethylation of aryl chlorides. Science 328, 1679–1681. https://doi.org/10.1126/science.1190524 (2010).
Leroux, F. R., Manteau, B., Vors, J.-P. & Pazenok, S. Trifluoromethyl ethers: Aynthesis and properties of an unusual substituent. Beilstein J. Org. Chem. https://doi.org/10.3762/bjoc.4.13 (2008).
Wang, Q., He, L., Li, K. K. & Tsui, G. C. Copper-mediated domino cyclization/trifluoromethylation/deprotection with TMSCF3: Synthesis of 4-(trifluoromethyl)pyrazoles. Org. Lett. 19, 658–661. https://doi.org/10.1021/acs.orglett.6b03822 (2017).
Artamonov, O. S. et al. Synthesis of trifluoromethyl-substituted 3-azabicyclo[n.1.0]alkanes: Advanced building blocks for drug discovery. Eur. J. Org. Chem. 2014, 3592–3598. https://doi.org/10.1002/ejoc.201402158 (2014).
Schiesser, S. et al. N-Trifluoromethyl amines and azoles: An underexplored functional group in the medicinal chemist’s toolbox. J. Med. Chem. 63, 13076–13089. https://doi.org/10.1021/acs.jmedchem.0c01457 (2020).
Krátký, M., Svrčková, K., Vu, Q. A., Štěpánková, Š & Vinšová, J. Hydrazones of 4-(trifluoromethyl)benzohydrazide as new inhibitors of acetyl- and butyrylcholinesterase. Molecules 26, 989. https://doi.org/10.3390/molecules26040989 (2021).
Wang, A.-Y., Lu, Y. I. N., Zhu, H.-L. & Jiao, Q.-C. URD12: A urea derivative with marked antitumor activities. Oncol. Lett. 3, 373–376. https://doi.org/10.3892/ol.2011.474 (2012).
Sroor, F. M. et al. Biochemical and histopathological studies of sulfonylurea derivative as a new chemotherapeutic agent against liver cancer in free- and nano-coated forms. Appl. Biol. Chem. https://doi.org/10.1186/s13765-022-00737-3 (2022).
Sroor, F. M., Othman, A. M., Aboelenin, M. M. & Mahrous, K. F. Anticancer and antimicrobial activities of new thiazolyl-urea derivatives: Gene expression, DNA damage, DNA fragmentation and SAR studies. Med. Chem. Res. 31, 400–415. https://doi.org/10.1007/s00044-022-02849-3 (2022).
Sroor, F. M., Basyouni, W. M., Aly, H. F., Ali, S. A. & Arafa, A. F. Design, synthesis and SAR of novel sulfonylurea derivatives for the treatment of Diabetes mellitus in rats. Med. Chem. Res. https://doi.org/10.1007/s00044-021-02829-z (2021).
Sroor, F. M., Abdelmoniem, A. M. & Abdelhamid, I. A. Facile synthesis, structural activity relationship, molecular modeling and in vitro biological evaluation of new urea derivatives with incorporated isoxazole and thiazole moieties as anticancer agents. ChemistrySelect 4, 10113–10121. https://doi.org/10.1002/slct.201901415 (2019).
Sroor, F. M., Othman, A. M., Tantawy, M. A., Mahrous, K. F. & El-Naggar, M. E. Synthesis, antimicrobial, anti-cancer and in silico studies of new urea derivatives. Bioorg. Chem. 112, 104953. https://doi.org/10.1016/j.bioorg.2021.104953 (2021).
Sikka, P. Role of aryl urea containing compounds in medicinal chemistry. Med. Chem. https://doi.org/10.4172/2161-0444.1000305 (2015).
Kumar Verma, S. et al. Antibacterial activities of sulfonyl or sulfonamide containing heterocyclic derivatives and its structure-activity relationships (SAR) studies: A critical review. Bioorg. Chem. 105, 104400. https://doi.org/10.1016/j.bioorg.2020.104400 (2020).
Listro, R. et al. Urea-based anticancer agents: Exploring 100-years of research with an eye to the future. Front. Chem. https://doi.org/10.3389/fchem.2022.995351 (2022).
Hawash, M. Recent advances of tubulin inhibitors targeting the colchicine binding site for cancer therapy. Biomolecules 12, 1843. https://doi.org/10.3390/biom12121843 (2022).
Pasello, G., Urso, L., Conte, P. & Favaretto, A. Effects of sulfonylureas on tumor growth: A review of the literature. Oncologist 18, 1118–1125. https://doi.org/10.1634/theoncologist.2013-0177 (2013).
Pulingam, T. et al. Antimicrobial resistance: Prevalence, economic burden, mechanisms of resistance and strategies to overcome. Eur. J. Pharm. Sci. 170, 106103. https://doi.org/10.1016/j.ejps.2021.106103 (2022).
Cassini, A. et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis. 19, 56–66. https://doi.org/10.1016/s1473-3099(18)30605-4 (2019).
Haenni, M. et al. Environmental contamination in a high-income country (France) by antibiotics, antibiotic-resistant bacteria, and antibiotic resistance genes: Status and possible causes. Environ. Int. 159, 107047. https://doi.org/10.1016/j.envint.2021.107047 (2022).
Klein, E. Y. et al. Global increase and geographic convergence in antibiotic consumption between 2000 and 2015. Proc. Natl. Acad. Sci. https://doi.org/10.1073/pnas.1717295115 (2018).
Goossens, H., Ferech, M., Vander Stichele, R. & Elseviers, M. Outpatient antibiotic use in Europe and association with resistance: A cross-national database study. Lancet 365, 579–587. https://doi.org/10.1016/s0140-6736(05)17907-0 (2005).
Wernli, D. et al. Antimicrobial resistance: The complex challenge of measurement to inform policy and the public. PLOS Med. 14, e1002378. https://doi.org/10.1371/journal.pmed.1002378 (2017).
de Kraker, M. E. A., Stewardson, A. J. & Harbarth, S. Will 10 million people die a year due to antimicrobial resistance by 2050?. PLOS Med. 13, e1002184. https://doi.org/10.1371/journal.pmed.1002184 (2016).
Hawash, M. et al. Molecular docking studies and biological evaluation of isoxazole-carboxamide derivatives as COX inhibitors and antimicrobial agents. 3 Biotech https://doi.org/10.1007/s13205-022-03408-8 (2022).
Garneau-Tsodikova, S. & Labby, K. J. Mechanisms of resistance to aminoglycoside antibiotics: Overview and perspectives. MedChemComm 7, 11–27. https://doi.org/10.1039/c5md00344j (2016).
Wright, G. D. Q&A: Antibiotic resistance: Where does it come from and what can we do about it?. BMC Biol. https://doi.org/10.1186/1741-7007-8-123 (2010).
Blair, J. M. A., Webber, M. A., Baylay, A. J., Ogbolu, D. O. & Piddock, L. J. V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13, 42–51. https://doi.org/10.1038/nrmicro3380 (2014).
Teleb, M., Sroor, F., Hassaneen, H. & Saleh, F. Synthesis of novel hydrazonoyl chlorides as useful precursor in synthesis of new thiadiazoles, selenadiazoles and triazolotriazines. Egypt. J. Chem. https://doi.org/10.21608/ejchem.2022.172184.7138 (2022).
Sroor, F. M., Basyouni, W. M., Tohamy, W. M., Abdelhafez, T. H. & El-awady, M. K. Novel pyrrolo[2,3-d]pyrimidine derivatives: Design, synthesis, structure elucidation and in vitro anti-BVDV activity. Tetrahedron 75, 130749. https://doi.org/10.1016/j.tet.2019.130749 (2019).
Helmy, M. T. et al. Design, synthesis and in-vitro evaluation of new furan-substituted thiadiazolyl hydrazone derivatives as promising antimicrobial agents. J. Heterocycl. Chem. https://doi.org/10.1002/jhet.4611 (2022).
Kamel, M. G. et al. Synthesis and biological evaluation of new 1,3,4-thiadiazole derivatives as potent antimicrobial agents. Monatsh. Chem. Chem. Mon. 153, 929–937. https://doi.org/10.1007/s00706-022-02967-z (2022).
Sroor, F. M. et al. Anticancer activity of new bis-(3-(thiophen-2-yl)-1h-pyrazol-4-yl)chalcones: Synthesis, in-silico, and in-vitro studies. Polycycl. Arom. Compd. 1, 1–18. https://doi.org/10.1080/10406638.2022.2046616 (2022).
Kamel, M. G. et al. Structure-based design of novel pyrazolyl–chalcones as anti-cancer and antimicrobial agents: Synthesis and in vitro studies. Monatsh. Chem. Chem. Mon. 153, 211–221. https://doi.org/10.1007/s00706-021-02886-5 (2022).
Jacobson, R. K., Notaro, M. J. & Carr, G. J. Comparison of Neisseria gonorrhoeae minimum inhibitory concentrations obtained using agar dilution versus microbroth dilution methods. J. Microbiol. Methods 157, 93–99. https://doi.org/10.1016/j.mimet.2019.01.001 (2019).
Saur, D. et al. CXCR4 expression increases liver and lung metastasis in a mouse model of pancreatic cancer. Gastroenterology 129, 1237–1250. https://doi.org/10.1053/j.gastro.2005.06.056 (2005).
Watanabe, T. et al. Comparison of lung cancer cell lines representing four histopathological subtypes with gene expression profiling using quantitative real-time PCR. Cancer Cell Int. 10, 2. https://doi.org/10.1186/1475-2867-10-2 (2010).
Yang, Q., Feng, M. H., Ma, X., Li, H. C. & Xie, W. Gene expression profile comparison between colorectal cancer and adjacent normal tissues. Oncol. Lett. 14, 6071–6078. https://doi.org/10.3892/ol.2017.6915 (2017).
Olive, P. L., Banáth, J. P. & Durand, R. E. Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the “comet” assay. Radiat. Res. 178, 35–42. https://doi.org/10.1667/rrav04.1 (2012).
Collins, A. et al. Comet assay in human biomonitoring studies: Reliability, validation, and applications. Environ. Mol. Mutag. 30, 139–146. https://doi.org/10.1002/(Sici)1098-2280(1997)30:2%3c139::Aid-Em6%3e3.0.Co;2-I (1997).
Yawata, A. et al. Prolonged cell survival enhances peritoneal dissemination of gastric cancer cells. Oncogene 16, 2681–2686. https://doi.org/10.1038/sj.onc.1201792 (1998).
Mohamed, M. F. et al. Novel [l,2,4]triazolo[3,4-a]isoquinoline chalcones as new chemotherapeutic agents: Block IAP tyrosine kinase domain and induce both intrinsic and extrinsic pathways of apoptosis. Investig. New Drugs 39, 98–110. https://doi.org/10.1007/s10637-020-00987-2 (2020).
Satoh, M. et al. In vitro evaluation of minimum inhibitory concentration of several antibacterial agents against Rickettsia japonica using a plaque reduction assay system. J. Infect. Chemother. 25, 917–919. https://doi.org/10.1016/j.jiac.2019.05.019 (2019).
Munita, J. M., Arias, C. A., Kudva, I. T. & Zhang, Q. Mechanisms of antibiotic resistance. Microbiol. Spectr. https://doi.org/10.1128/microbiolspec.VMBF-0016-2015 (2016).
Villarroel, M. C. et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol. Cancer Ther. 10, 3–8. https://doi.org/10.1158/1535-7163.mct-10-0893 (2011).
Venkitaraman, A. R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 108, 171–182. https://doi.org/10.1016/s0092-8674(02)00615-3 (2002).
Eklund, E. A. et al. KRAS mutations impact clinical outcome in metastatic non-small cell lung cancer. Cancers 14, 2063. https://doi.org/10.3390/cancers14092063 (2022).
Sigismund, S., Avanzato, D. & Lanzetti, L. Emerging functions of theEGFRin cancer. Mol. Oncol. 12, 3–20. https://doi.org/10.1002/1878-0261.12155 (2017).
Fhu, C. W. & Ali, A. Fatty acid synthase: An emerging target in cancer. Molecules 25, 3935. https://doi.org/10.3390/molecules25173935 (2020).
Zhang, J. H. & Xu, M. DNA fragmentation in apoptosis. Cell Res. 10, 205–211. https://doi.org/10.1038/sj.cr.7290049 (2000).
Bergler, H., Fuchsbichler, S., Hogenauer, G. & Turnowsky, F. The enoyl-[acyl-carrier-protein] reductase (FabI) of Escherichia coli, which catalyzes a key regulatory step in fatty acid biosynthesis, accepts NADH and NADPH as cofactors and is inhibited by palmitoyl-CoA. Eur. J. Biochem. 242, 689–694. https://doi.org/10.1111/j.1432-1033.1996.0689r.x (1996).
Zhang, J. et al. Unique dependence on Sos1 in KrasG12D-induced leukemogenesis. Blood 132, 2575–2579. https://doi.org/10.1182/blood-2018-09-874107 (2018).
Acknowledgements
A part of this work was supported by the National Research Centre (NRC) under Project No. 12010102. Farid Sroor greatly appreciates the financial support of this work by the National Research Centre (NRC) under Project No. 12010102.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
The study was conceptualized by F.S. The writing-original draft was written by F.S. with the contribution of all the authors. Data acquisition and data analysis were performed by F.S., A.O., N.S., K.M., H.A.E.-K.. The final manuscript, Writing–review and editing were checked by F.S.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sroor, F.M., Mahrous, K.F., El-Kader, H.A.M.A. et al. Impact of trifluoromethyl and sulfonyl groups on the biological activity of novel aryl-urea derivatives: synthesis, in-vitro, in-silico and SAR studies. Sci Rep 13, 17560 (2023). https://doi.org/10.1038/s41598-023-44753-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-44753-9
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
